

Abstract
We identified a novel class of aryl-substituted triazine compounds as potent non-nucleoside reverse transcriptase inhibitors (NNRTIs) during a high-throughput screening campaign that evaluated more than 200000 compounds for antihuman immunodeficiency virus (HIV) activity using a cell-based full replication assay. Herein, we disclose the optimization of the antiviral activity in a cell-based assay system leading to the discovery of compound 27, which possessed excellent potency against wild-type HIV-1 (EC50 = 0.2 nM) as well as viruses bearing Y181C and K103N resistance mutations in the reverse transcriptase gene. The X-ray crystal structure of compound 27 complexed with wild-type reverse transcriptase confirmed the mode of action of this novel class of NNRTIs. Introduction of a chloro functional group in the pyrazole moiety dramatically improved hERG and CYP inhibition profiles, yielding highly promising leads for further development.
Keywords: HIV-1, reverse transcriptase, non-nucleoside reverse transcriptase inhibitors, synthesis, X-ray crystal structure
According to global estimates of the WHO/UNAIDS in 2009, more than 33 million people are infected with the human immunodeficiency virus (HIV), the causative agent of acquired immunodeficiency syndrome (AIDS), which currently accounts for the highest number of deaths by any single infectious agent.1 The global HIV/AIDS pandemic triggered intensive drug discovery efforts, and the first FDA-approved antiretroviral drug, Zidovudine (AZT), was available in 1987. Currently, 26 drugs belonging to six different inhibitor classes have been approved for the treatment of HIV infection: nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs/NtRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), integrase inhibitors (INIs), and entry (CCR5 coreceptor antagonist) and fusion inhibitors (FIs).2,2b Highly active antiretroviral therapy (HAART)—a regimen combining 3–4 antiretrovirals from different inhibitor classes—has dramatically improved the life quality of infected people by delaying the progression of the disease and reducing disabilities, transforming HIV/AIDS into a chronic manageable disease.3,4 NNRTIs that interact noncompetitively with an allosteric binding pocket in the vicinity of the reverse transcriptase's (RT's) polymerase active site are an important component of first-line regimens.5 Although there are five-FDA approved NNRTIs for clinical use (Figure 1), alternatives are still needed due to the tendency of HIV-1 to rapidly mutate. Prolonged HAART treatment leads to the emergence of drug-resistant viral mutants.6,6b Also, the undesired side effects of combination therapy have limited their clinical effectiveness.7,7b Therefore, further development of novel NNRTIs with acceptable toxicity and an improved drug resistance profiles is undoubtedly required.8
Figure 1.

Structures of the FDA-approved NNRTI drugs.
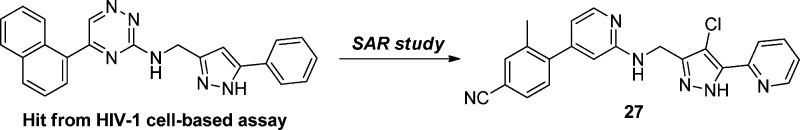
In a high-throughput screening campaign for the discovery of novel antiretrovirals employing a HIV full replication assay based on reporter cells harboring an EGFP expression cassette under the control of the HIV promoter, we identified hit compound 6 containing a triazine scaffold (Figure 2) that exhibited inhibitory activities against HIV replication at submicromolar concentrations. Compound 6 also showed moderate inhibitory activity in an in vitro RT polymerase assay, indicating that HIV RT is the potential target. Here, we report a detailed structure–activity relationship (SAR) study of this series of compounds that led to compound 27, which has excellent antiviral activity against wild-type (WT) and NNRTI-resistant HIV-1. Also, its X-ray cocrystal structure in complex with WT RT is included.
Figure 2.

Hit compound from the cell-based HIV-1 replication assay.
