

Abstract
We report the design, synthesis, and biological evaluation of the first macrocyclic peptoid-containing histone deacetylase (HDAC) inhibitors. The compounds selectively inhibit human class I HDAC isoforms in vitro, with no inhibition of the tubulin deacetylase activity associated with class IIb HDAC6 in cultured Jurkat cells. Compared to the natural product apicidin (1), one inhibitor (compound 10) showed equivalent potency against K-562 cells, but was more cytoselective across a panel of cancer cell lines.
Keywords: HDAC inhibitors, peptides, peptoids, macrocycles, apicidin
We have recently described the design of conformationally homogeneous macrocyclic tetrapeptides as scaffolds to display four amino acid side chain functionalities in predictable three-dimensional arrangements (Figure 1).1−6 The structure-guided studies relied on peptide backbone modifications, such as the incorporation of beta amino acids or triazole moieties, to simultaneously promote conformational homogeneity and provide efficient synthetic routes to the macrocycles. Our studies included systematic investigations of the importance of stereochemical changes and N-methylation of backbone amides, as well as the effect of various side chains.1−6 With the goal of further expanding the repertoire of unique side chain spatial arrangements and backbone conformations beyond those that are available with simple α- or β-peptides, here we incorporate peptoid (N-alkylglycine) residues into cyclic tetrapeptide histone deacetylase (HDAC) inhibitors. In addition to accessing unique structure space, we anticipated that the inclusion of peptoid residues could impart useful biological properties such as proteolytic resistance or improved cell permeability.
Figure 1.
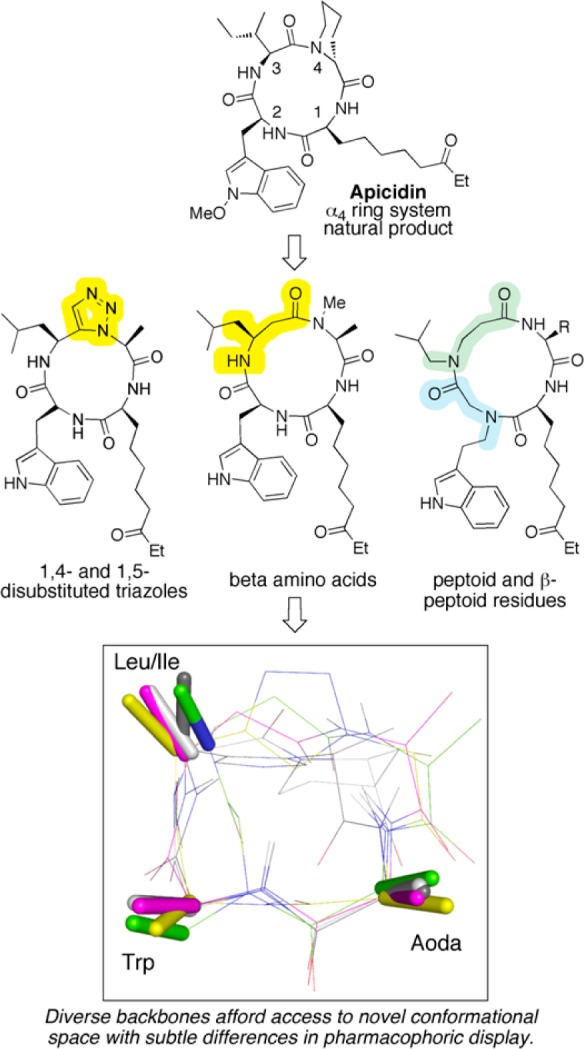
Peptoid moieties provide access to conformational space not available to α- or β-peptides, so they complement efforts in designing conformationally distinct macrocyclic HDAC inhibitors. The structure overlay highlights the overlap of pharmacophoric side chains between the global minimum energy molecular conformation of a peptoid–peptide hybrid obtained in a conformational search (green) with the NMR structures of apicidin (white),38 a 1,4-disubstituted triazole peptide (yellow),3 a 1,5-disubstituted triazole peptide (gray),3 and a peptide containing a β-amino acid (magenta).1 Aoda, (S)-2-amino-8-oxodecanoic acid.
