

Abstract

Herein, we describe the discovery of potent and highly selective inhibitors of both CDK4 and CDK6 via structure-guided optimization of a fragment-based screening hit. CDK6 X-ray crystallography and pharmacokinetic data steered efforts in identifying compound 6, which showed >1000-fold selectivity for CDK4 over CDKs 1 and 2 in an enzymatic assay. Furthermore, 6 demonstrated in vivo inhibition of pRb-phosphorylation and oral efficacy in a Jeko-1 mouse xenograft model.
Keywords: CDK4/6, pRb phosphorylation, mantle cell lymphoma, fragment-based screening, structure-guided optimization
Progression through the cell cycle is a highly regulated process. In the absence of appropriate growth signals, a family of pocket proteins including retinoblastoma protein (pRb) prevents cells from entering the DNA replication phase (S phase).1,2 The replication cycle begins when mitogens trigger signal transduction pathways, leading to increase of cellular levels of D-cyclins. D-cyclins, in turn, activate cyclin-dependent kinases 4/6 (CDK4/6), which phosphorylate and inactivate pRb.1,3,4
Uncontrolled cell proliferation is one of the hallmarks of cancer, and pRb inactivation is the key event that enables tumor cells to progress through the cell cycle unchecked. While some tumors delete the pRb gene itself, the majority maintain a functional pRb and instead activate CDK4/6 kinase activity.5−11 Ablation of CDK4 kinase activity led to complete tumor growth inhibition in CDK4/cyclin D1-dependent tumors such as those found in breast cancer that overexpresses Her2/neu.12 Furthermore, normal fibroblast cells were shown to overcome the absence of CDK4/6 due to compensation by CDK1, whose absence is not tolerated.13 Taken together, this evidence suggests that a selective inhibitor of CDK4/6 may have a wider therapeutic window than pan-CDK inhibitors.
A variety of CDK inhibitors have been evaluated preclinically and clinically.1,14,15 Given the evidence described above, many research groups have embarked on the discovery of a CDK4/6 selective inhibitor, with the most well-documented being PD-0332991.16,17
Previously, we reported on the identification and structure-guided optimization of a series of 4-(pyrazol-4-yl) pyrimidines as selective CDK4/6 inhibitors. Several potency and selectivity determinants were established based on the X-ray crystallographic analysis of representative compounds bound to CDK6.18 Herein, we describe the structure-guided optimization of a CDK6 fragment-based screening hit leading to the discovery of compound 6, a potent, selective, and orally bioavailable CDK4/6 inhibitor.
The starting point for this work was provided by the CDK6 fragment-based screening hit A [Scheme 1, CDK6 IC50 = 7.2 μM, ligand efficiency (LE)19 = 0.44]. The X-ray crystallographic structure of CDK6 in complex with fragment A (Figure 1a) was solved using a novel back-soakable form of CDK6. The crystal structure revealed that the ligand binds in the ATP pocket and forms hydrogen bonds with the backbone NH and the backbone carbonyl of Val101. Further inspection of the crystal structure suggested that the two heterocyclic rings offered multiple “growth” vectors and therefore the opportunity to interact with many of the residues within the ATP pocket known to be important for potency and selectivity.18,20−22 Some of these key residues are highlighted in Figure 1. In particular, His100 (CDK6) and Thr107 (CDK6) are replaced with Phe and Lys, respectively, in CDKs 1 and 2. Exploiting these two differences is important for establishing selectivity over CDK1/2. Hydrogen bonding between the ligands and the side chain of Lys43 and the backbone of Asp163 also seems to contribute to a potent and selective CDK4/6 profile.21,23
Scheme 1. Identification of Benzimidazole 1, Starting from Fragment A.

IC50 values are given as the average of two or more determinations unless otherwise indicated by an asterisk (*) (where n = 1).
Figure 1.