The target phenylaminopyridine (PAP) compounds (7–27 in Tables 1 and 2) were synthesized according to the general routes.9 The lead compound 27 was prepared efficiently in a convergent manner by coupling two subunits as depicted in Scheme 1. The formylated pyrazole 1s required for the reductive amination was prepared from 1-(pyridin-2-yl)ethanone. The addition of enolate to diethyl oxalate, followed by the cyclization with hydrazine for the pyrazole ring formation under refluxing condition, gave the desired ester 1o in 69% yield over two steps.10 Halogenation on the pyrazole with NCS produced chlorinated pyrazole 1p in good yield, and the resulting pyrazole was protected with THF group to give intermediate 1q. The ester moiety was then converted into the aldehyde 1s in a typical two-step reduction with LAH and oxidation with Dess-Martin reagent sequence. After the Suzuki coupling reaction to produce compound 1t, the resulting LHS subunit was then subjected to the final coupling step with compound 1s involving reductive amination, followed by deprotection of the THF group under acidic conditions to give the desired lead compound 27 in good yield.
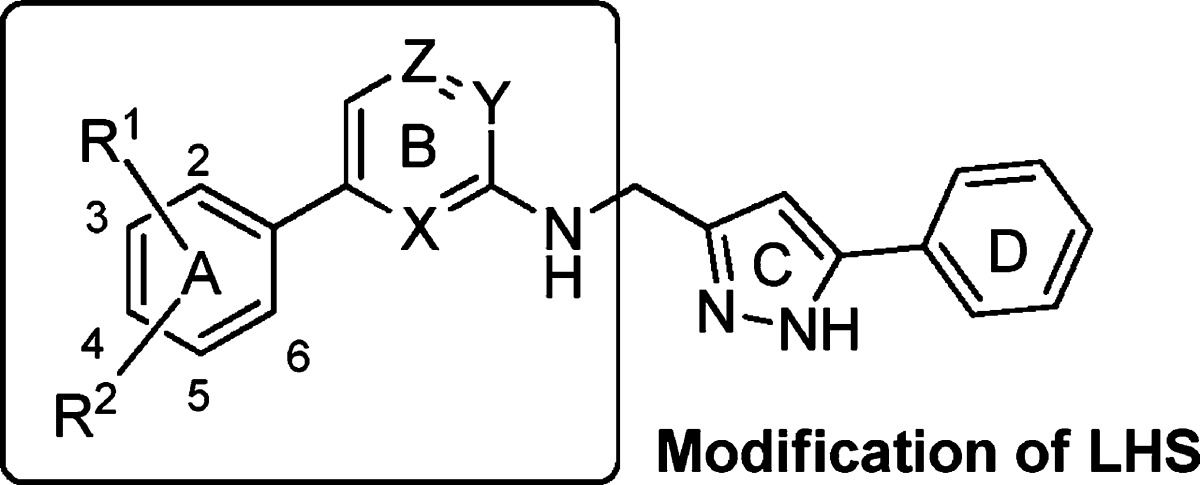
Table 1. Cell-Based Antiviral Activity of PAP Derivatives 7–21 with LHS Modifications.
| compd | R1 | R2 | X | Y | Z | EC50 (μM)a | CC50 (μM)b |
|---|---|---|---|---|---|---|---|
| 7 | H | H | N | N | N | 1.29 | >10 |
| 8 | 2-Me | H | N | N | N | 0.124 | >10 |
| 9 | 3-Me | H | N | N | N | 2.30 | >10 |
| 10 | 4-Me | H | N | N | N | >10 | >10 |
| 11 | 2-Me | 6-Me | N | N | N | 0.0032 | >10 |
| 12 | 2-Me | H | CH | CH | CH | 2.20 | >10 |
| 13 | 2-Me | H | CH | CH | N | 1.20 | >10 |
| 14 | 2-Me | H | N | CH | CH | >10 | >10 |
| 15 | 2-Me | H | CH | N | CH | 0.149 | >10 |
| 16 | 2-Me | H | N | N | CH | 0.366 | >10 |
| 17 | 2-Me | H | N | CH | N | 0.910 | >10 |
| 18 | 2-Me | 4-CN | N | N | N | 0.0017 | >10 |
| 19 | 2-Me | 4-CN | CH | CH | N | 0.016 | >10 |
| 20 | 2-Me | 4-CN | CH | N | CH | 0.0008 | >10 |
| 21 | 2-Me | 4-CN | N | N | CH | 0.0043 | >10 |
| NVPc | 0.150 | >10 |
EC50 is the concentration of compound that inhibits HIV-1 replication by 50%. For compounds 7–21, the values are the geometric mean of two determinations; all individual values are within 25% of the mean.
