

Abstract
NAD+-dependent histone deacetylases (sirtuins) play important roles in epigenetic regulation but also through nonhistone substrates for other key cellular events and have been linked to the pathogenesis of cancer, neurodegeneration, and metabolic diseases. The subtype Sirt5 has been shown recently to act as a desuccinylating and demalonylating enzyme. We have established an assay for biochemical testing of Sirt5 using a small labeled succinylated lysine derivative. We present a comparative study on the profiling of several established sirtuin inhibitors on Sirt1–3 as well as Sirt5 and also present initial results on a screening for new compounds that block Sirt5. Thiobarbiturates were identified as new Sirt5 inhibitors in the low micromolar range, which are selective over Sirt3 that can be found in the same cell compartment as Sirt5.
Keywords: sirtuin, Sirt5, desuccinylation, sirtinol, AMC assay
Sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent protein deacylases that are involved in many fundamental cellular processes. For some of them, adenosine diphosphate (ADP)-ribosyltransferase activity has also been shown.1−4 Inhibitors and activators of sirtuins have been proposed as potential new drugs for the treatment of a variety of diseases. Sirtuins have mainly been characterized as deacetylases, but recent examples show that selected subtypes are able to cleave off other acyl groups. This has been shown on small molecule substrates for the phenacetyl group5 but also in vivo for long chain aliphatic acids for the Plasmodium falciparum Sir2A.6 Especially interesting is the fact that Sirt5 recently has been shown to have demalonylase and desuccinylase activity in vitro and in vivo.7,8 Initial inhibitor studies have shown that thiosuccinylated peptides act as inhibitors9 similar to thioacetylated congeners for other sirtuins.10−12 Also, the standard sirtuin inhibitor suramin seems to possess some activity against Sirt5, which was shown in assays using acetylated, respectively, succinylated substrates.9,13 Because of the involvement of Sirt5 in primary metabolism, development of selective inhibitors of that isotype is an attractive area for characterizing the physiological function and therapeutical potential. In this paper, we present a modified in vitro assay that has been optimized for Sirt5 and results for profiling of a set of reference sirtuin inhibitors as well as initial results of a screening campaign.
Various assay formats have been used for the detection of Sirt5 activity and its inhibition in vitro. This involves the use of a small molecule lysine derivative,14 mass spectrometry,7 HPLC/absorbance,9 and 32P-NAD-thin-layer chromatography.7 We have synthesized an analogue of a known small molecule substrate, namely, Z-Lys(succ)-aminomethyl coumarin (AMC) {here termed 3-[(5-{[(benzyloxy)carbonyl]amino}-5-[(4-methyl-2-oxo-2H-chromen-7-yl)carbamoyl]pentyl)carbamoyl]propanoic acid (ZK(s)A)} (synthesis scheme and spectral data for intermediate compounds can be found in the Supporting Information).
We used a Z (CbZ) protecting group rather than a Boc derivative that has already been used by another group in a succinyl lysine derivative,14 allowing us a better comparison of the inhibition values of Sirt1–3, which have been obtained with the same inhibitors using an acetylated analogue as the substrate. We compared the ability of trypsin to cleave ZK(s)A and its desuccinylated metabolite benzyl N-{5-amino-1-[(4-methyl-2-oxo-2H-chromen-7-yl)carbamoyl]pentyl}carbamate (ZKA) and discovered that ZK(s)A was very stable even under a high trypsin concentration (1.5 mg/mL for over 20 min). In contrast, ZKA as shown before can easily be cleaved by trypsin, and the amount of released AMC can be measured because of the shift in fluorescence wavelength. It has been shown before that εN-modifications, like acetyl or succinyl on this kind of lysine substrates, prevent tryptic cleavage15,16 (see Scheme 1).
Scheme 1. Sirt5 Activity Assay Using a Small Molecule Substrate.

We used a concentration of 200 μM ZK(s)A and an enzyme concentration leading to a conversion of 5–10% in 1 h. Converting more than 5% of ZK(s)A to AMC led to a very stable and strong fluorescence signal, which could be measured at λEx = 390 nm and λEm = 460 nm in a microtiter plate reader.