HDAC inhibitors are promising agents for anticancer chemotherapy,7−9 with several candidates in clinical trials,7 and vorinostat (SAHA)10 and romidepsin (FK-228)11 approved to treat cutaneous T-cell lymphoma. A notable group of HDAC inhibitors are macrocyclic compounds, such as apicidin (1),12 bearing an extended Zn2+-coordinating unit that interacts with the active site zinc ion of the class I, II, and IV HDAC enzymes.13,14
Peptoid residues15,16 offer the potential for extensive modular diversification of ligands due to the ready availability of the synthetic subunits.17 Peptoid-based peptidomimetics have found utility in various biological applications,18−26 and N-alkyl-β-alanine residues (“β-peptoids”) have also been successfully incorporated into biologically active oligomers.27−31 We envisioned that installing peptoid or β-peptoid residues in our cyclic tetrapeptides would provide access to distinct conformational space compared to traditional α- or β-peptides. For instance, mixing peptide and peptoid residues affords the opportunity to vary the number of backbone bonds between side chains, and thereby change their relative distances. Further, the secondary amides found in peptides are overwhelmingly populated by the trans conformation, whereas both the cis and trans conformers are generally accessible in the tertiary amides found in peptoids.32−35 Indeed, a force field based conformational search of a peptoid–peptide hybrid (vide infra) indicated that its lowest energy conformation would contain a cis amide but would nevertheless closely match the pharmacophoric model for compounds containing all trans amide bonds determined in our previous studies (Figure 1).
By applying a combination of standard Fmoc solid-phase synthesis and peptoid subunit synthesis (Figure S1, Supporting Information), we initially prepared analogs 3–6, in which the amino acids of our parent compound 2(1) (Figure 2) were gradually substituted with peptoid units (Figure 2b). In contrast to the low yields obtained in the macrolactamization of 12-membered ring peptides,36 the efficiency of the ring-closure was generally good as judged by LC-MS analysis, similar to that observed for 13-membered ring α3β peptides (typical overall isolated yields were 10–18%, Table S1).1−6
Figure 2.
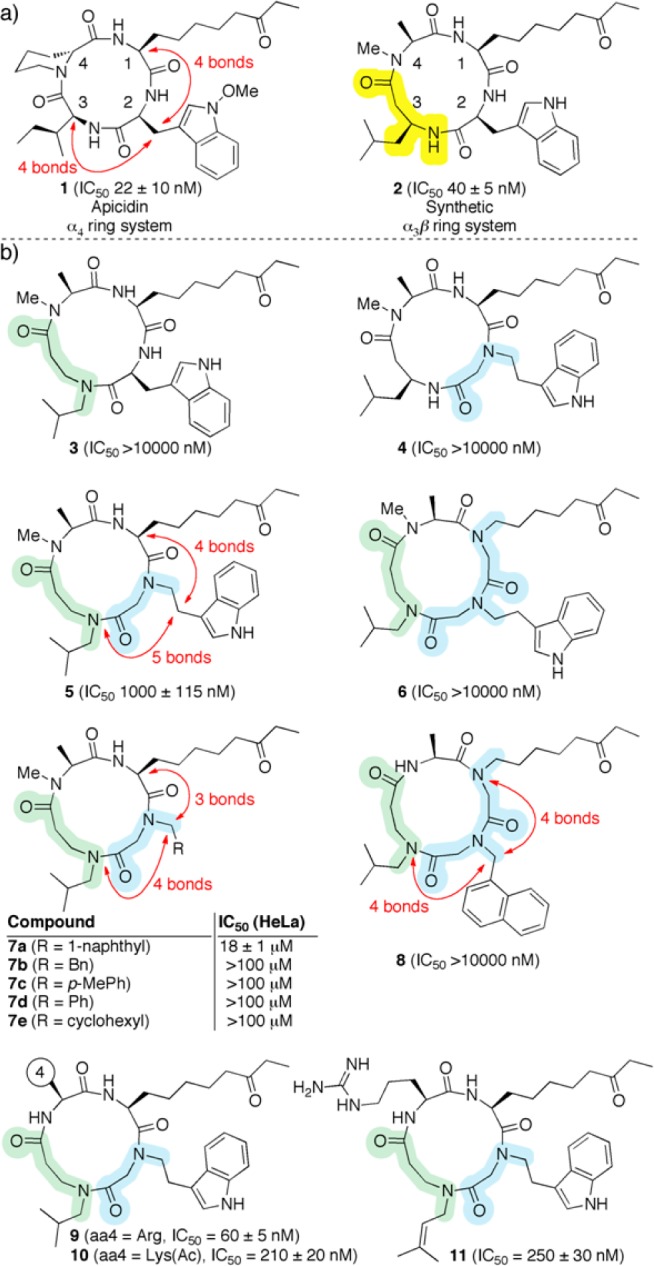
Compound structures and their corresponding IC50 values measured against the HDAC activity in HeLa cell extract. (a) Parent compounds 1 and 2. (b) Structures of analogs 3–11. Peptoid residues are highlighted in blue, and β-peptoid residues are highlighted in green.