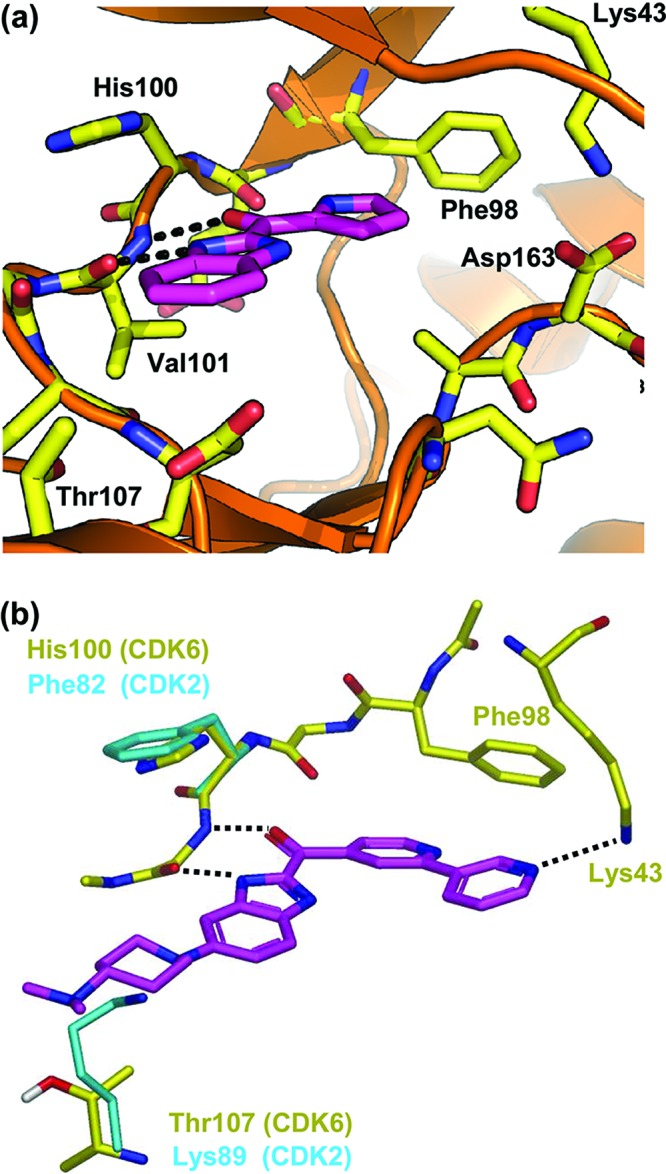
(a) X-ray crystal structure of benzimidazole fragment A bound to CDK6, showing hydrogen bonds to the kinase hinge region (dotted black lines). (b) Model of compound 1 (magenta) bound to CDK6 (yellow) overlaid with key amino acid differences from CDK2 (1hck; shown in cyan). The pyridin-3-yl nitrogen aims to hydrogen bond with the side chain of Lys43 (CDK6). The 4-dimethylaminopiperidine group is designed to make unfavorable electrostatic and steric repulsion with Lys89 (CDK2) but to be tolerated by Thr107 (CDK6). Docking performed using GOLD.25,26
Examination of the structure in Figure 1 suggested that optimal growth vectors to target Lys43 would be provided by replacing the pyrrole with a pyridine ring. The pyridine analogue B (Scheme 1, 57% inhibition at 30 μM, LE = ca. 0.36) retained acceptable LE and provided a better starting point for further optimization.
