

Abstract
From the 13 533 chemical structures published by GlaxoSmithKline in 2010, we identified 47 quality starting points for lead optimization. One of the most promising hits was the TCMDC-139046, a molecule presenting an indoline core, which is well-known for its anxiolytic properties by interacting with serotonin antagonist receptors 5-HT2. The inhibition of this target will complicate the clinical development of these compounds as antimalarials. Herein, we present the antimalarial profile of this series and our efforts to avoid interaction with this receptor, while maintaining a good antiparasitic potency. By using a double-divergent structure–activity relationship analysis, we have obtained a novel lead compound harboring an indoline core.
Keywords: indoline, malaria, Tres Cantos Antimalarial set, divergent SAR, open-innovation
Malaria is a major global disease caused by parasites of the genus Plasmodium, which are transmitted to people when female anopheles mosquitoes feed on human blood. The reasons for the mortality and morbidity are varied; however, probably the most remarkable is the existence of Plasmodium falciparum-resistant strains to nearly all standard antimalarial drugs.1 As a consequence, there is an urgent requirement for novel antimalarial drugs, and numerous programs from public and private institutions or public–private partnerships are being developed.2,3
The criteria for new antimalarial drugs are very demanding; first, the drug must be safe and efficacious.4 Then, the profile of a new molecule should be better than any of the existing drugs, and it should be affordable (less than $1/treatment for an adult), should be active against resistant strains, and preferably should exert its antimalarial effects through a new mode of action. Moreover, with the eradication strategy in mind, one component of the profile should ideally include activity against sexual stages and mosquito stages of the parasite lifecycle or also be effective against hypnozoites (the hard-to-control dormant liver stages of this parasite).
In 2010, St. Jude's Children's Research Hospital,5 Novartis,6 and GlaxoSmithKline7 published the structures of thousands of compounds that inhibit parasite intraerythrocytic growth. This represents a big step change in the number of hits available for drug discovery programs (the three sets of compounds are available for download from the Chembl-NTD database: http://www.ebi.ac.uk/chemblntd).
Very recently, we published our initial strategy to mine the GSK set, the TCAMS (Tres Cantos Antimalarial Set), which includes the identification of quality starting points to start lead optimization programs.8 Different mining approaches have already been published; however, our priority was to identify quality starting points suitable for the discovery of oral bioavailable drugs. These criteria have dictated the mining and filtering processes that we used. This is just one approach for mining TCAMS and one that may deprioritize compounds that could represent perfectly good starting points if a different mining or filtering process is carried out. Authors also included a top-five favorites series. The efforts for one of this series, the cyclopropyl carboxamides, have been already published.9,10 In this communication, we intend to update our efforts in exploring one of the other four series, the indolines.
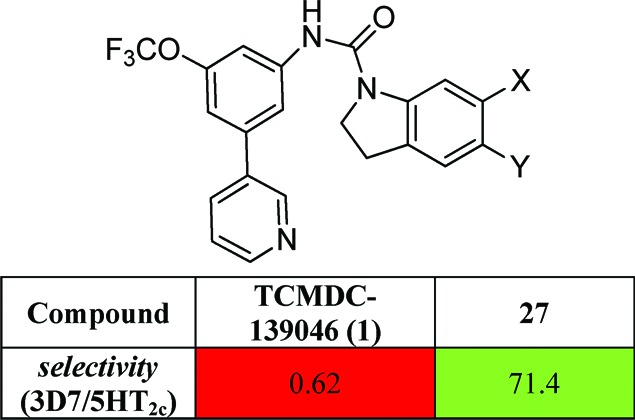
The primary hit for this series, TCMDC-139046 (1) (Figure 1), is a compound that shows a good in vitro antimalarial activity with IC50 values of 80, 40, and 25 nM against P.falciparum 3D7A at 48, 72, and 96 h assays, respectively. This indicates that compound 1 kills the parasite in the first generation, and it does not present a delayed death phenotype. When parasite cultures are treated with this compound (at a concentration of 10-fold IC50 value), the erythrocytic cycle is stopped, and pyknotic forms are visible after 24 h of exposure (Figure 1). The appearance of treated parasites is similar to phenotypes produced in the parasites by chloroquine or artemisinin treatments (both are fast-killing antimalarials), and these are very different to the phenotypes produced by atovaquone (bc1 complex inhibitor),11,12 which stops the cell cycle in the trophozoite stage. In vitro efficacy was also determined using P. falciparum strains HB3 (moderately resistant to pyrimethamine) and Dd2 (chloroquine- and pyrimethamine-resistant). Compound 1 retains its in vitro activity against P. falciparum HB3 (IC50 value 90 nM); however, a more than 10-fold decreased is observed against multidrug-resistant strain Dd2 (IC50 > 1 μM).