The kinetic constants were measured by end-point determination using the same assay with ZK(s)A concentrations ranging from 1 to 1000 μM. The KM value for ZK(s)A was 17.4 ± 1.8 μM, and the Vmax was 5.3 × 10–3 μM. A published succinylated H3K9 peptide is reported to have a KM value of 5.8 ± 2.7 μM for Sirt5,7 whereas other AMC derivatives like H410–12-K(succ)-AMC or Ac-Lys(succ)-AMC showed slightly lower values with 33 ± 1.8 and 84 ± 22 μM, respectively.17
We then tested a variety of reference sirtuin inhibitors for Sirt5 inhibition and compared the results to the inhibition values for Sirt1–3 using the reported acetylated substrate. The reference inhibitors are Nicotinamide, Sirtinol,18 Cambinol,19 Suramin,20 EX-527 (Selisistat),21 a splitomicin derivative (HR 103),22 and Ro31-822015 (see Chart 1).
Chart 1. Standard Sirtuin Inhibitors.

As shown in Table 1, the relative potency of the reference compounds on Sirt5 varies. While Cambinol shows similar activity on Sirt5 as on Sirt1 and -2, the splitomicin derivative HR 103 seems to be selective on its major target Sirt2. Suramin is weaker on Sirt5 (in an assay using a nonfluorogenic peptide as a substrate, an IC50 of 25 μM was measured9) and Ro31-8220 that is potent on Sirt1–3 did not show an effect on Sirt5. Among all of the reference inhibitors, only Ro31-8220 shows significant potency against Sirt3.
Table 1. IC50 Values of Standard Sirtuin Inhibitors (μM): Inhibitory Values for Sirt1–3 from Ref (23) and Sirt5 from This Study.
| compd | Sirt1 | Sirt2 | Sirt3 | Sirt5 |
|---|---|---|---|---|
| nicotinamide | 50–100 | 1.2–100 | 30 | 46.6 ± 3.0 |
| sirtinol | 37–131 | 38–58 | 24% at 50 μM | 48.9 ± 6.3 |
| cambinol | 56 | 59 | NI | 42.5 ± 1.5 |
| suramin | 0.3–2.6 | 1.1–20 | NI | 46.6 ± 4.8 |
| EX-527 | 0.1–1 | 20–33 | 49 | NI |
| HR 103 (5a in lit.22) | NI | 1–5 | NI | NI |
| Ro 31-8220 | 3.5 | 0.8 | 3.7 | NI |
We then screened our internal library of thiourea inhibitors, which mainly contains thiobarbiturates. The results are presented in Table 2, and structures can be found in Chart 2.
Table 2. IC50 Values of Thiobarbiturate Compounds.
| compd | Sirt1 | Sirt2 | Sirt3 | Sirt5 |
|---|---|---|---|---|
| 1 | 3.4 ± 0.3 | 10.4 ± 3.0 | 30% at 50 μM | 3.6 ± 0.2 |
| 2 | 5.3 ± 0.7 | 9.7 ± 1.6 | 41% at 50 μM | 2.3 ± 0.2 |
| 3 | 5.9 ± 0.3 | 20.3 ± 1.5 | 14% at 50 μM | 46.5 ± 8.5 |
| 4 | 42.4 ± 7.2 | 20% at 50 μM | 17% at 50 μM | 27.0 ± 4.0 |
| 5 | 89.3 ± 11.3 | 10% at 50 μM | NI at 50 μM | 55.9 ± 6.2 |
| 6 | 28.4 ± 1.44 | 20% at 50 μM | 116.3 ± 10.1 | 16.6 ± 0.4 |
| 7 | 10.5 ± 0.4 | 9.8 ± 0.8 | 29.3 ± 3.2 | 12.6 ± 0.4 |
| 8 | 56.5 ± 3.4 | 10.0 ± 1.3 | 22% at 50 μM | 17.8 ± 1.2 |
| 9 | 53.2 ± 1.6 | 14.4 ± 2.3 | 25% at 50 μM | 12.9 ± 0.3 |
| 10 | 36.8 ± 2.3 | NI at 50 μM | 13% at 50 μM | 67.3 ± 5.9 |
| 11 | 9.9 ± 0.7 | 3.4 ± 0.8 | 30.3 ± 2.2 | 6.2 ± 0.8 |
| 12 | 6.7 ± 1.1 | 7.5 ± 0.9 | 46.4 ± 4.6 | 12.4 ± 5.1 |
| 13 | 50.5 ± 9.1 | 8.7 ± 0.7 | 40.3 ± 3.7 | 30.0 ± 4.1 |
| 14 | 12.4 ± 0.5 | 14.7 ± 2.1 | 13% at 50 μM | 39.4 ± 12.2 |
| 15 | 6.0 ± 0.6 | 11.7 ± 2.5 | 43.3 ± 5.0 | 22.4 ± 3.8 |
Chart 2. Sirtuin Inhibitors with a Thiobarbiturate Scaffold.