Structure-based design and previous experience with CDK4/6 described above18,20−22 enabled rapid elaboration of the scaffold as exemplified by compound 1 (CDK6 IC50 = 1.1 μM, LE = 0.25).24 The rationale for this compound design is illustrated by the model of 1 bound to CDK6 (Figure 1b). First, the pyridin-3-yl group was introduced with the aim of establishing a hydrogen bond with the side chain of Lys43. Second, a 4-(dimethylamino) piperidine group was introduced at the 5-position of the benzimidazole, with the intention of placing a bulky amine in the vicinity of Thr107. This residue is less sterically demanding and less basic than the corresponding lysine in CDK1/2, and so, it was hoped that this substitution would improve both selectivity over CDK1/2 and solubility.18,20−22
Subsequent optimization around the pyridin-3-yl motif of compound 1 led to the identification of the isoquinoline analogue 2, which demonstrated ca. 20-fold increase in affinity for CDK6 (IC50 = 0.049 μM, LE = 0.28) as compared with 1 (Table 1). Compound 2 also exhibited good potency against CDK4 (IC50 = 0.006 μM, LE = 0.31) and moderate selectivity over CDKs 1 and 2.
Table 1. IC50 values for compounds in the CDK enzyme assaysa,b.
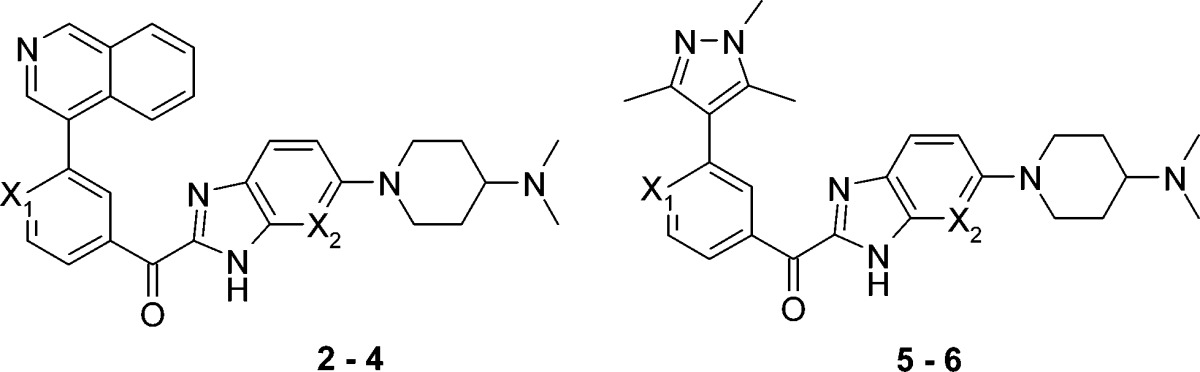
| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| X1 | X2 | CDK4/Cyclin D1 | CDK6/Cyclin D3 | CDK1/Cyclin B | CDK2/Cyclin A | |
| 2 | N | CH | 0.006 ± 0.001 | 0.049 ± 0.006 | 1.2 ± 0.2 | 0.27 ± 0.06 |
| 3 | N | N | 0.012 ± 0.002 | 0.3 ± 0.1 | >15 | 6.2 ± 0.4 |
| 4 | CCN | N | 0.003 ± 0.000 | 0.026 ± 0.003 | 5.6 ± 0.4 | 2.4 ± 0.2 |
| 5 | N | CH | 0.25 ± 0.03 | NT | >15 | >15 |
| 6 | CCN | N | 0.015 ± 0.001 | 0.12 | >15 | >15 |
Results are expressed as the mean ± standard deviation of 1–2 IC50 determinations. For each determination, concentration–inhibition curves were obtained in duplicate and then averaged to afford a single IC50 curve with a ≥95% confidence interval.
70% homology shared between CDK4 and -6 contributes to comparable SAR profiles of two enzymes.
The next strategy to improve selectivity relied on the CDK6 hinge residue His100, an amino acid that is rarely conserved in other kinases. Previously reported structural studies with selective inhibitors bound to CDK6 propose an aromatic sp2 nitrogen in proximity to His100 (His92 in CDK4) as an important determinant for selectivity over other kinases.16,18,22
Analysis of the structural information in Figure 1 suggested that replacing the benzimidazole with 7-azabenzimidazole would provide an opportunity to test this hypothesis. Indeed, the 7-azabenzimidazole analogue 3 was the first compound to exhibit greater than 10-fold selectivity for CDKs 4 and 6 over CDKs 1 and 2. The structure of 3 bound to CDK6 is shown in Figure 2a. As with fragment A, two hydrogen-bonding interactions are observed between 3 and the backbone NH/carbonyl of Val101. The pyridine forms a favorable edge-to-face aromatic–aromatic contact with the gatekeeper residue Phe98. The isoquinoline ring nitrogen is positioned toward the side chain of Lys43, indicating a hydrogen bond.
