

Abstract
High DGAT1 expression levels in the small intestine highlight the critical role this enzyme plays in nutrient absorption. Identification of inhibitors which predominantly inhibit DGAT1 in the gut is an attractive drug discovery strategy with anticipated benefits of reduced systemic toxicity. In this report we describe our discovery and optimization of DGAT1 inhibitors whose plasma exposure is minimized by the action of transporters, including the P-glycoprotein transporter. The impact of this unique absorption profile on efficacy in rat and dog efficacy models is presented.
Keywords: DGAT1, triglyceride synthesis, efflux
Orally ingested triglycerides (TG) undergo hydrolysis and then are reassembled within enterocytes into TG-rich chylomicrons destined for systemic circulation. The final committed step in triglyceride biosynthesis is known to be mediated by at least two distinct intracellular acyl-coA diacylglycerol acyltransferases (DGATs), namely DGAT11 and DGAT2.2 Since the development of whole-body knockout models of these enzymes, there has been intense evaluation of pharmacological approaches to modulate their activity.3−8 For DGAT1, this interest is inspired by the favorable metabolic phenotype of DGAT1–/– mice,9 which are resistant to diet-induced body weight gain,10 are more insulin-sensitive relative to wild-type littermates,11 and exhibit a reduced rate of chylomicrons formation when challenged with lipid nutrients.12
Interestingly, all aspects of this phenotype are lost when DGAT1 is reintroduced via a tissue-specific promoter into the intestines of female DGAT1–/– mice, implying that intestinal DGAT1 plays a crucial role in the effects observed in the whole-body knockout model.13 Indeed, DGAT1 mRNA expression levels have been shown to be high in regions of the small intestine in mice and humans.14,15 Selective inhibition of intestinal DGAT1 therefore becomes an intriguing drug discovery approach to recapitulate aspects of the DGAT1–/– mouse, especially if this gut-specific inhibition reduces the potential risk of on- and off-target activity for candidate molecules. Particularly relevant for DGAT1 pharmacological inhibition is the observation of functional and morphological abnormalities in the fur and sebaceous glands of DGAT1–/– mice.16 In this report we describe a novel approach to specifically inhibit intestinal DGAT1, and demonstrate the potential viability of this strategy with regard to efficacy and safety in multiple preclinical models.
High-throughput screening efforts using recombinant human DGAT1 enzyme identified the benzimidazole 1 (DGAT1 IC50 = 1.3 μM; DGAT2 IC50 > 20 μM; Figure 1) as a potential starting point for optimization. Initial structural modifications demonstrated that both the ethyl carbamate and the 2,6-dichlorophenyl substituents on the benzimidazole core could be replaced without substantial loss of activity (i.e., 2, DGAT1 IC50 = 1.4 μM), and in fact introducing an additional substituent at the 4-position of the 2,6-dimethylphenyl ring led to an improvement in potency (3, IC50 = 0.5 μM).
Figure 1.

Benzimidazole-based DGAT1 inhibitors.
As several reported DGAT1 inhibitor scaffolds feature a carboxylic acid group, we developed a synthetic strategy to access a variety of carboxylic acid trajectories via the synthetic routes described in Scheme 1. Transmetalation of 8a followed by reaction with DMF furnished aldehyde 8b. After hydrolytic removal of the trifluoroacetate group, diazotization of aniline 9 was immediately followed by Heck coupling with methyl acrylate and then hydrogenation to afford 11.17 The aniline 9 could also be alkylated with methyl bromoacetate to afford 13, and the oxygen-linked analogue 14 was prepared in a similar fashion.18 A convergent assembly of the 1,2-phenylene diamine 15 and C4-functionalized benzaldehyde derivatives under oxidative cyclization conditions afforded the fully elaborated benzimidazole carboxylic acid targets 4–6.19,20
Scheme 1. Assembly of C-4 Functionalized Benzimidazole Derivatives.