HDAC inhibitory activity was initially assessed using HeLa cell nuclear extract.37 Compound 5 showed modest inhibition of HDAC activity (∼1 μM), whereas the other three compounds were inactive. Based on the structure of 5, we prepared analogs 7a–e to probe the tryptamine subunit more carefully (Figure 2b); unfortunately, compound 7a showed a decrease in potency and 7b–e were inactive. We also prepared analog 8, in which the number of bonds between the aromatic group and the 1- and 3-position side chains, respectively, is the same as in compound 2 and apicidin. This analog did not inhibit HDAC activity in the tested concentration range.
Next, we constructed compounds 9 and 10, in which amino acid positions 1–3 of the macrocycle were kept as present in compound 5, while the N-Me-Ala residue was substituted with arginine (9) or ε-N-acetyl-lysine (10) (Figures 2 and S1). We had previously identified these substitutions as being beneficial for HDAC inhibition in a library screen of α3β-peptides.2 In the present context, these substitutions gave rise to nanomolar inhibition of the HeLa cell HDAC activity, with the potency measured for 9 being close to that observed for parent compound 2 (58 nM and 40 nM, respectively). Finally, position three was probed by preparing the more sterically demanding N-dimethylallyl analog 11 (Figures S2 and S3), which turned out to be less potent than 9 or 10 (Figure 2).
The 1H NMR spectra of compounds 9 and 10 recorded in DMSO-d6 at 295 K revealed a single set of signals (Figures S8 and S9). In contrast, both N-Me-Ala analogs 2(1) and 5, like apicidin,3,38 exhibited multiple conformations by NMR (Figures S6 and S7). The multiple conformations of apicidin are known to stem from cis/trans isomerization of the pipecolic acid residue (position 4);3,38 by analogy, compound 2 also presumably isomerizes at the alkylated amide at position 4. Likewise, considering that compounds 5, 9, and 10 all contain peptoid moieties at residues 2 and 3, but only compound 5 is alkylated at position 4, it appears that the multiple conformations observed for 5 are due to isomerization at the alkylated position 4 amide.
To further explore the possible conformations of compounds 9 and 10, we carried out a force field based conformational search of a representative structure having an Ala residue at position 4. The ten lowest energy conformations (Figure S4, Table S2) contained a cis amide at the tryptamine subunit; the rest of the amides were in the trans configuration (Figure 3). An inspection of these low energy structures suggests that adoption of the cis configuration reduces unfavorable steric interactions between the indole group and the side chains at positions 1 and 3. Interestingly, there is good overlap of this backbone conformer with the NMR structure of apicidin in the pharmacophoric region (rmsd of 0.4 Å, Figures 3 and S4), suggesting a basis for the observed potencies of compounds 9 and 10. Due to the additional methylene unit between the backbone and the indole functionality as compared to the natural product, the active peptoid–peptide compounds are more flexible in this important region of the pharmacophore, which may explain the somewhat lower potencies observed. Some higher-energy modeled conformations contained all-trans amide configurations; while the cis-trans-trans-trans conformations overlay well with the pharmacophoric region of apicidin (Figure S4), the all-trans conformations do not.
Figure 3.