Figure 2.
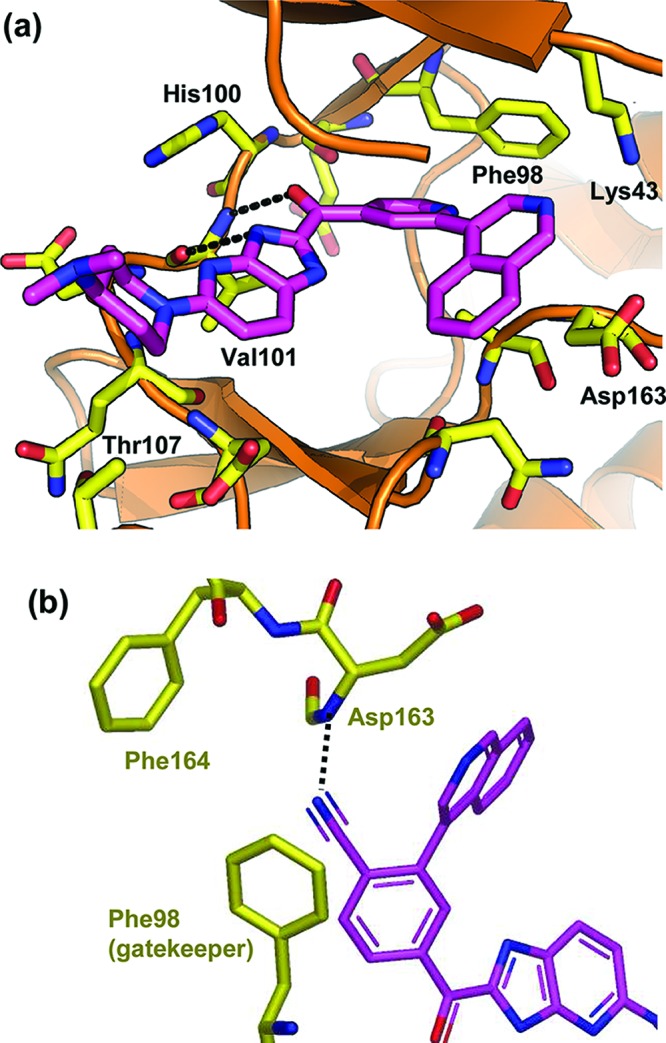
(a) X-ray crystal structure of compound 3 bound to CDK6. (b) Model of compound 4 bound to CDK6 created using GOLD-based software. Projection is rotated (∼90°) and expanded to illustrate the putative hydrogen bond between the nitrile and the backbone NH of Asp163. Lys43 has been removed for clarity.
Interestingly, the isoquinoline ring also shows ca. 30° twist relative to the plane of the pyridine, which appears to provide a better orientation for hydrogen bonding and the resulting increased potency of the isoquinoline analogues 2 and 3 compared to the pyridyl analogue 1. A close examination of the structure in Figure 2a also revealed an opportunity to form a hydrogen bond with the backbone of NH of Asp163 (part of the DFG kinase motif) by placing a nitrile group in the back of the pocket. This strategy is illustrated by the docking model in Figure 2b. Replacing the pyridine ring of compound 3 with a benzonitrile motif gave compound 4, which showed increased potency for CDK4/6 as well as improved selectivity over CDKs 1 and 2.