Figure 1.

Complete profile12 of TCMDC-139046 as a representative compound of the indoline series showing antimalarial activity, cytotoxicity, affinity for 5HT2c, in vitro DMPK profile, and chemical profile (table on the left,). The structure of TCMDC-139046 and the effects on the parasites are shown on the right.
This compound showed a good metabolic stability in vitro when it was tested in mouse and human microsome assays.12 To determine the cytotoxicity profile, it was evaluated using five different mammalian cell lines [HepG2, L1210, Neuro2A, H9c2(2-1), and MDCK].12 Results showed TOX50 values around 10 μM, probably due to its moderate solubility in aqueous media (less than 0.1 μg mL–1 in PBS).12 This representative compound has been profiled in a secondary pharmacology panel of assays relevant for clinical development, resulting inactive in 40 of 42 assays, showing that the main issue of this series is their high affinity for the 5-hydroxytryptamine subtype 2c (5HT2c) receptor (Figure 1).
The 5HT receptors are part of the G-protein-coupled receptor (GPCR) superfamily and are classified into seven main types, 5-HT1–7. They can be localized in both the peripheral and the central nervous system (CNS), and they mediate in numerous physiological functions. The serotonin 5-HT2 receptor family is comprised of 5-HT2A, 5-HT2B, and 5-HT2C subtypes. These are grouped together on the basis of their primary structure, secondary messenger system, and operational profile.13−15 In fact, sequence analysis of these receptors has revealed an approximately 80% of similarity, so it is not surprising that compounds with affinity for one of the receptors also bind with high affinity to the others. 5-HT2A and 5-HT2B receptors are widely distributed in the peripheral (also can be found in the CNS but lower quantity), while the 5-HT2C receptor has been found only in the CNS.
A complete structure–activity relationship (SAR) study was published 12 years ago16,17 presenting the biarylcarbamoylindolines 2 and the bispyridyl ether carbamoylindolines 3 (Figure 2) as very promising novel and selective 5-HT2C receptor inverse agonists, with antidepressant and anxiolytic properties. This work is justified by the significant number of therapeutic opportunities for the treatment of different CNS disorders such as depression,18 schizophrenia,18,19 migraine,20 and Parkinson's disease21 offered by 5-HT2C antagonism.
Figure 2.

5-HT2c inhibitor hits.
Both hits can be related easily to our lead compound 1, especially compound 2 (IC50 5-HT2C = 0.041 μM), which presents a biarylcarbamoyl-5-methoxy-6-trifluoromethylindoline in the right-hand side and a fluorine atom in the aniline ring directly attached to the urea instead of a trifluoromethoxy group (compound 1). This potent inhibition of the 5-HT2c receptors can complicate the clinical development of this series as antimalarials, so we wondered: would it be possible to reduce the high human 5-HT2c affinity while increasing the antimalarial activity?
To face this challenge, we designed a double SAR study aimed at determining which features can drive to a more potent antimalarial in both in P. falciparum 3D7 and P. falciparum Dd2, while avoiding interaction with 5-HT2c receptors. In addition, the optimization should lead to a compound with an improved physicochemical profile because 1 presents a poor developability profile (high cLog P and low solubility in aqueous media, Figure 1).
If we insert the indoline scaffold A (Figure 3) in the Chembl-NTD compound structure search page (https://www.ebi.ac.uk/chemblntd/compound/structure_home), 76 hits are obtained with potencies 0.040–1.3 μm in 3D7 at 72 h, but no clear SAR conclusions can be achieved. We decided to start our analysis using classical SAR techniques by studying the aniline moiety by testing if the lipophilic trifluoromethoxy can be replaced for a more desirable group such as F, H, CF3, or Br (Table 1, entries 2, 3, 4, and 5).
Figure 3.
Scaffold A.
Table 1. SAR Analysis on the Left-Hand Side of the Molecule.