Several thiobarbiturates showed inhibition of Sirt5 in the micromolar region, usually with similar potency than for Sirt1 and -2. These inhibitors were generally less potent on Sirt3, but some of them showed an inhibition of Sirt3, which was rarely seen among the reference inhibitors. Inhibitor 6 shows the best selectivity for Sirt5 and might be used as a lead structure for further selectivity optimizations.
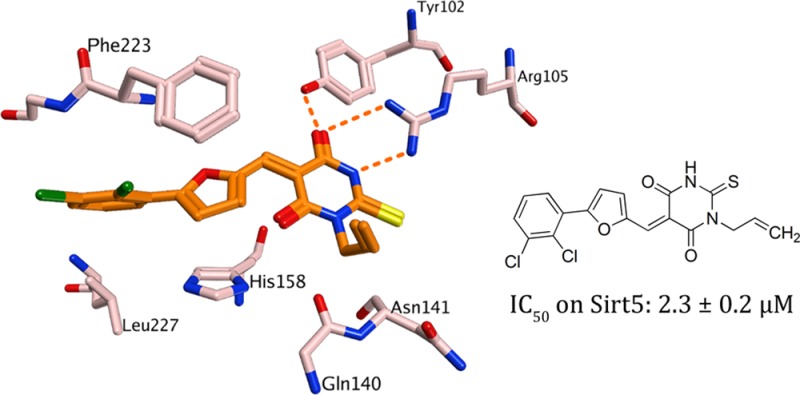
To rationalize our findings for further optimization studies, docking studies were carried out using the available X-ray structures of Sirt5.7 The docking showed that the thiobarbiturate ring is mimicking the succinyl group of the substrate and shows hydrogen bonds with Tyr102, Arg105, and Gln140 (Figure SI1 in the Supporting Information). The interaction is stabilized by strong electrostatic interactions between acidic thiobarbiturate and basic guanidinium group of Arg105 (exemplarily shown for the most active thiobarbiturate 2 in Figure 1). This binding mode was observed for all docked thiobarbiturates (see Figure SI2 in the Supporting Information). Thiobarbiturates containing a flexible side chain and an aromatic ring interact with aromatic residues located in the lysine substrate channel of Sirt5 (His158 and Phe223), thus resulting in high inhibitory activity. The docking results rationalized the finding that substituting the thiobarbituric −NH– function by methyl, ethyl, or allyl groups is tolerated. These can be accommodated by the binding pocket but result in slightly different orientation of the inhibitor (see Figure SI2 in the Supporting Information). Still, similar inhibition of Sirt2 is observed while this N-alkylation leads to an increased potency as compared to Sirt3 and partially also Sirt1 (compare 7 vs 8 and 9).
Figure 1.