While demonstrating good potency and modest selectivity, the isoquinoline analogues typically showed limited oral exposure. In an attempt to improve the pharmacokinetic profile, analogues of 2 [plasma clearance (CLp) = 144 mL/min/kg at 2 mg/kg iv, AUC 250 nM h at 5 mg/kg po, Sprague–Dawley rats] were prepared, as these provided the most convenient way to explore the effect of replacing the isoquinoline ring with alternative heterocycles. This led to the identification of the trimethylpyrazole 5 (Table 1), which exhibited moderate potency (CDK4 IC50 0.25 μM, LE = 0.26), reduced in vivo clearance (27 mL/min/kg), and improved oral exposure (AUC 2526 nM h, 5 mg/kg po) in rat.
Experience gained with compounds 2–4 was then used to further optimize potency and selectivity. Thus, introducing the azabenzimidazole group and replacing the pyridine motif with benzonitrile led to the identification of compound 6 (CDK4 IC50 = 0.015 μM, LE = 0.30), which exhibited a ≥1000-fold selectivity for CDK4 over CDKs 1 and 2. Furthermore, 6 demonstrated an oral bioavailability of 26% in rat (AUC 1147 nM h, 5 mg/kg po) with acceptable plasma clearance (37 mL/min/kg, 1 mg/kg iv).
Compound 6 was also shown to be selective for CDK4/6 over a panel of 35 serine-threonine and tyrosine kinases (Supporting Information). These results indicated a comparable or improved selectivity profile relative to currently published CDK4/6 selective inhibitors.16,18
A critical aim of this project was to generate compounds with a high degree of selectivity for CDK4/6 over CDK1/2. During the optimization process, certain modifications were made to the molecule, not solely to increase CDK4/6 potency but to drive the high selectivity over CDK1/2. Examples include the (dimethylamino)piperidine group and the azabenzimidazole modification, which actually reduces CDK4/6 potency (compare 2 and 3). One consequence of this is that compound 6 has a lower LE (CDK4 LE = 0.31, CDK6 LE = 0.26) than the starting fragment A (CDK6 LE = 0.44).
Selective CDK4/6 inhibition prevents phosphorylation of pRb, consequently blocking the G1 to S phase transition. Compound 6 demonstrated a normalized cellular IC5018 value of 0.26 μM in a Jeko-1 mantle cell lymphoma assay measuring inhibition of pRb phosphorylation.9,27 Fluorescence-activated cell sorting (FACS) demonstrated a dose-dependent increase in exclusive G1 arrest with increasing concentrations of compound (Figure 3). Clean G1 block was maintained over a range of 0.625–2.5 μM. At a higher concentration of 10 μM, block in non-G1 phases of the cell cycle emerged.
Figure 3.
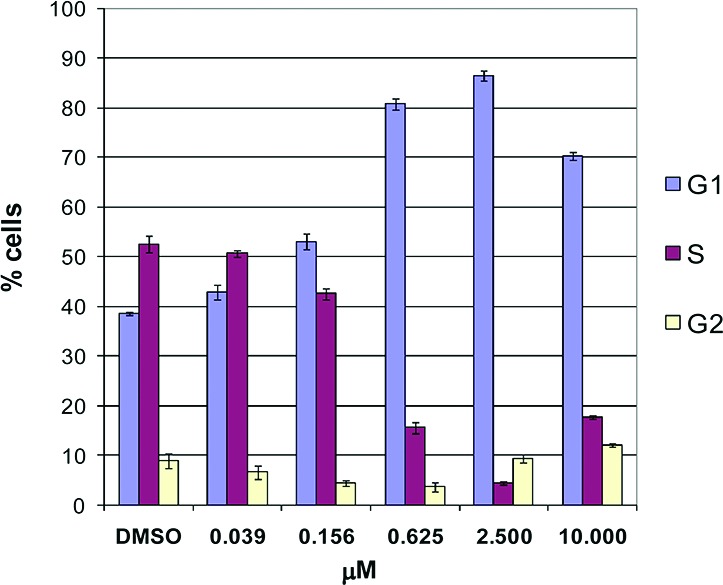
Cell cycle flow cytometry analyses of compound 6.