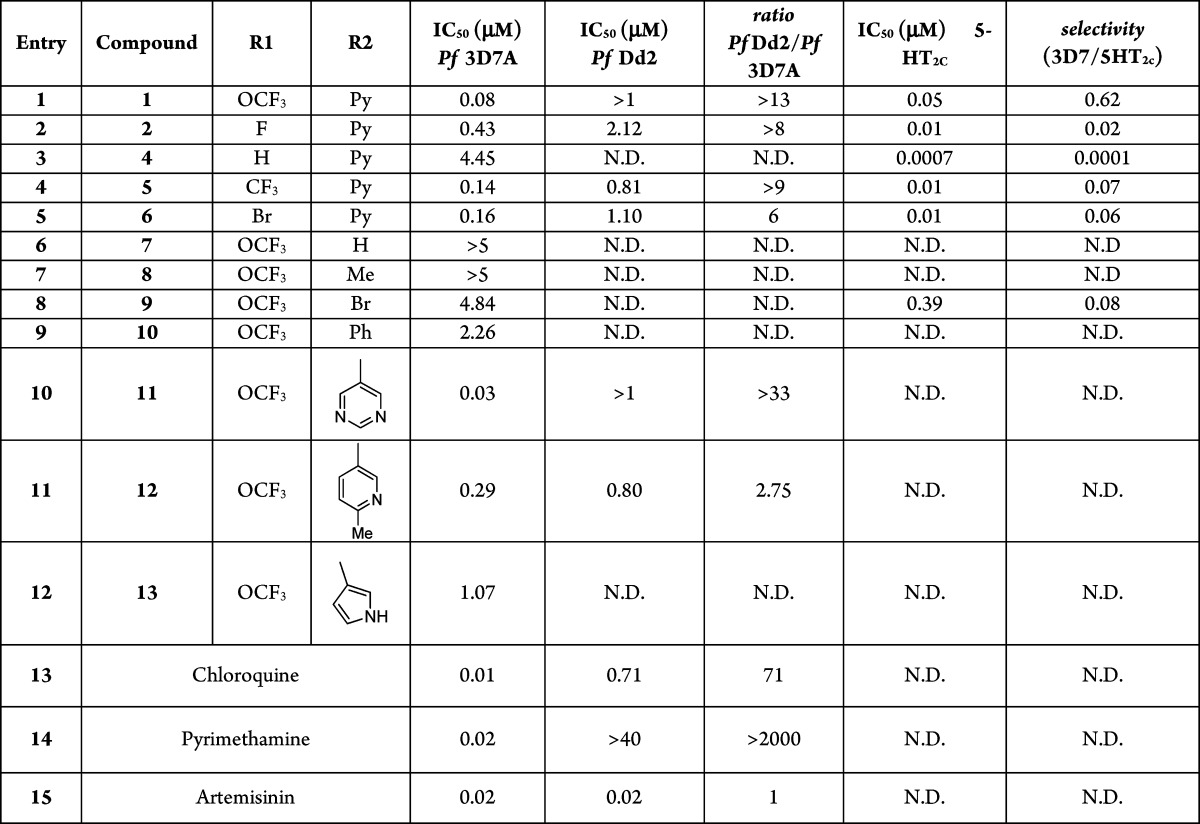
However, the trifluoromethoxy was shown to be the best substitution because the rest of the groups tested presented a reduced antimalarial activity, and moreover, a notable increase in the 5-HT2c affinity was observed. In fact, the results suggested that small substituents, or even no substitution, at this position drive to compounds with high affinity to 5-HT2c while the antiplasmodial potency, and consequently selectivity, can be achieved with bulky substituents. In addition, all of them lose potency when they are tested against P. falciparum Dd2.
Next, we drove our attention to the pyridyl ring (R2). When we changed the pyridyl ring for a H, Me, and Br (Table 1, entries 6, 7, and 8), a loss in the antiplasmodial activity was observed. The same result was obtained when this moiety was substituted for different aromatic entities such as phenyl ring and related heterocycles (Table 1, entries 9–12). Although no great improvements were obtained in terms of potency, we realized that the pyridyl ring can be substituted by a pyrimidyl, which presents a slight improvement in the physicochemical profile (clog P = 5.25). However, following the docking analysis of compound 2 described by Bromidge et.al.,16,17 where in the proposed binding site, the 3-pyridyl ring occupies a quite deep lipophilic pocket that can be occupied by the pyrimidine ring or related aromatic moieties, a drastic change in this part of the molecule would be needed to decrease the 5-HT2c selectivity.