Reagents and condtions: (a) MeLi/LiBr, THF, s-BuLi, DMF; (b) 1 N NaOH, MeOH; (c) 48% HBF4, NaNO2, water, Pd(OAc)2, MeOH, methyl acrylate; (d) H2, Pd/C, CH2Cl2, 1 atm; (e) BrCH2COOMe, K2CO3, DMF; (f) 11, 13, or 14, Yb(OTf)3, DMSO.
The introduction of a carboxylic acid at the 4-position of the 2,6-dimethylphenyl ring led to a dramatic enhancement of inhibitor potency, with the oxyacetic acid derivative 4 among the most potent analogues in this series (IC50 = 28 nM, Table 1). Despite exceptional biochemical potency, however, carboxylic acid derivatives 4–6 only weakly inhibited triglyceride synthesis in a C2C12 mouse myoblast cellular assay, whereas more permeable inhibitors such as 3 (PAMPA; cFA = 89%) showed comparable if not improved cellular potency relative to the enzymatic assay. Carboxylate inhibitors including 4 (measured pKa21 = 4.8 and 10.7) are zwitterionic at physiological pH and presumably unable to passively penetrate cell membranes (PAMPA; cFA < 32%).
Table 1. In Vitro Potency and Permeability Profile of Compounds 3-6.
| 3 | 4 | 5 | 6 | |
|---|---|---|---|---|
| DGAT1 IC50 (nM) | 500 | 28 | 56 | 28 |
| Cell IC50 (nM) | 120 | >10,000 | 2,000 | 6,500 |
| PAMPA cFAa (%) | 89 | 17 | <32 | 22 |
| Caco-2 (A–B) (10–6 cm/s) | 1.3 | 0.65 | 0.55 | |
| Caco-2 (B–A) (10–6 cm/s) | 2.1 | 1.2 | 10.0 | |
| pKa | 4.8 | 5.1 | 4.3 |
cFA = calculated fraction absorbed. See the Supporting Information for a description of the cellular TG synthesis assay.
However, additional in vitro profiling of 4–6 revealed an unanticipated difference among these benzimidazole carboxylate inhibitors. In the bidirectional Caco-2 monolayer assay, compounds 4 and 5 showed approximately equal permeability in the absorptive (A–B) and nonabsorptive (B–A) directions, whereas the carbon-linked analogue 6 uniquely exhibited 18-fold higher permeability in the nonabsorptive direction. The high (B–A)/(A–B) ratio suggests the potential for transporter-mediated efflux to influence the intestinal disposition of 6, whereas compounds 4 and 5 are predicted by the Caco-2 assay to be passively transported. It is remarkable that a single atom replacement (O and N vs C) leading to a subtle but measurable increase in carboxylic acid pKa (pKa difference of +0.5 and +0.8 with respect to 6) can potentially account for such dramatic differences in the Caco-2 permeability assay.
The pharmacological relevance of this distinct Caco-2 permeability data was assessed in an acute rat model of intestinal DGAT1 activity (Figure 2). Male Sprague–Dawley rats were dosed with inhibitors 4–6 (5 mg/kg) 2 h prior to receiving an oral Intralipid challenge (10 mL/kg). In vehicle-treated control animals, a robust plasma triglyceride excursion was observed with peak levels reached 2 h after caloric challenge (see the Supporting Information for the time course). Interestingly, only compound 6 blunted the Intralipid-induced triglyceride excursion (73% reduction relative to vehicle at t = 2 h) whereas plasma triglyceride levels following oral administration of 4 or 5 were indistinguishable from vehicle. Higher doses of 4 or 5 were not investigated in this model.
Figure 2.

Plasma triglyceride levels 2 h following Intralipid challenge for compounds 4–6 (5 mg/kg). *p< 0.05 for compound 6.
A potential explanation for these divergent pharmacodynamic effects is that transporter-mediated efflux effectively recycles 6 in the intestine, leading to significantly higher local concentrations of 6 compared to compound 5 in DGAT1-expressing enterocytes. This unique recycling allows for effective blunting of intracellular triglyceride synthesis despite the intrinsic low permeability of 6 and weaker cellular potency relative to nontransported inhibitors such as 5.