This favorable balance of PK properties with biochemical and cellular potency/selectivity profile led us to evaluate 6 in Jeko-1 xenograft tumor-bearing severe combined immunodeficiency (SCID) mice. Modulation of pRb phosphorylation was examined at several time points (0, 1, 4, and 7 h post the last dose) after orally administering 6 for 2.5 days twice daily at 125 and 250 mg/kg (Figure 4a,b). Dose-dependent increases in Cmax and inhibition of pRb were observed. A dose level of 250 mg/kg/day with 6 resulted in a maximum 80% inhibition of phosphorylation, which declined to 60% at 7 h. At the 500 mg/kg/day dose level, the pharmacodynamic (PD) readout reached 100% 1 h postdosing and was sustained above 85% over 7 h. We then verified the maximum tolerated dose (MTD) following 7 days of oral treatment of naive SCID mice at 100, 200, and 400 mg/kg/day bid. The MTD was established as the maximum dose that resulted in less than 15% mean body weight loss and no treatment-related deaths. On the basis of these criteria, the MTD of 6 was determined to be between 200 and 400 mg/kg/day. Efficacy was then assessed in a Jeko-1 xenograft model, where tumor-bearing mice were treated with 6 twice daily for 10 days (Figure 4c). Results were reported as T/C, determined by the ratio of the change in tumor volume of the treated animals to the change in tumor volume of the control animals. Following oral dosing at 75, 150, and 250 mg/kg/day, compound 6 exhibited dose-related antiproliferative effects with T/C values of 41, 25, and 4%, respectively. These results indicated that 6 was able to demonstrate dose-dependent tumor growth inhibition, with the 250 mg/kg/day dose significantly blocking tumor growth. The 250 mg/kg/day dose was well tolerated and did not exceed the MTD criteria described above. Coupled with the PD data obtained, the CDK4/6 selective inhibitor 6 exhibited sufficient in vivo exposure to block CDK4/6 specific pRb phosphorylation in the tumors and a robust antitumor activity in this mouse tumor model.
Figure 4.
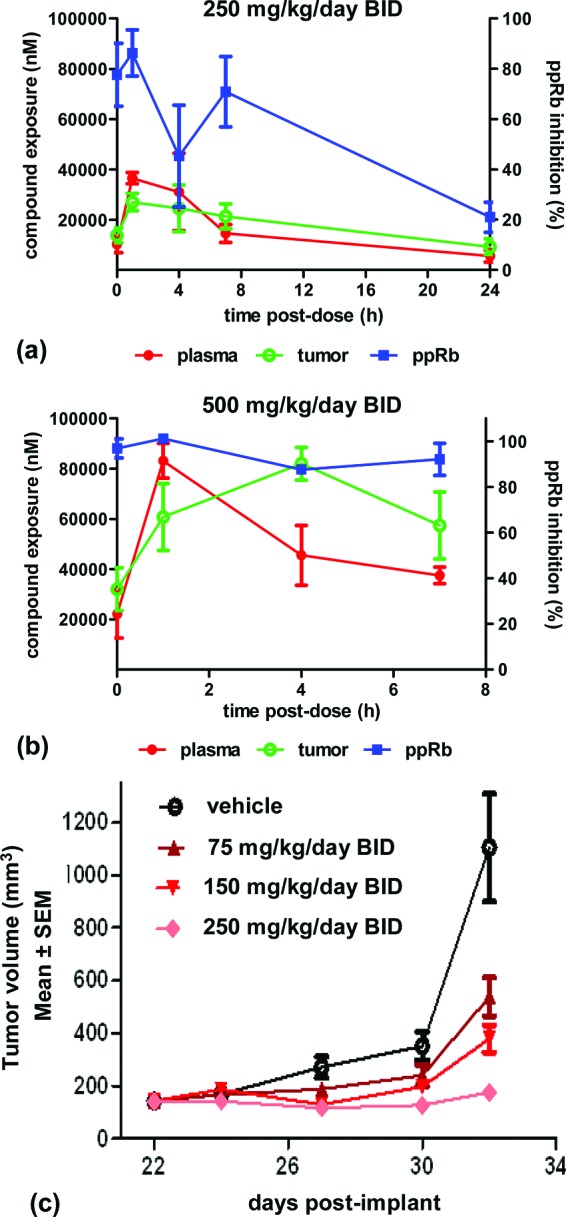
(a and b) Inhibition of phosphorylation of pRb in Jeko-1 xenograft tumor bearing SCID mice treated with 6. (c) Efficacy of compound 6 in Jeko-1 xenograft tumor bearing SCID mice.