So, finally, we focused our efforts in the indoline ring where a wide variety of substituents are allowed based on the diversity found in the TCAMS. That gave us confidence of the real chances for getting selectivity while maintaining antiplasmodial activity plus improving the druglike properties. Searching the literature, we found three main opportunities to avoid 5-HT2c affinity in the indoline core: unsubstituted indolines, introduction of substituents at position 7, and introduction of polar groups.15,16
We started by synthesizing the unsubstituted indoline 14 where we saw that an increase in the antimalarial activity was achieved [low tens of nanomolar in both 3D7 and Dd2 (Table 2, entry 2)], but also, we achieved a selectivity ratio of 125. Moreover, no less important is the fact that this change drove us to a more attractive compound in terms of physicochemical properties (cLog P = 4.9), although it showed a weak metabolic stability [in vitro clearance12 mouse/human, 6.2/1.0 mL/min g]. This issue was solved by just introducing a fluorine atom at position 6 (compound 15, Table 2, entry 3; in vitro clearance mouse/human, 2.1/0.5 mL/min g).12 The substitution by Cl, Br, and methylene (Table 2, entries 4–6) resulted in loss of activity as compared with 14 and 15. Neither combination Cl/Me (Table 2, entry 7) nor monosubstitution at position 5 (Table 2, entries 8–10) resulted in any improvement.
Table 2. SAR Analysis on the Right-Hand Side of the Molecule.
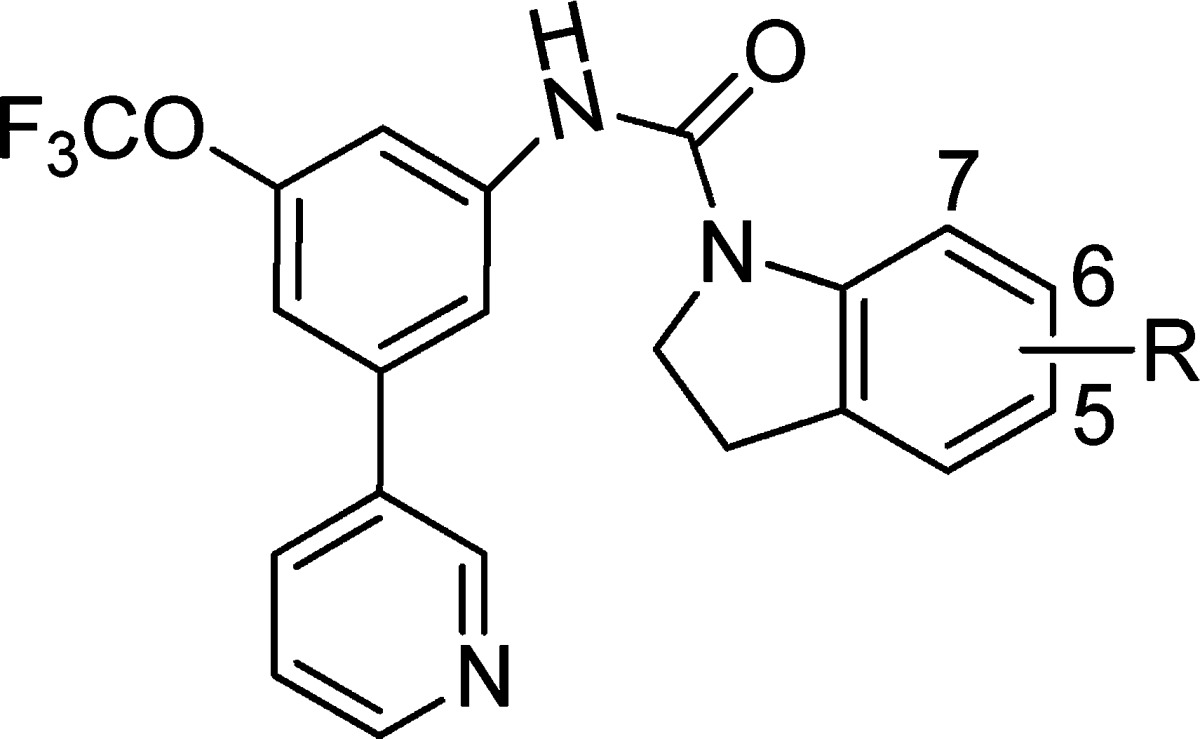

Although compounds 14 and 15 were satisfactory to our purpose, we tested the two other hypotheses, so compound 23 (table 2, entry 11), which presents a fluorine in position 7, was prepared. This compound showed a good selectivity window although a loss in potency against both 3D7A and Dd2 was detected. Finally, analogues 24, 25, 26, and 27 presenting polar groups were prepared to test the third hypothesis. As expected, better selectivity was obtained, highlighting the piperidine derivatives 26 and 27 where no interaction with 5-HT2c was detected (Table 2, entry 12–15) and the activity against multidrug resistance strains Dd2 was maintained.