CC50 is the cytotoxic concentration of compound that reduces viability of uninfected cells by 50%.
Nevirapine (NVP) was used as a positive control.
Table 2. Cell-Based Antiviral Activity of PAP Derivatives 22–27 with RHS Modifications.
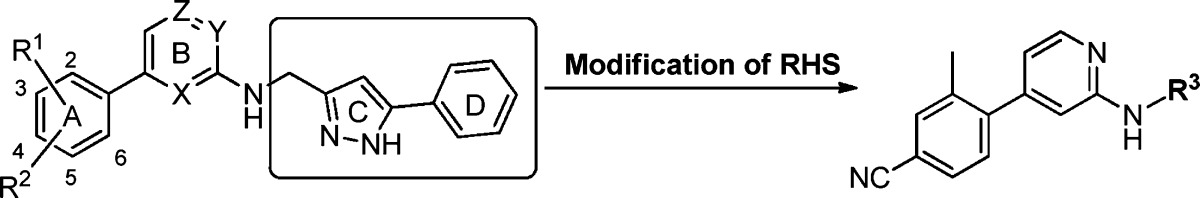
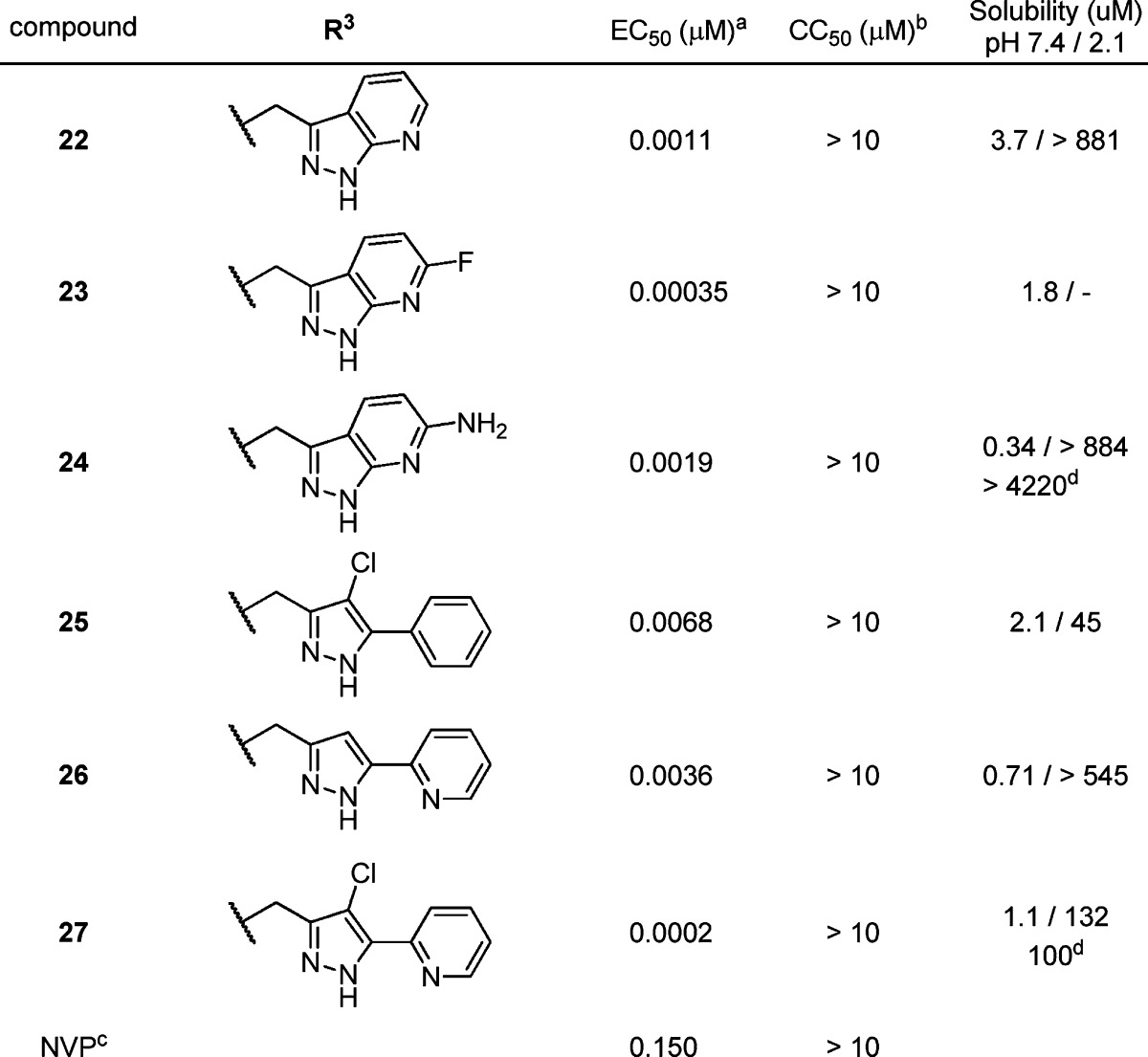
EC50 is the concentration of compound that inhibits HIV-1 replication by 50%. For compounds 22–27, the values are the geometric mean of two determinations; all individual values are within 25% of the mean.
CC50 is the cytotoxic concentration of compound that reduces viability of uninfected cells by 50%.
Nevirapine (NVP) was used as a positive control.
Solubility of HCl salt in distilled water.
Scheme 1. Synthesis of Compound 27.

Reagents and conditions: (a) (i) NaOEt, EtOH, diethyl oxalate, 25 °C, 20 h. (ii) H2NNH2, H2O/EtOH, reflux, 2 h, 69% over two steps. (b) NCS, CCl4, DMF, 55 °C, 15 h, 86%. (c) DHP, TFA, CH3CN/toluene, 100 °C, 6 h, 42%. (d) LAH, THF, 0 °C, 30 min, 58%. (e) Dess-Martin Periodinane, CH2Cl2, 25 °C, 3 h, 91%. (f) Pd(dppf)Cl2, Na2CO3, DME/H2O, 140 °C, 2 h, 70%. (g) Compound 1s, NaBH(OAc)3, AcOH, 1,2-dichloroethane, 25 °C, 24 h, 50%. (h) HCl, MeOH, 40 °C, 15 h, 82%.
The first series of LHS modifications (7–21) were evaluated for their inhibitory activity against HIV-1 WT replication in a cell-based assay, and Nevirapine was used as a positive control. Assay results of compounds (7–21) are summarized in Table 1.