In summary, we have identified a potent, highly selective, and orally bioavailable CDK4/6 inhibitor 6, starting from benzimidazole fragment A. CDK6 structure-guided design and pharmacokinetic data were utilized for optimization toward compound 6. CDK4/6 specific activity of this compound was apparent in both biochemical assays and cellular functional readouts. Multiday treatment of Jeko-1 tumors in mice demonstrated dose-dependent inhibition of pRb phosphorylation as well as tumor growth delay.
Acknowledgments
We thank Dr. Timothy Ramsey and Dr. Chris Johnson for helpful suggestions in the preparation of the manuscript.
Supporting Information Available
Enzyme assay conditions for CDK1, -2, -4, and -6, CDK4 cellular assay conditions, fluorescence-activated cell-sorting conditions, crystallography conditions, in vivo experiments method, experimental details for the synthetic procedures and characterization data of A, B, and 1–6, and further in vitro and in vivo pharmacokinetic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Malumbres M.; Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Rev. Cancer 2009, 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Weinberg R. A. The retinoblastoma protein and cell-cycle control. Cell 1995, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Sherr C. J.; Roberts J. M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Lundberg A. S.; Weinberg R. A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 1998, 18, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro G. I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [DOI] [PubMed] [Google Scholar]
- Ortega S.; Malumbres M.; Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta, Rev. Cancer 2002, 1602, 73–87. [DOI] [PubMed] [Google Scholar]
- Shapiro G. I.; Park J. E.; Edwards C. D.; Mao L.; Merlo A.; Sidransky D.; Ewen M. E.; Rollins B. J. Multiple mechanisms of p16 (Ink4A) inactivation in non-small-cell lung-cancer cell-lines. Cancer Res. 1995, 55, 6200–6209. [PubMed] [Google Scholar]
- Kamb A.; Gruis N. A.; Weaverfeldhaus J.; Liu Q. Y.; Harshman K.; Tavtigian S. V.; Stockert E.; Day R. S.; Johnson B. E.; Skolnick M. H. A cell-cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264, 436–440. [DOI] [PubMed] [Google Scholar]
- Amin H. M.; McDonnell T. J.; Medeiros L. J.; Rassidakis G. Z.; Leventaki V.; O’Connor S. L.; Keating M. J.; Lai R. Characterization of 4 mantle cell lymphoma cell lines - Establishment of an in vitro study model. Arch. Pathol. Lab. Med. 2003, 127, 424–431. [DOI] [PubMed] [Google Scholar]
- Sutherland R. L.; Musgrove E. A. Cyclins and breast cancer. J. Mammary Gland Biol. Neoplasia 2004, 9, 95–104. [DOI] [PubMed] [Google Scholar]
- Sirvent N.; Coindre J. M.; Maire G.; Hostein I.; Keslair F.; Guillou L.; Ranchere-Vince D.; Terrier P.; Pedeutour F. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: Utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am. J. Surg. Pathol. 2007, 31, 1476–1489. [DOI] [PubMed] [Google Scholar]
- Yu Q. Y.; Sicinska E.; Geng Y.; Ahnstrom M.; Zagozdzon A.; Kong Y.; Gardner H.; Kiyokawa H.; Harris L. N.; Stal Q.; Sicinski P. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006, 9, 23–32. [DOI] [PubMed] [Google Scholar]