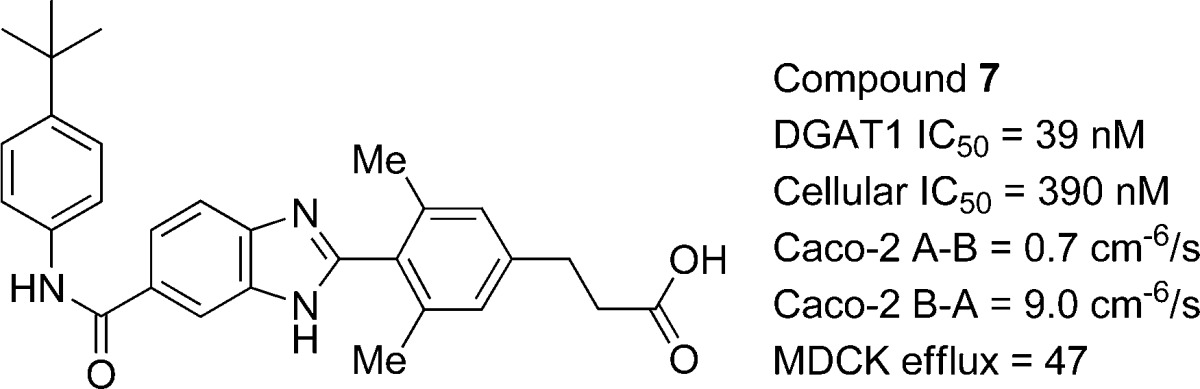
During the course of our optimization efforts, we identified the closely related analogue 7, which was 17-fold more potent than 6 in inhibiting cellular triglyceride synthesis (DGAT1 IC50 = 39 nM; cell IC50 = 390 nM; see Table 2). Consistent with our earlier findings, the propanoic acid derivative 7 also exhibited strong efflux potential in Caco-2 cells, and this disposition was independently confirmed in a MDR1-MDCK assay where the P-glycoprotein transporter is specifically overexpressed (MDCK efflux ratio = 47).22 We cannot rule out the participation of other transporters such as MRP2 and BCRP, which are known to also function on the apical membrane of enterocytes.23
Table 2. Rat Enterocyte and Plasma Concentrations of Compound 7 Following a 5 mg/kg Oral Dose.
| time (h) | duodenum (nM) | jejunum (nM) | p. veina (nM) | plasma (nM) |
|---|---|---|---|---|
| 2 | 500 | 930 | 22 | 16 |
| 6 | 630 | 2020 | 8 | 7 |
| 17 | 610 | 1400 | 5 | BDLb |
Portal vein.
BDL = below detection limit (<3 nM).
To better understand the potential role of transporter-mediated efflux on local enterocyte concentrations in vivo, we measured enterocyte and portal vein concentrations of 7 following a 5 mg/kg oral dose to rats (0.5% methyl cellulose/0.1% Tween-80 suspension vehicle). Tissue samples were extensively washed to minimize the contribution of unabsorbed compound to enterocyte concentration measurements. Two hours after an Intralipid challenge, the concentration of 7 (500 nM) in duodenal enterocytes was well above the intrinsic potency of 7 (39 nM) and also 23-fold higher than the level detected in portal vein and plasma (Table 2). At this time point, intracellular triglyceride levels were significantly lower in rats dosed with 7 (67.8% reduction compared to vehicle), confirming robust inhibition of DGAT1 at this time point. The sustained enterocyte levels of 7 even 17 h following compound oral administration support the notion of repeated cycling of the compound throughout the length of the small intestine. Interestingly, concentrations of 7 were uniformly higher in enterocytes harvested from the jejunum, providing additional evidence for recycling of 7. The high lipophilicity of 7 (log D(21) = 2.6 at pH 6.8) may enrich concentration in membrane structures including the endoplasmic reticulum, the presumed intracellular site of action for DGAT1 inhibition. Meanwhile, the low portal vein concentrations highlight that the unique absorption characteristics of 7, rather than first-pass metabolism, underly the vanishingly low plasma concentrations of 7. Compounds 4–7 all show clearance from plasma well below hepatic blood flow following intravenous administration (Clp for 7 = 10 mL/min/kg in rats; Clp for 4–6 all <19 mL/min/kg in rats), which argues against hepatic extraction contributing to the low circulating levels of 7 following oral dosing.