As seen in Table 1, it was clear that the anti-HIV activities of PAP compounds are sensitive to structural perturbations. Compounds 7–10 with common triazine core B ring exhibited considerable variation in the inhibitory activity (EC50 = 0.124 μM to >10 μM). Ortho-substitution on the A ring markedly improved their potency, indicating that increasing the angle between the phenyl A ring and the triazine B ring is favorable. The effect of ortho-substitution is further exemplified by compound 11, which showed over 400-fold improved antiviral activity as compared to compound 7. We next investigated the effect of nitrogen atoms in the B ring in PAP derivatives 12–17, and the results showed a broad range of EC50 values. Compound 15 with a pyridine B ring is the most active among these analogues. However, the complete removal of nitrogen atoms from the B ring induced a significant reduction in inhibitory activity in compound 12. Pyrimidine 16 and pyrazine 17 derivatives displayed 2- or 6-fold reduced potencies as compared to compound 15. Further optimization on the A ring was based on a computational approach by overlaying with known NNRTIs (Etravirine and MK-4965). Gratifyingly, the introduction of a cyano group in the para-position on the A ring significantly enhanced HIV-1 inhibitory activity more than 75-fold in derivatives 18–21 as compared with derivatives lacking the cyano group (8, 13, 15, and 16).11 This dramatic effect was not seen in ortho- and meta- positions (data not shown). These results indicate that the binding mode of PAP derivatives might be similar to that of diarylpyrimidine (DAPY) compounds (e.g., etravirine 4, rilpivirine 5).12,13
After the evaluation of LHS subunits in the PAP scaffold, we investigated the modification of the RHS subunits by synthesizing derivatives 22–27, and the results are presented in Table 2. Structural modification on the RHS subunits was directed toward the improvement of physicochemical properties, especially solubility of this series by the introduction of fused heteroaryls or substituted pyrazoles with a polar moiety. Azaindazole 22–24 and pyrazole 25–27 derivatives maintained excellent HIV-1 inhibitory activities along with good solubility profiles. As shown in derivatives 24 and 27, the formation of HCl salt could be utilized to further increase absorption for pharmacokinetic (PK) studies.
Intrinsic enzymatic inhibitory activity against WT and key mutant RTs (K103N, Y181C) and also hERG inhibition profiles of PAP derivatives (22–27) are summarized in Table 3. In the enzymatic assay, the majority of the tested compounds except compound 26 showed single digit nanomolar IC50 values against WT RT and also displayed significant inhibitory activity against the K103N and Y181C mutated RTs, with only minimal changes in IC50 values with respect to WT RT. Results from hERG inhibition assays revealed the intriguing characteristics of PAP derivatives. Although rather moderate inhibition values were observed in azaindazoles 22–24, the halogenated pyrazole derivatives (25 and 27) exhibited acceptable safety margins, which could be beneficial for long-term treatment.14,14b
Table 3. HIV-1 RT Inhibitory Activity and hERG Inhibition of PAP Derivatives 22–27.
| IC50a nM (fold resistance)b |
||||
|---|---|---|---|---|
| compd | WT | K103N | Y181C | hERGc (μM) |
| 22 | 1.2 | 10 (8.3) | 5.6 (4.7) | 1.9 |
| 23 | 1.9 | 10 (5.3) | 7.7 (4.1) | 2.7 |
| 24 | 5.9 | 36 (6.1) | 16 (2.7) | 4.3 |
| 25 | 5.1 | 11 (2.2) | 6.3 (1.2) | 30 |
| 26 | 10 | 134 (13) | 84 (8.4) | 1.1 |
| 27 | 3.0 | 16 (5.3) | 7.9 (2.6) | >30 |
| MK-4965d | 3.0 | 32 (11) | 9.5 (3.2) | |
Compounds were evaluated in a standard SPA assay. Inhibition of RNA-dependent DNA polymerase activity using the WT, K103N, and Y181C polymerases. For compounds 22–27, the values are the geometric mean of two determinations; all individual values are within 25% of the mean. Assay protocols are detailed in the Supporting Information.
Fold resistance is defined by IC50 (mutant)/IC50 (WT).
hERG inhibition was measured in CHO cells stably expressing the recombinant human hERG channel subunit (IKr) using an automated patch clamp platform (QPatch-Sophion Biosciences).
MK-4965 was used as a positive control.15
The PK parameters of key compounds (24, 25, and 27) were measured in rats and are tabulated in Table 4. All tested compounds showed bioavailability above 70% and moderate clearance rates and half-lives. Notably, compound 27 exhibited an improved AUC value as compared to other analogues, which suggested that once daily dosing in humans might be achievable after further optimization. Also, the in vitro drug interaction profile showed that compound 27 was not inhibitory against a panel of five isoforms of the cytochrome P450 (CYP) enzymes and did not induce CYP 3A4 at concentrations up to 10 μM.16−17b
Table 4. PK Parameters for 24, 25, and 27 in Rat.