- Santamaria D.; Barriere C.; Cerqueira A.; Hunt S.; Tardy C.; Newton K.; Caceres J. F.; Dubus P.; Malumbres M.; Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–818U8. [DOI] [PubMed] [Google Scholar]
- Malumbres M.; Pevarello P.; Barbacid M.; Bischoff J. R. CDK inhibitors in cancer therapy: What is next?. Trends Pharmacol. Sci. 2007, 29, 16–21. [DOI] [PubMed] [Google Scholar]
- Hirai H.; Kawanishi N.; Iwasawa Y. Recent advances in the development of selective small molecule inhibitors for cyclin-dependent kinases. Curr. Top. Med. Chem. 2005, 5, 167–179. [DOI] [PubMed] [Google Scholar]
- Toogood P. L.; Harvey P. J.; Repine J. T.; Sheehan D. J.; VanderWel S. N.; Zhou H.; Keller P. R.; McNamara D. J.; Sherry D.; Zhu T.; Brodfuehrer J.; Choi C.; Barvian M. R.; Fry D. W. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [DOI] [PubMed] [Google Scholar]
- Fry D. W.; Harvey P. J.; Keller P. R.; Elliott W. L.; Meade M.; Trachet E.; Albassam M.; Zheng X.; Leopold W. R.; Pryer N. K.; Toogood P. L. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [PubMed] [Google Scholar]
- Cho Y. S.; Borland M.; Brain C.; Chen C. H.-T.; Cheng H.; Chopra R.; Chung K.; Groarke J.; He G.; Hou Y.; Kim S.; Kovats S.; Lu Y.; O'Reilly M.; Shen J.; Smith T.; Trakshel G.; Vögtle M.; Xu M.; Xu M.; Sung M. J. 4-(Pyrazol-4-yl)-pyrimidines as selective inhibitors of cyclin-dependent kinase 4/6. J. Med. Chem. 2010, 53, 7938–7957. [DOI] [PubMed] [Google Scholar]
- LE = −ΔG/HAC ≈ –RT ln(IC50)/HAC.Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: A useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Day P. J.; Cleasby A.; Tickle I. J.; O'Reilly M.; Coyle J. E.; Holding F. P.; McMenamin R. L.; Yon J.; Chopra R.; Lengauer C.; Jhoti H. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4166–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt D. J.; Bentley J.; Jewsbury P.; Boyle F. T.; Endicott J. A.; Noble M. E. M. Dissecting the determinants of cyclin-dependent kinase 2 and cyclin-dependent kinase 4 inhibitor selectivity. J. Med. Chem. 2006, 49, 5470–5477. [DOI] [PubMed] [Google Scholar]
- Lu H.; Schulze-Gahmen U. Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J. Med. Chem. 2006, 49, 3826–3831. [DOI] [PubMed] [Google Scholar]
- Honma T.; Yoshizumi T.; Hashimoto N.; Hayashi K.; Kawanishi N.; Fukasawa K.; Takaki T.; Ikeura C.; Ikuta M.; Suzuki-Takahashi I.; Hayama T.; Nishimura S.; Morishima H. A novel approach for the development of selective Cdk4 inhibitors: Library design based on locations of Cdk4 specific amino acid residues. J. Med. Chem. 2001, 44, 4628–4640. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for a detailed preparation of compounds A, B, and 1–6.
- Jones G.; Willett P.; Glen R. C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Tchakarska G.; Lan-Leguen A.; Roth G.; Sola B. The targeting of the sole cyclin D1 is not adequate for mantle cell lymphoma and myeloma therapies. Haematologica 2009, 94121781–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.