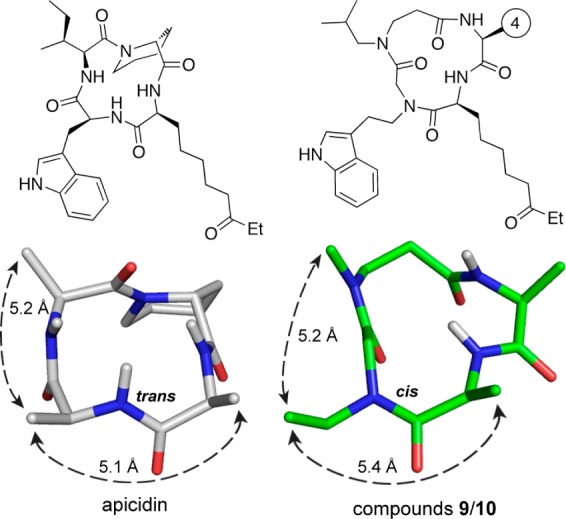
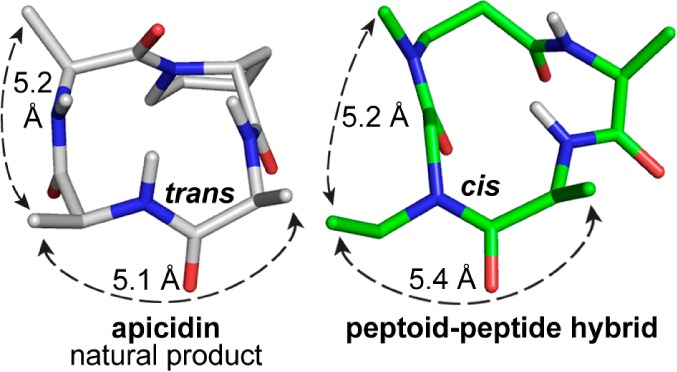
Comparison of the NMR structure of apicidin (left) with one of the low energy calculated structures of compounds 9/10 (right). Side chain atoms have been removed for clarity. Even though the amide bond between positions 1 and 2 adopts a different cis/trans configuration, the distances and vectors between pharmacophoric side chains are nearly identical.
Selected compounds were further tested against several recombinant human HDACs in a standard trypsin-coupled fluorogenic assay (Table 1). The potencies recorded against the class I HDACs 1–3 generally followed the trends observed for apicidin and 2, with the most potent candidates (9 and 10) being ∼6-fold less potent than 2 against HDAC1. Because ethylketone-containing apicidin analogs do not appear to inhibit class IIa HDACs,39,40 we excluded enzymes of this class in the present investigation. As expected based on inhibition profiles of other ligands containing the ethylketone Zn2+-coordinating amino acid,1−3 all the compounds were inactive against HDAC6, a representative member of the HDAC class IIb (Table 1). HDAC8 is a class I HDAC but diverges significantly from the other class I isoforms41 and was recently shown to be similar to class IIa isoforms in terms of inhibitor specificity.39 It was therefore not surprising that compound 9 was 4-fold less potent against HDAC8 as compared to HDAC1. In contrast, compound 9 was ∼3-fold more potent than parent compound 2 against HDAC8, which corresponds with a possible interaction of arginine residues with HDAC8 previously proposed by Mrksich and co-workers based on experiments with histone substrates.42
Table 1. IC50 Values (nM) for Selected Compounds against Recombinant Human HDACs 1–3 and 6a.
| class I |
class IIb | ||||
|---|---|---|---|---|---|
| compd | HDAC1 | HDAC2 | HDAC3b | HDAC8 | HDAC6 |
| apicidin | 11 ± 3 | 34 ± 2 | 13 ± 5 | 750 | >10000 |
| 2 | 30 ± 5 | 70 ± 24 | 30 ± 7 | 2200 | >10000 |
| 5 | 540 ± 60 | 2000 ± 500 | 650 ± 140 | N.D. | >10000 |
| 9 | 180 ± 16 | 300 ± 16 | 100 ± 14 | 750 | >10000 |
| 10 | 200 ± 72 | 370 ± 48 | 320 ± 21 | N.D. | >10000 |
| 11 | 700 ± 160 | 1200 ± 140 | 270 ± 4 | N.D. | >10000 |
IC50 values are means of at least two assays performed in duplicate.
In complex with nuclear receptor corepressor 2 (NCoR-2). N.D. = not determined.