| compd | Vd (L/kg) | AUCpo (μM h) | F (%) | Cl [(mL/min)/kg] | t1/2 (h) |
|---|---|---|---|---|---|
| 24a | 1.26 | 20 | 72 | 14 | 2.5 |
| 25b | 2.08 | 34 | 71 | 8.7 | 3.1 |
| 27c | 1.10 | 67 | 127 | 8.2 | 2.0 |
iv dosing at 1.10 mg/kg and oral dosing at 10.7 mg/kg. iv and po formulation: 1.0 mg/mL in 50% PEG.
iv dosing at 1.19 mg/kg and oral dosing at 10.9 mg/kg. iv and po formulation: 1.0 mg/mL in 40% PEG.
iv dosing at 0.94 mg/kg and oral dosing at 9.44 mg/kg. iv and po formulation: 1.0 mg/mL in 20% HP-β-CD.
To understand the mode of action of PAP compounds, the lead compound 27 was subjected to cocrystallization with WT RT.18 Indeed, compound 27 was bound to the non-nucleoside reverse transcriptase inhibitor binding pocket (NNIBP) as shown in Figure 3. The X-ray crystal structure revealed that the RT-bound conformation of compound 27 resembled a U-shape, which is similar to the binding modes of Etravirine as well as MK-4965. This U-shape orientation ideally adapts a combination of torsional flexibility (“wiggling”) and rotational and translational shifts (“jiggling”) of the inhibitor within the binding pocket to have potency against WT and a wide range of drug-resistant HIV-1 RTs.19
Figure 3.
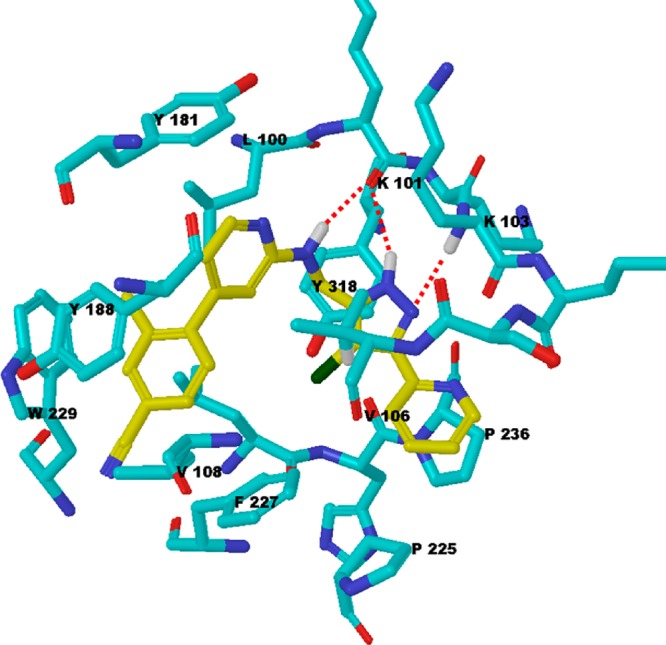
Cocrystal structure of HIV-1 WT RT with compound 27. Three H-bonds (red dotted line) were formed between residues K101 and K103 (atom colored blue stick) and compound 27 (atom colored yellow stick).
Compound 27 interacted favorably with NNIBP residues. The LHS subunit of compound 27 is positioned in the hydrophobic pocket surrounded by aromatic amino acids such as Y181, Y188, F227, and W229. The 4-cyano-2-methyl-phenyl A ring makes a π–π interaction with Y188 and an edge−π interaction with W229. The 2-methyl on the A ring orients toward the side chain of L100 contributing to additional hydrophobic interactions. The pyridine B ring makes van der Waals interactions with Y181. The nitrogen linker made a hydrogen bond (H-bond) interaction with the main chain carbonyl oxygen of K101. One additional H-bond was found between the same atom of K101 and one of the nitrogens in the chloropyrazole C ring as the RHS subunit was positioned in the flexible loop region. The other nitrogen in the pyrazole formed an H-bond with the main chain nitrogen of K103. The chloropyrazole C ring also makes van der Waals interactions with Y318, and the chloride of this ring is pointing in the same direction as the hydroxyl group of Y318. The pyridine D ring is surrounded by V106, P225, and P236 (Figure 3).