As result of the SAR analysis, compound 27 has shown to be the most promising compound, demonstrating that a divergent SAR between in vitro antimalarial activity and affinity for 5-HT2c is possible. This compound has not lost significant antimalarial potency against the 3D7 sensitive strain as compared to our initial hit 1, it is active against P. falciparum Dd2 (chloroquine- and pyrimethamine-resistant), and treated parasites retain the same phenotypic effect (supporting the same mode of action).12 Compound 27 was tested in vivo against P. berghei. Compound 27 was administered by the oral route at 50 mg/kg, once a day for two consecutive days. In this assay, compound 27 inhibited parasitemia at day 4 after infection by 35% with respect to vehicle group. However, despite this weak in vivo efficacy, this new derivative has been able to reduce the initial number of viable parasites in a culture, from 105 to less than 10 parasites, when this culture was treated during 72 h at 10-fold the IC50 concentration. This result is similar to cultures treated at bioequivalent concentrations of chloroquine or artemisinin. Compound 27 presents a compatible profile with oral absorption in terms of physicochemical properties (CHI log D pH 6.5/7.4 3.9/4.2)12 and in vitro intrinsic clearance (mouse/human, 4.0/1.5 mL/min g). Compound 27 was tested as 5-HT2c agonist showing a selectivity (3D7/5HT2c) > 70.
In conclusion, the SAR analysis reported in this paper describes our strategy to convert a well-known 5-HT2c serotonin receptor inhibitor into a promising antimalarial lead compound presenting a potent whole cell activity, fast killing profile, and no cross-resistance with chloroquine- and pyrimethamine-resistant strains. The next milestone will be to improve its in vivo efficacy.
Acknowledgments
We thank our colleagues from the Computational, Analytical and Structural Chemistry (CASC) department for the measurement of the physicochemical properties of compounds described in this paper, GSK Screening R&D group for the 5-HT2c assays, Steve Bromidge for his contribution in the understanding of the previous work carried out with the indolines, the Cell Biology Group for the in vitro toxicity studies, and Timothy J. Miles for his useful help.
Supporting Information Available
Experimental procedures and characterization data for the synthesis of all compounds and phenotypes for assays for compounds 14, 27, and 25. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Ferrer R&D Center C, Juan de Sada, 32, E-08028 Barcelona, Spain.
Author Present Address
∥ School of Chemistry, University of Edinburgh, Joseph Black Building, King's Buildings West Mains Road, Edinburgh, EH9 3JJ, United Kingdom.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
GlaxoSmithKline acknowledges financial support from Medicines for Malaria Venture (MMV).
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. Global Report on Antimalarial Drug Efficacy and Drug Resistance: 2000–2010; WHO Press: Geneva, Switzerland, 2010. [Google Scholar]
- Leroy D.; Doerig C. Drugging the Plasmodium Kinome: The Benefits of Academia–Industry Synergy. Trends Pharmacol. Sci. 2008, 29, 241–249. [DOI] [PubMed] [Google Scholar]
- Nishtar S. Public–Private “Partnerships” in Health—A Global Call to Action. Health Res. Policy Syst. 2004, 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Leroy D.; Lotharius J.; Waterson D. Challenges in Antimalarial Drug Discovery. Future Med. Chem. 2011, 3, 1401–1412. [DOI] [PubMed] [Google Scholar]
- Guiguemde W. A; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jimenez-Diaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; Mazouni F. E.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; DeRisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. 2008, 105, 9059–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V. S.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of Chemical Starting Points for Antimalarial Lead Identification. Nature 2010, 465, 305–312. [DOI] [PubMed] [Google Scholar]