Encouraged by the efficacy profile of 7 in rodents, the pharmacodynamic effect of compound 7 was further evaluated in a dog model of hypertriglyceridemia. In this model, beagle dogs were given a high-fat challenge 30 min after receiving an oral 1 mg/kg dose of compound 7. The plasma triglyceride level at baseline was 35.4 ± 4.7 mg/dL in four dogs treated with vehicle, and a marked increase in triglycerides ensued for at least 6 h following caloric challenge (Figure 3). Oral preadministration of compound 7 (0.5% carboxymethylcellulose suspension vehicle) robustly suppressed the plasma TG excursion compared to vehicle alone, leading to a mean AUC0–6h reduction of 75 ± 6% (p < 0.005, paired t test). Consistent with our observations for 7 in rats, robust pharmacodynamic effects were seen up to 6 h in dogs despite plasma levels of 7 that were below the intrinsic biochemical potency (39 nM) and cellular potency (390 nM) of 7 throughout the postprandial period. Despite the complete blunting of postprandial triglycerides in plasma, no visible steatorrhea was observed in the any of the studies described above.
Figure 3.
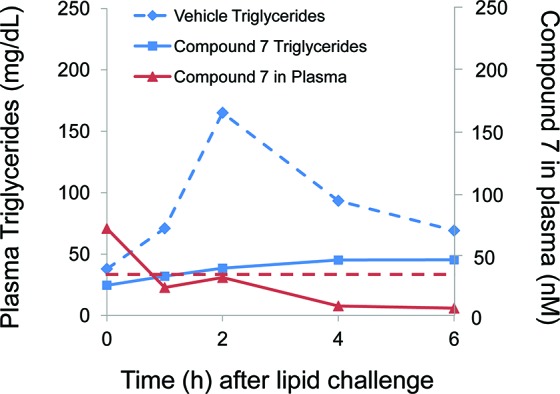
Plasma triglycerides in dogs administered compound 7 (1 mg/kg, 0.5% carboxymethylcellulose suspension, solid line) or vehicle (dotted line) followed by lipid challenge. Plasma concentrations of 7 shown in red. Intrinsic biochemical potency of 7 (39 nM) indicated with dashed red line.
A potential advantage of our gut-targeting approach is to reduce risk of on- or off-target effects that may be driven by systemic exposure. We studied the toxicological effects of 7 over 2 weeks in rats at doses exceeding the pharmacologically active range (10, 30, and 100 mg/kg; 5% hydroxypropyl β-cyclodextrin solution vehicle). At the 100 mg/kg dose, which is at least 20-fold higher than the dose required for a pharmacodynamic effect in rats, no adverse effects were observed following in-life observation, hematology, and clinical chemistry. Thus, intestinally targeted DGAT1 inhibitors have the potential to favorably impact postprandial metabolism with a large therapeutic margin. Interestingly, appreciable plasma levels of 7 were observed at the highest dose level examined (AUC0–24h = 9800 nM·h), suggesting that the mechanism(s) which limits the systemic exposure of 7 can be saturated at superpharmacological doses.
We have thus demonstrated that intestinally restricted DGAT1 inhibitors can exert robust effects in rodent and nonrodent preclinical models of hypertriglyceridemia. While this single inhibitor cannot definitively establish the contribution of each DGAT1-expressing tissue to overall energy metabolism, the profound postprandial effects of an intestinally restricted inhibitor certainly underscore the importance of intestinal DGAT1 in modulating nutrient handling. A better understanding of the role of nongut tissues expressing DGAT1 awaits the development of inhibitors with diverse pharmacokinetic signatures to potentially modulate DGAT1 activity in a tissue-specific fashion.