To confirm that compound 9 could inhibit HDACs in a cellular context, we treated cultured Jurkat cells with various concentrations of 9 for 24 h. Western blot analysis of the cell lysates showed high levels of acetylated-histone H3 (K9Ac+K14Ac) when compound 9 was present at 2 μM or 20 μM, which indicates that 9 inhibited class I HDACs in the cells (Figure S5). On the other hand, the level of acetylated α-tubulin (substrate for HDAC6) was not elevated by any tested concentration of compound 9, indicating that HDAC6 was not inhibited. These results are in agreement with the recombinant enzyme assays (Table 1).
Finally, we investigated the ability of the peptoid–peptide hybrids to inhibit growth of selected cancer cell lines (Table 2). The moderate potencies observed for 5 correlate well with its lower potencies against the various HDACs as compared to apicidin and 2. Compounds 9 and 11 showed no activity in the tested concentration range, possibly due to poor cell permeability caused by the cationic guanidinium functionality in arginine. Compound 10 showed moderate activity against HeLa and Hct-116 cells but intermediate and good activities against the leukemic cell lines KYO-1 and K-562, respectively. Interestingly, when comparing the activities to those of apicidin (1), the less potent HDAC inhibitor 10 is equipotent against K-562 cells but shows a much higher degree of cytoselectivity, which may be desirable with respect to the future development of therapeutic agents.
Table 2. Growth Inhibition of Selected Cancer Cell Linesa.
| cell growth
inhibition, GI50 (μM) |
|||||
|---|---|---|---|---|---|
| compd | HeLa | Hct-116 | MCF-7 | KYO-1 | K-562 |
| apicidin | 2 | 1 | 5 | 2.7 | 1.5 |
| 2 | 4b | N.D. | 18b | 7.5b | 7b |
| 5 | 30 | 28 | >50 | 18 | 15 |
| 9 | >50 | >50 | >50 | >50 | >50 |
| 10 | 38 | 50 | >50 | 10 | 1.5 |
| 11 | >50 | >50 | >50 | N.D. | N.D. |
GI50 values are based on at least two assays performed in triplicate. N.D. = not determined.
From a previous report.1
In summary, we have successfully constructed cyclic peptoid-containing ligands with potent HDAC inhibitory activity in HeLa cell extract, comparable to the parent compound. The choice of an ethyl ketone Zn2+-coordinating amino acid enabled high class I selectivity against recombinant enzyme isoforms 1–3 as well as in Jurkat cells. The introduction of a position-4 Arg residue, which caused a considerable increase in HDAC inhibitory activity (5 vs 9), had the opposite effect on cytotoxicity, whereas the Lys(Ac) residue in the same position yielded submicromolar class I HDAC inhibition as well as growth inhibition of selected cancer cell lines at micromolar GI50 values. This work should serve as a foundation for further exploration of HDAC inhibitors with an apicidin-like molecular scaffold.
Acknowledgments
We thank the laboratories of Prof. P. S. Baran and Prof. D. L. Boger for use of their microwave reactors, Prof. J. M. Gottesfeld and Dr. D. Herman for discussions and assistance with the Western blot development, and Dr. B. E. Maryanoff for valuable comments on the manuscript. Dr. P. Fristrup (Technical University of Denmark) is thanked for performing the force field calculations.
Supporting Information Available
Experimental procedures, compound characterization data, supplementary figures, PDB structures of the 25 lowest energy modeled conformations, and selected NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, The Technical University of Denmark, Kemitorvet 207, DK-2800 Kgs. Lyngby, Denmark.
This work was supported by the Benzon Foundation (C.A.O.), the Lundbeck Foundation (C.A.O.), the Danish Council for Independent Research | Natural Sciences Grant 10-080907 (C.A.O.), and the Skaggs Institute for Chemical Biology (M.R.G).
The authors declare no competing financial interests.