- Calderón F.; Barros D.; Bueno J. M.; Coterón J. M.; Fernández E.; Gamo F. J.; Lavandera J. L.; León M. L.; Macdonald S. J. F.; Mallo A.; Manzano P.; Porras E.; Finador J. M.; Castro J. An Invitation to Open Innovation in Malaria Drug Discovery: 47 Quality Starting Points from the TCAMS. ACS Med. Chem. Lett. 2011, 2, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda L.; Castellote I.; Castro-Pichel J.; Chaparro M. J.; de la Rosa J. C.; García-Perez A.; Gordo M.; Jimenez-Díaz M. B.; Kessler A.; MacDonald S. J. F.; Santos Martínez M.; Sanz L. M.; Gamo F. J.; Fernandez E. Cyclopropyl Carboxamides: A New Oral Antimalarial Series Derived from the Tres Cantos Anti-Malarial Set (TCAMS). ACS Med. Chem. Lett. 2011, 2, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L. M..; Jiménez-Díaz M. B.; Crespo B.; De-Cozar C.; Almela M. J.; Angulo-Barturen I.; Castañeda P.; Ibañez J.; Fernández E.; Ferrer S.; Herreros E.; Lozano S.; Martínez M. S.; Rueda L.; Burrows J. N.; García-Bustos J. F.; Gamo F. J. Cyclopropyl Carboxamides, a Chemically Novel Class of Antimalarial Agents Identified in a Phenotypic Screen. Antimicrob. Agents Chemother. 2011, 55, 5740–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Lange B. B.; Merbitz-Zahradnik T.; Zwicker K.; Hill P.; Meunier B.; Pálsdottir H.; Hunte C.; Meshnick S.; Trumpower B. Molecular Basis for Atovaquone Binding to the Cytochrome bc1 Complex. J. Biol. Chem. 2003, 278, 31312–31318. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information.
- Humphrey P. P. A; Hartig P.; Hoyer D. A Proposed New Nomenclature of 5-HT Receptors. Trends Pharmacol. Sci. 1993, 14, 233–236. [DOI] [PubMed] [Google Scholar]
- Nelson D. L. The 5-HT2 Subfamily of Receptors: Pharmacological Challenges. Med. Chem. Res. 1993, 3, 306–316. [Google Scholar]
- Baxter G.; Kennett G. A.; Blaney F.; Blackburn T. 5-HT2 Receptor Subtypes: A Family Re-united?. Trends Pharmacol. Sci. 1995, 16, 105–110. [DOI] [PubMed] [Google Scholar]
- Bromidge S. M.; Dabbs S.; Davies D. T.; Malcolm Duckworth D.; Forbes I. T.; Ham P.; Jones G. E.; King F. D.; Saunders D. V.; Starr S.; Thewlis K. M.; Wyman P. A.; Blaney F. E.; Naylor C. B.; Bailey F.; Blackburn T. P.; Holland V.; Kennett G. A.; Riley G. J.; Wood M. D. J. Med. Chem. 1998, 41, 1598–1612. [DOI] [PubMed] [Google Scholar]
- Bromidge S. M.; Dabbs S.; Davies D. T.; Davies S.; Malcolm Duckworth D.; Forbes I. T.; Gaster L. M.; Ham P.; Jones G. E.; King, King F. D.; Mulholland K. R.; Saunders D. V.; Wyman P. A.; Blaney F. E.; Clarke S. E.; Blackburn T. P.; Holland V.; Kennett G. A.; Lightowler S.; Middlemiss D. N.; Trail B.; Riley G.; Wood M. D. J. Med. Chem. 2000, 43, 1123–1134. [DOI] [PubMed] [Google Scholar]
- Kennett G. A.; Lightowler S.; De Biasi V.; Stevens N. C.; Wood M. D.; Tulloch I. F.; Blackburn T. P. Effect of chronic administration of selective 5-hydroxytryptamine and noradrenaline uptake inhibitors on a putative index of 5-HT2C/2B receptor function. Neuropharmacology 1994, 33, 1581–1588. [DOI] [PubMed] [Google Scholar]
- Di Giovanni G.; De Deurwaerdere P.; Di Mascio M.; Di Matteo V.; Esposito E.; Spampinato U. Selective blockade of serotonin-2C/2B receptors enhances mesolimbic and mesostriatal dopaminergic function: A combined in vivo electrophysiological and microdialysis study. Neuroscience 1999, 91, 587–597. [DOI] [PubMed] [Google Scholar]
- Kalkman H. O. Is migraine prophylactic activity caused by 5-HT2B or 5-HT2C receptor blockade. Life Sci. 1994, 54, 641–644. [DOI] [PubMed] [Google Scholar]
- Fox S. H.; Moser B.; Brotchie J. M. Behavioural effects of 5-HT2C receptor antagonism in the substantia nigra zona reticulate of the 6-hydroxydopamine-lesioned rat model of Parkinson's disease. Exp. Neurol. 1998, 151, 35–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.