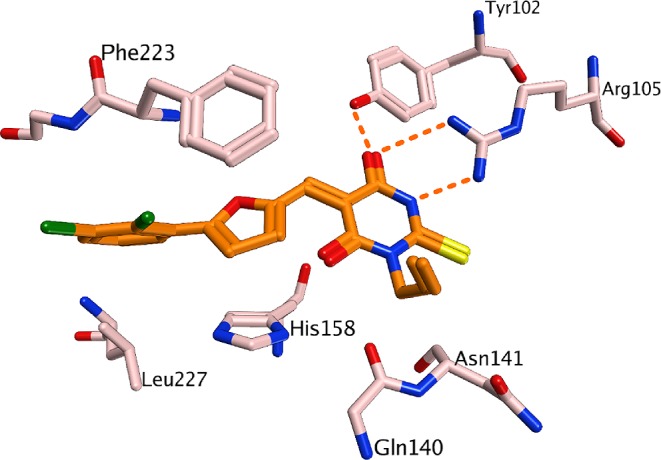
Docking result for 2 at Sirt5. Hydrogen bonds are shown as dashed orange lines.
With ZK(s)A, we have synthesized a small substrate that works well for Sirt5 and allows for high-throughput screening to discover Sirt5 inhibitors. Among sirtuin reference inhibitors, Suramin, Sirtinol, and Cambinol did not show selectivity toward the different sirtuins, while EX-527 is Sirt1 selective also in comparison to Sirt5, and HR 103 works best on Sirt2. We also tested a library of Sirt inhibitors with a thiobarbiturate scaffold on Sirt5 and discovered several potent hits with activity in the low micromolar region, and some of them inhibit Sirt5 better than the other subtypes. With N-alkylation, we have identified a structural feature that favors Sirt5 inhibition. As Sirt3 and Sirt5 are present in the same compartment of the cell (mitochondria), the observed difference in affinity among these is especially of interest. These results in view of the docking analyses may guide further development of optimized inhibitors of Sirt5.
Experimental Procedures
The Supporting Information contains the procedures for the expression and purification of recombinant sirtuins and the synthesis of the precursors of ZK(s)A. Compounds 1, 3–5, 8–11, and 13–15 were synthesized in our lab according to the Supporting Information. Compounds 2, 6, 7, and 12 were ordered from Chembridge (San Diego, CA) (Chembridge and lab-internal numbers can be found in Table SI1 in the Supporting Information). High analytical grade chemicals and solvents were purchased from Acros, Aldrich, Sigma, or Fluka. When necessary, solvents were dried by standard techniques and distilled. After extraction from aqueous layers, the organic solvents were dried over anhydrous sodium sulfate.
Thin-layer chromatography (TLC) was performed on aluminum sheets precoated with silica gel 60 F254 (0.2 mm) (E. Merck). Chromatographic spots were visualized by UV light. Purification of crude compounds was carried out by flash column chromatography on Merck Silica Gel 60 (Kieselgel, 0.040–0.063 mm, E. Merck). 1H NMR spectra were recorded in DMSO-d6 on a Bruker Avance DRX 400 MHz spectrometer and 13C NMR on a Varian 100 MHz. Chemical shifts (δ scale) are reported in parts per million (ppm) relative to the central peak of the solvent. Coupling constant (J values) are expressed in Hertz (Hz). Spin multiplicities are given as s (singlet), b s (broad singlet), d (doublet), t (triplet), dd (double doublet), dt (double triplet), or m (multiplet). EI- and CI-mass spectra were measured with a TSQ700 mass spectrometer (Thermoelectron). ESI- and PCI-mass spectra were recorded with a LCQ-Advantage mass spectrometer. In all cases, spectroscopic data are in agreement with known compounds and assigned structures.