Supplementary Material
References
- Montero A.; Beierle J. M.; Olsen C. A.; Ghadiri M. R. Design, synthesis, biological evaluation, and structural characterization of potent histone deacetylase inhibitors based on cyclic alpha/beta-tetrapeptide architectures. J. Am. Chem. Soc. 2009, 131, 3033–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C. A.; Ghadiri M. R. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic alpha3beta-tetrapeptides. J. Med. Chem. 2009, 52, 7836–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne W. S.; Olsen C. A.; Beierle J. M.; Montero A.; Ghadiri M. R. Probing the bioactive conformation of an archetypal natural product HDAC inhibitor with conformationally homogeneous triazole-modified cyclic tetrapeptides. Angew. Chem., Int. Ed. 2009, 48, 4718–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierle J. M.; Horne W. S.; van Maarseveen J. H.; Waser B.; Reubi J. C.; Ghadiri M. R. Conformationally homogeneous heterocyclic pseudotetrapeptides as three-dimensional scaffolds for rational drug design: receptor-selective somatostatin analogues. Angew. Chem., Int. Ed. 2009, 48, 4725–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt D. M.; Olsen C. A.; Vickers C. J.; Herman D.; Chalfant M.; Montero A.; Leman L. J.; Burkle R.; Maryanoff B. E.; Balch W. E.; Ghadiri M. R. Potential agents for treating cystic fibrosis: Cyclic tetrapeptides that restore trafficking and activity of DeltaF508-CFTR. ACS Med. Chem. Lett. 2011, 2, 703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers C. J.; Olsen C. A.; Leman L. J.; Ghadiri M. R. Discovery of HDAC inhibitors that lack an active site Zn2+-binding functional group. ACS Med. Chem. Lett. 2012, 3, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris M.; Porcelloni M.; Binaschi M.; Fattori D. Histone deacetylase inhibitors: from bench to clinic. J. Med. Chem. 2008, 51, 1505–1529. [DOI] [PubMed] [Google Scholar]
- Minucci S.; Pelicci P. G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [DOI] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- Furumai R.; Matsuyama A.; Kobashi N.; Lee K. H.; Nishiyama M.; Nakajima H.; Tanaka A.; Komatsu Y.; Nishino N.; Yoshida M.; Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [PubMed] [Google Scholar]
- Darkin-Rattray S. J.; Gurnett A. M.; Myers R. W.; Dulski P. M.; Crumley T. M.; Allocco J. J.; Cannova C.; Meinke P. T.; Colletti S. L.; Bednarek M. A.; Singh S. B.; Goetz M. A.; Dombrowski A. W.; Polishook J. D.; Schmatz D. M. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13143–13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [DOI] [PubMed] [Google Scholar]
- Biel M.; Wascholowski V.; Giannis A. Epigenetics--an epicenter of gene regulation: histones and histone-modifying enzymes. Angew. Chem., Int. Ed. 2005, 44, 3186–3216. [DOI] [PubMed] [Google Scholar]
- Zuckermann R. N.; Kodadek T. Peptoids as potential therapeutics. Curr. Opin. Mol. Ther. 2009, 11, 299–307. [PubMed] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K.; et al. Peptoids: a modular approach to drug discovery. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckermann R. N.; Kerr J. M.; Kent S. B. H.; Moos W. H. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar]
- Nguyen J. T.; Turck C. W.; Cohen F. E.; Zuckermann R. N.; Lim W. A. Exploiting the basis of proline recognition by SH3 and WW domains: design of N-substituted inhibitors. Science 1998, 282, 2088–2092. [DOI] [PubMed] [Google Scholar]
- Hara T.; Durell S. R.; Myers M. C.; Appella D. H. Probing the structural requirements of peptoids that inhibit HDM2-p53 interactions. J. Am. Chem. Soc. 2006, 128, 1995–2004. [DOI] [PubMed] [Google Scholar]
- Udugamasooriya D. G.; Dineen S. P.; Brekken R. A.; Kodadek T. A peptoid “antibody surrogate” that antagonizes VEGF receptor 2 activity. J. Am. Chem. Soc. 2008, 130, 5744–5752. [DOI] [PubMed] [Google Scholar]