In summary, our search for novel anti-HIV compounds led to the discovery of a highly potent NNRTI with a PAP scaffold. The lead compound 27 possessed excellent antiviral activity against WT and key RT mutants. The binding mode of compound 27 was unambiguously confirmed by the X-ray cocrystal structure with WT RT. Introduction of a chloro functional group in the pyrazole moiety markedly improved hERG and CYP inhibition profiles. Altogether, the results presented here suggested that further development of this series has the potential to generate a valuable drug candidate for the treatment of HIV-1 infected patients.
Glossary
Abbreviations
- AIDS
acquired immunodeficiency syndrome
- HAART
highly active antiretroviral therapies
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- RT
reverse transcriptase
- SAR
structure–activity relationship
- WT
wild-type
- NNIBP
non-nucleoside reverse transcriptase inhibitor binding pocket
- PK
pharmacokinetic
Supporting Information Available
Experimental procedures for the synthesis and characterization of 27 and 7–27 and details for biological methods. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the National Research foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2012-00011), Gyeonggi-do and KISTI.
The authors declare no competing financial interest.
Supplementary Material
References
- Xu H.; Lv M. Developments of indoles as anti-HIV-1 inhibitors. Curr. Pharm. Des. 2009, 15, 2120–2148. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Highlights in the discovery of antiviral drugs: A personal retrospective. J. Med. Chem. 2010, 53, 1438–1450. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; De Clercq E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go?. J. Med. Chem. 2010, 53, 521–538. [DOI] [PubMed] [Google Scholar]
- Mocroft A.; Ledergerber B.; Katlama C.; Kirk O.; Reiss P.; d'Arminio Monforte A.; Knysz B.; Dietrich M.; Phillips A. N.; Lundgren J. D. Decline in the AIDS and death rates in the EuroSIDA study: An observational study. Lancet 2003, 362, 22–29. [DOI] [PubMed] [Google Scholar]
- Panos G.; Samonis G.; Alexiou V. G.; Kavarnou G. A.; Charatsis G.; Falagas M. E. Mortality and morbidity of HIV infected patients receiving HAART: A cohort study. Curr. HIV Res. 2008, 6, 257–260. [DOI] [PubMed] [Google Scholar]
- de Bethune M.-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antiviral Res. 2010, 85, 75–90. [DOI] [PubMed] [Google Scholar]
- Paredes R.; Clotet B. Clinical management of HIV-1 resistance. Antiviral Res. 2010, 85, 245–265. [DOI] [PubMed] [Google Scholar]
- Kiertiburanakul S.; Sungkanuparph S. Emerging of HIV Drug Resistance: Epidemiology, Diagnosis, Treatment and Prevention. Curr. HIV Res. 2009, 7, 273–278. [DOI] [PubMed] [Google Scholar]
- Esplugues J. V.; Blas-Garcia A.; Apostolova N. Twenty Years of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Time to Reevaluate their Toxicity. Curr. Med. Chem. 2011, 18, 2186–2195. [DOI] [PubMed] [Google Scholar]
- Hawkins T. Understanding and managing the adverse effects of antiretroviral therapy. Antiviral Res. 2010, 85, 201–209. [DOI] [PubMed] [Google Scholar]