Compound ZKA (see SI4 in the Supporting Information) (300 mg, 0.69 mmol) was solved at 0 °C in 10 mL of dry THF, together with diisopropylethylamine (DIPEA) (249 μL, 1.37 mmol). Succinic anhydride (82 mg, 0.82 mmol) was added portionwise, and the reaction was stirred for 1 h at the same temperature, then left overnight at room temperature. When TLC indicated complete consumption of the starting material, the reaction mixture was evaporated, and the residue was solved in dichloromethane (DCM) and extracted by a 5% aqueous solution of NaHCO3. The aqueous phase was collected and acidified (pH ∼ 2), then poured into a separating funnel containing DCM, and extracted. The organic layer was dried over MgSO4, filtered, and evaporated to give a crude reaction mixture, which was purified by flash column chromatography on silica gel with 5% methanol in DCM as the eluent to furnish the titled compound ZK(s)A (Scheme 1) as a white solid (280 mg, 76%): mp 148 °C. 1H NMR (DMSO-d6) δ: 12.02 (bs, 1H), 10.52 (s, 1H), 7.83 (t, J = 5.4 Hz, 1H), 7.78 (d, J = 1.6 Hz, 1H), 7.73 (d, J = 8.7 Hz, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.51 (dd, J = 1.6, 8.7 Hz, 1H), 7.40–7.15 (m, 5H), 5.04 (s, 2H), 4.19–4.08 (m, 1H), 3.05–2.98 (m, 2H), 2.44–2.33 (m, 5H), 2.27 (t, J = 7.5 Hz, 2H), 1.75–1.56 (m, 2H), 1.47–1.26 (m, 4H). 13C NMR (DMSO-d6) δ: 174.3, 172.4, 171.2, 160.5, 156.6, 154.1, 153.5, 142.7, 137.4, 128.8, 128.3, 128.2, 126.3, 115.7, 115.5, 112.7, 106.1, 65.9, 56.0, 38.7, 31.7, 30.5, 29.7, 29.2, 23.4, 18.4. LRMS (ESI) m/z 538 [M + H]+.
Inhibition of recombinant Sirt1/2/3 was determined using a homogeneous fluorescence deacetylase assay.16 Sirt1/2/3 was mixed with the fluorescent substrate ZMAL (assay concentration, 10.5 μM), NAD+ (500 μM), the inhibitor, DMSO [5% (v/v)], and assay buffer (25 mM Tris·HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0) to a volume of 60 μL, incubated for 4 h (37 °C, 180 rpm), then treated with 60 μL of trypsin solution [50 mM Tris·HCl, 100 mM NaCl, 8 mM nicotinamide, 5.5 U/μL trypsin, 6.7% DMSO (v/v)] and incubated (37 °C, 180 rpm, 20 min). The fluorescence intensity was then measured with a microplate reader (BMG Polarstar, λEx = 390 nm, λEm = 460 nm). To ensure initial state conditions, the conversion of ZMAL was adjusted to 10–30% substrate conversion without inhibitor. A mixture with only DMSO was used as the control. Inhibition rates were calculated in reference to the DMSO control. All inhibition experiments were run at least three times independently. IC50 values were determined with Graphpad Prism 4.0 software (La Jolla, CA).
Sirt5 was mixed with the fluorescent substrate ZK(s)A (assay concentration, 200 μM), NAD+ (500 μM), the inhibitor, DMSO [<2% (v/v)], and assay buffer (25 mM Tris·HCl, 130 mM NaCl, 3 mM KCl, 1 mM MgCl2, pH 8.0, and 0.1% PEG8000) to a volume of 51 μL, incubated for 1 h (37 °C, 750 rpm), then treated with 10 μL of trypsin solution (50 mM Tris·HCl, 130 mM NaCl, and 6 mg/mL trypsin) and incubated (37 °C, 1250 rpm, 2 min). The fluorescence intensity was then measured with a microplate reader (BMG Polarstar, λEx = 390 nm, λEm = 460 nm). To ensure initial state conditions, the conversion of ZK(s)A was adjusted to 4–10% substrate conversion without inhibitor. A mixture with only DMSO was used as the control. Inhibition rates were calculated in reference to the DMSO control. All inhibition experiments were run at least two times independently. IC50 values were determined with Graphpad Prism 4.0 software.