Acknowledgments
We thank Brian Jones and Jason Elliott for helpful suggestions to this manuscript, as well as members of the MAP-ADME group for profiling support.
Supporting Information Available
Characterization data for all new compounds and biological assay information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors contributed to the writing of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Cases S.; Smith S. J.; Zheng Y.-W.; Myers H. M.; Lear S. R.; Sande E.; Novak S.; Collins C.; Welch C. B.; Lusis A. J.; Erickson S. K.; Farese R. V. Jr. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases S.; Stone S. J.; Zhou P.; Yen E.; Tow B.; Lardizabal K. D.; Voelker T.; Farese R. V. Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 2001, 276, 38870–38876. [DOI] [PubMed] [Google Scholar]
- Hubbard B. K.; Enyedy I.; Gilmore T. A.; Serrano-Wu M. H. Antisense and small-molecule modulation of diacylglycerol acyltransferase. Expert Opin. Ther. Pat. 2007, 17, 1331–1339. [Google Scholar]
- Birch A. M.; Buckett L. K.; Turnbull A. V. DGAT1 inhibitors as anti-obesity and anti-diabetic agents. Curr. Opin. Drug Discovery Dev. 2010, 13, 489–96. [PubMed] [Google Scholar]
- Matsuda D.; Tomoda H. DGAT inhibitors for obesity. Curr. Opin. Invest. Drugs 2007, 8, 836–841. [PubMed] [Google Scholar]
- For recent examples of DGAT1 inhibitor design, see:; Yeh V. S.; Beno D. W.; Brodjian S.; Brune M. E.; Cullen S. C.; Dayton B. D.; Dhaon M. K.; Falls H. D.; Gao J.; Grihalde N.; Hajduk P.; Hansen T. M.; Judd A. S.; King A. J.; Klix R. C.; Larson K. J.; Lau Y. Y.; Marsh K. C.; Mittelstadt S. W.; Plata D.; Rozema M. J.; Segreti J. A.; Stoner E. J.; Voorbach M. J.; Wang X.; Xin X.; Zhao G.; Collins C. A.; Cox B. F.; Reilly R. M.; Kym P. R.; Souers A. J. Identification and Preliminary Characterization of a Potent, Safe, and Orally Efficacious Inhibitor of Acyl-CoA:Diacylglycerol Acyltransferase 1. J. Med. Chem. 2012, 55, 1751–1757. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Wertheimer S. J.; Ahmad M.; Cheung A. W.-H.; Firooznia F.; Hamilton M. M.; Hayden S.; Li S.; Marcopulos N.; McDermott L.; Tan J.; Yun W.; Guo L.; Pamidimukkala A.; Chen Y.; Huang K.-S.; Ramsey G. B.; Whittard T.; Conde-Knape K.; Taub R.; Rondinone C. M.; Tilley J.; Bolin D. Discovery of orally active carboxylic acid derivatives of 2-phenyl-5-trifluoromethyloxazole-4-carboxamide as potent diacylglycerol acyltransferase-1 inhibitors for the potential treatment of obesity and diabetes. J. Med. Chem. 2011, 54, 2433–2446. [DOI] [PubMed] [Google Scholar]
- Birch A. M.; Birtles S.; Buckett L. K.; Kemmitt P. D.; Smith G. J.; Smith T. J. D.; Turnbull A. V.; Wang S. J. Y. Discovery of a potent, selective, and orally efficacious pyrimidinooxazinyl bicyclooctaneacetic acid diacylglycerol acyltransferase-1 inhibitor. J. Med. Chem. 2009, 52, 1558–1568and references cited therein.. [DOI] [PubMed] [Google Scholar]
- Chen H. C.; Farese R. V. Jr. Inhibition of triglyceride synthesis as a treatment strategy for obesity, lessons from DGAT1-deficient mice. Arterioscler., Thromb., Vasc. Biol. 2005, 25, 482–486. [DOI] [PubMed] [Google Scholar]