- Chongsiriwatana N. P.; Patch J. A.; Czyzewski A. M.; Dohm M. T.; Ivankin A.; Gidalevitz D.; Zuckermann R. N.; Barron A. E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler S. A.; Stacy D. M.; Blackwell H. E. Design and synthesis of macrocyclic peptomers as mimics of a quorum sensing signal from Staphylococcus aureus. Org. Lett. 2008, 10, 2329–2332. [DOI] [PubMed] [Google Scholar]
- Levine P. M.; Imberg K.; Garabedian M. J.; Kirshenbaum K. Multivalent peptidomimetic conjugates: a versatile platform for modulating androgen receptor activity. J. Am. Chem. Soc. 2012, 134, 6912–6915. [DOI] [PubMed] [Google Scholar]
- Comegna D.; Benincasa M.; Gennaro R.; Izzo I.; De Riccardis F. Design, synthesis and antimicrobial properties of non-hemolytic cationic alpha-cyclopeptoids. Bioorg. Med. Chem. 2010, 18, 2010–2018. [DOI] [PubMed] [Google Scholar]
- Huang M. L.; Shin S. B.; Benson M. A.; Torres V. J.; Kirshenbaum K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem 2012, 7, 114–122. [DOI] [PubMed] [Google Scholar]
- Demmer O.; Frank A. O.; Hagn F.; Schottelius M.; Marinelli L.; Cosconati S.; Brack-Werner R.; Kremb S.; Wester H. J.; Kessler H. A Conformationally Frozen Peptoid Boosts CXCR4 Affinity and Anti-HIV Activity. Angew. Chem., Int. Ed. Engl. 2012, 51, 8110–8113. [DOI] [PubMed] [Google Scholar]
- Olsen C. A. Peptoid-Peptide hybrid backbone architectures. ChemBioChem 2010, 11, 152–160. [DOI] [PubMed] [Google Scholar]
- Olsen C. A.; Ziegler H. L.; Nielsen H. M.; Frimodt-Moller N.; Jaroszewski J. W.; Franzyk H. Antimicrobial, hemolytic, and cytotoxic activities of beta-peptoid-peptide hybrid oligomers: improved properties compared to natural AMPs. ChemBioChem 2010, 11, 1356–1360. [DOI] [PubMed] [Google Scholar]
- Olsen C. A.; Bonke G.; Vedel L.; Adsersen A.; Witt M.; Franzyk H.; Jaroszewski J. W. Alpha-peptide/beta-peptoid chimeras. Org. Lett. 2007, 9, 1549–1552. [DOI] [PubMed] [Google Scholar]
- Vedel L.; Bonke G.; Foged C.; Ziegler H.; Franzyk H.; Jaroszewski J. W.; Olsen C. A. Antiplasmodial and prehemolytic activities of alpha-peptide-beta-peptoid chimeras. ChemBioChem 2007, 8, 1781–1784. [DOI] [PubMed] [Google Scholar]
- Shuey S. W.; Delaney W. J.; Shah M. C.; Scialdone M. A. Antimicrobial beta-peptoids by a block synthesis approach. Bioorg. Med. Chem. Lett. 2006, 16, 1245–1248. [DOI] [PubMed] [Google Scholar]
- Shin S. B.; Yoo B.; Todaro L. J.; Kirshenbaum K. Cyclic peptoids. J. Am. Chem. Soc. 2007, 129, 3218–3225. [DOI] [PubMed] [Google Scholar]
- Roy O.; Faure S.; Thery V.; Didierjean C.; Taillefumier C. Cyclic beta-peptoids. Org. Lett. 2008, 10, 921–924. [DOI] [PubMed] [Google Scholar]
- Stringer J. R.; Crapster J. A.; Guzei I. A.; Blackwell H. E. Extraordinarily robust polyproline type I peptoid helices generated via the incorporation of alpha-chiral aromatic N-1-naphthylethyl side chains. J. Am. Chem. Soc. 2011, 133, 15559–15567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caumes C.; Roy O.; Faure S.; Taillefumier C. The click triazolium peptoid side chain: a strong cis-amide inducer enabling chemical diversity. J. Am. Chem. Soc. 2012, 134, 9553–9556. [DOI] [PubMed] [Google Scholar]
- White C. J.; Yudin A. K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]
- Enzo Life Sciences, HDAC fluorometric activity assay kit, BML-AK500-0001.
- Kranz M.; Murray P. J.; Taylor S.; Upton R. J.; Clegg W.; Elsegood M. R. J. Solution, solid phase and computational structures of apicidin and its backbone-reduced analogs. J. Pept. Sci. 2006, 12, 383–388. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.; Steinkuhler C. From natural products to small molecule ketone histone deacetylase inhibitors: development of new class specific agents. Curr. Pharm. Des. 2008, 14, 545–561. [DOI] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- Gurard-Levin Z. A.; Mrksich M. The activity of HDAC8 depends on local and distal sequences of its peptide substrates. Biochemistry 2008, 47, 6242–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.