Glossary
Abbreviations
- ADP
adenosine diphosphate
- AMC
aminomethyl coumarin
- NAD+
nicotinamide adenine dinucleotide
- ZKA
benzyl N-{5-amino-1-[(4-methyl-2-oxo-2H-chromen-7-yl)carbamoyl]pentyl}carbamate
- ZK(s)A
3-[(5-{[(benzyloxy)carbonyl]amino}-5-[(4-methyl-2-oxo-2H-chromen-7-yl)carbamoyl]pentyl)carbamoyl]propanoic acid
- TLC
thin-layer chromatography
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
Supporting Information Available
Description of computational methods and docking results, recombinant sirtuin expression and purification, additional synthetic procedures and spectral data, and HPLC purity of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank the Deutsche Forschungsgemeinschaft (Ju295/8-1, Si868/6-1) and the EU (Nr. 241865-FP7 Health) for funding and A. Walter for supporting inhibitor synthesis.
The authors declare no competing financial interest.
Author Present Address
∥ Freiburg Institute of Advanced Studies (FRIAS), Universität Freiburg, Germany.
Supplementary Material
References
- Tanny J. C.; et al. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 1999, 997735–745. [DOI] [PubMed] [Google Scholar]
- Frye R. A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 2601273–279. [DOI] [PubMed] [Google Scholar]
- Haigis M. C.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 1265941–954. [DOI] [PubMed] [Google Scholar]
- Van Meter M.; et al. Repairing split ends: SIRT6, mono-ADP ribosylation and DNA repair. Aging (Albany NY) 2011, 39829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heltweg B.; et al. Subtype selective substrates for histone deacetylases. J. Med. Chem. 2004, 47215235–5243. [DOI] [PubMed] [Google Scholar]
- Zhu A. Y.; et al. Plasmodium falciparum Sir2A Preferentially Hydrolyzes Medium and Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2012, 71155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 3346057806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey M. D. Old enzymes, new tricks: sirtuins are NAD(+)-dependent de-acylases. Cell Meta. 2011, 146718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B.; Du J.; Lin H. Thiosuccinyl peptides as sirt5-specific inhibitors. J. Am. Chem. Soc. 2012, 13441922–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatkins D. G.; Monnot A. D.; Zheng W. Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg. Med. Chem. Lett. 2006, 16143651–3656. [DOI] [PubMed] [Google Scholar]
- Smith B. C.; Denu J. M. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 2007, 465014478–14486. [DOI] [PubMed] [Google Scholar]
- Hawse W. F.; et al. Structural insights into intermediate steps in the Sir2 deacetylation reaction. Structure 2008, 1691368–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A.; et al. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure 2007, 153377–389. [DOI] [PubMed] [Google Scholar]
- Peng C.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics 2011, 1012M111012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp J.; et al. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J. Med. Chem. 2006, 49257307–7316. [DOI] [PubMed] [Google Scholar]
- Heltweg B.; Trapp J.; Jung M. In vitro assays for the determination of histone deacetylase activity. Methods 2005, 364332–337. [DOI] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. Substrates for Efficient Fluorometric Screening Employing the NAD-Dependent Sirtuin 5 Lysine Deacylase (KDAC) Enzyme. J. Med. Chem. 2012, 55115582–5590. [DOI] [PubMed] [Google Scholar]
- Grozinger C. M.; et al. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 2764238837–38843. [DOI] [PubMed] [Google Scholar]
- Heltweg B.; et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006, 6684368–4377. [DOI] [PubMed] [Google Scholar]
- Howitz K. T.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 4256954191–196. [DOI] [PubMed] [Google Scholar]
- Napper A. D.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48258045–8054. [DOI] [PubMed] [Google Scholar]
- Neugebauer R. C.; et al. Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J. Med. Chem. 2008, 5151203–1213. [DOI] [PubMed] [Google Scholar]
- Lawson M.; et al. Inhibitors to understand molecular mechanisms of NAD(+)-dependent deacetylases (sirtuins). Biochim. Biophys. Acta 2010, 179910–12726–739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.