- Smith S. J.; Cases S.; Jensen D. R.; Chen H. C.; Sande E.; Tow B.; Sanan D. A.; Raber J.; Eckel R. H.; Farese R. V. Jr. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking DGAT. Nat. Genet. 2000, 25, 87–90. [DOI] [PubMed] [Google Scholar]
- Chen H. C.; Smith S. J.; Ladha Z.; Jensen D. R.; Ferreira L. D.; Pulawa L. K.; McGuire J. G.; Pitas R. E.; Eckel R. H.; Farese R. V. Jr. Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglycerol acyltransferase 1. J. Clin. Invest. 2002, 109, 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhman K. K.; Smith S. J.; Stone S. J.; Repa J. J.; Wong J. S.; Knapp F. F. Jr.; Burri B. J.; Hamiltons R. L.; Abumrad N. A.; Farese R. V. Jr. DGAT1 is not essential for intestinal triacylglycerol absorption or chylomicron synthesis. J. Biol. Chem. 2002, 277, 25474–25479. [DOI] [PubMed] [Google Scholar]
- Lee B.; Fast A. M.; Zhu J.; Buhman K. K. Intestine-specific expression of acyl CoA:diacylglycerol acyltransferase 1 reverses resistance to diet-induced hepatic steatosis and obesity in Dgat1–/– mice. J. Lipid Res. 2010, 51, 1770–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramine Y.; Tanabe T. Characterization of acyl-coenzyme A:diacylglycerol acyltransferase (DGAT) enzyme of human small intestine. J. Physiol. Biochem. 2011, 67, 259–264. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Iqbal J.; Devenny J.; Chu C. H.; Chen L.; Dong J.; Seethala R.; Keim W. J.; Azzara A. V.; Lawrence R. M.; Pelleymounter M. A.; Hussain M. M. Acylation of acylglycerols by acyl coenzyme A:diacylglycerol acyltransferase 1 (DGAT1). Functional importance of DGAT1 in the intestinal fat absorption. J. Biol. Chem. 2008, 283, 29802–29811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. C.; Smith S. J.; Tow B.; Elias P. M.; Farese R. V. Jr. Leptin modulates the effects of acyl CoA:diacylglycerol acyltransferase deficiency on murine fur and sebaceous glands. J. Clin. Invest. 2002, 109, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S.; Bhattacharyya S. Aqueous Heck reaction of amino acid derived arenediazonium salts: semisynthetic modification of phenylalanine and tyrosine. Tetrahedron Lett. 1995, 36, 4475–4478. [Google Scholar]
- Coppola G. M.; Gong Y. A reliable, high-yielding preparation of 2,6-dimethyl-4-hydroxybenzaldehyde. Org. Prep. Proced. Int. 2007, 39, 199–202. [Google Scholar]
- Curini M.; Epifano F.; Montanari F.; Rosati O.; Taccone S. Ytterbium triflate promoted synthesis of benzimidazole derivatives. Synlett 2004, 10, 1832–1834. [Google Scholar]
- Beaulieu P. L.; Haché B.; von Moos E. A practical oxone®-mediated, high-throughput, solution-phase synthesis of benzimidazoles from 1,2-phenylenediamines and aldehydes and its application to preparative scale synthesis. Synthesis 2003, 1683–1692. [Google Scholar]
- pKa and log D measurements were conducted via potentiometric titration.
- Tang F.; Horie K.; Borchardt R. T. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa?. Pharm. Res. 2002, 19, 765–72. [DOI] [PubMed] [Google Scholar]
- Zolk O.; Fromm M. F. Transporter-mediated drug uptake and efflux: important determinants of adverse drug reactions. Clin. Pharmacol. Ther. 2011, 89, 798–805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.