- Li D.; Zhan P.; De Clercq E.; Liu X. Strategies for the Design of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Lessons from the Development of Seven Representative Paradigms. J. Med. Chem. 2012, 55, 3595–3613. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- Heller S. T.; Natarajan S. R. 1,3-Diketones from acid chlorides and ketones: A rapid and general one-pot synthesis of pyrazoles. Org. Lett. 2006, 8, 2675–2678. [DOI] [PubMed] [Google Scholar]
- Fleming F. F.; Yao L.; Ravikumar P. C.; Funk L.; Shook B. C. Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. Y.; Chen X. W.; Zhan P.; Li D. Y.; De Clercq E. Recent Advances in DAPYs and Related Analogues as HIV-1 NNRTIs. Curr. Med. Chem. 2011, 18, 359–376. [DOI] [PubMed] [Google Scholar]
- Lansdon E. B.; Brendza K. M.; Hung M.; Wang R.; Mukund S.; Jin D. B.; Birkus G.; Kutty N.; Liu X. H. Crystal Structures of HIV-1 Reverse Transcriptase with Etravirine (TMC125) and Rilpivirine (TMC278): Implications for Drug Design. J. Med. Chem. 2010, 53, 4295–4299. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Kazmierski W. M.; Anderson D. L.; Aquino C.; Chauder B. A.; Duan M.; Ferris R.; Kenakin T.; Koble C. S.; Lang D. G.; McIntyre M. S.; Peckham J.; Watson C.; Wheelan P.; Spaltenstein A.; Wire M. B.; Svolto A.; Youngman M. Novel 4,4-disubstituted piperidine-based C-C chemokine receptor-5 inhibitors with high potency against human immunodeficiency virus-1 and an improved human ether-a-go-go related gene (hERG) profile. J. Med. Chem. 2011, 54, 3756–3767. [DOI] [PubMed] [Google Scholar]
- Tucker T. J.; Sisko J. T.; Tynebor R. M.; Williams T. M.; Felock P. J.; Flynn J. A.; Lai M.-T.; Liang Y.; McGaughey G.; Liu M.; Miller M.; Moyer G.; Munshi V.; Perlow-Poehnelt R.; Prasad S.; Reid J. C.; Sanchez R.; Torrent M.; Vacca J. P.; Wan B.-L.; Yan Y. Discovery of 3-{5-[(6-Amino-1H-pyrazolo[3,4-b]pyridine-3-yl)methoxy]-2-chlorophenoxy}-5-chlorobenzonitrile (MK-4965): A Potent, Orally Bioavailable HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitor with Improved Potency against Key Mutant Viruses. J. Med. Chem. 2008, 51, 6503–6511. [DOI] [PubMed] [Google Scholar]
- Dickinson L.; Khoo S.; Back D. Pharmacokinetics and drug-drug interactions of antiretrovirals: An update. Antiviral Res. 2010, 85, 176–189. [DOI] [PubMed] [Google Scholar]
- Mills J. B.; Rose K. A.; Sadagopan N.; Sahi J.; de Morais S. M. Induction of drug metabolism enzymes and MDR1 using a novel human hepatocyte cell line. J. Pharmacol. Exp. Ther. 2004, 309, 303–309. [DOI] [PubMed] [Google Scholar]
- Ripp S. L.; Mills J. B.; Fahmi O. A.; Trevena K. A.; Liras J. L.; Maurer T. S.; de Morais S. M. Use of immortalized human hepatocytes to predict the magnitude of clinical drug-drug interactions caused by CYP3A4 induction. Drug Metab. Dispos. 2006, 34, 1742–1748. [DOI] [PubMed] [Google Scholar]
- The co-crystal structure was obtained from Proteros Biostructures; http://www.proteros.de/.
- Das K.; Clark A. D.; Lewi P. J.; Heeres J.; de Jonge M. R.; Koymans L. M. H.; Vinkers H. M.; Daeyaert F.; Ludovici D. W.; Kukla M. J.; De Corte B.; Kavash R. W.; Ho C. Y.; Ye H.; Lichtenstein M. A.; Andries K.; Pauwels R.; de Béthune M. -P.; Boyer P. L.; Clark P.; Hughes S. H.; Janssen P. A. J.; Eddy A. Roles of Conformational and Positional Adaptability in Structure-Based Design of TMC125-R165335 (Etravirine) and Related Non-nucleoside Reverse Transcriptase Inhibitors That Are Highly Potent and Effective against Wild-Type and Drug-Resistant HIV-1 Variants. J. Med. Chem. 2004, 47, 2550–2560. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.