

Abstract

A new class of HCV NS3/4a protease inhibitors containing a P2 to P4 macrocyclic constraint was designed using a molecular modeling-derived strategy. Building on the profile of previous clinical compounds and exploring the P2 and linker regions of the series allowed for optimization of broad genotype and mutant enzyme potency, cellular activity, and rat liver exposure following oral dosing. These studies led to the identification of clinical candidate 15 (MK-5172), which is active against genotype 1–3 NS3/4a and clinically relevant mutant enzymes and has good plasma exposure and excellent liver exposure in multiple species.
Keywords: hepatitis C, HCV, MK-5172, macrocycle, genotype 3a, mutant enzymes
Hepatitis C virus (HCV) is a chronic liver infection that affects an estimated 130–170 million people worldwide.1,2 HCV displays a high degree of genetic heterogeneity and can be classified into six major genotypes with different geographic distributions: genotypes 1, 2, and 3 account for more than 90% of the infections in the developed world. Treatment for HCV is based on combination therapy with pegylated interferon-α and ribavirin.3 Sustained viral response is seen in ∼45% of HCV genotype 1-infected patients treated for 48 weeks and in ∼80% of genotype 2- and 3-infected patients treated for 24 weeks. Interferon and ribavirin therapy is also associated with a number of serious side effects, limiting the number of patients who may be treated.4
There is a compelling medical need for new oral therapeutic agents with improved efficacy and tolerability. Several promising antiviral targets for HCV have emerged,5 with NS3/4a protease inhibitors showing perhaps the most dramatic antiviral effects.6 Clinical proof of concept for this mechanism was first achieved with BILN-2061.7 Other compounds have entered clinical trials, including telaprevir8 and boceprevir,9 both of which are now marketed treatments for use in combination with a standard of care. Compounds currently in development include TMC-43510 and ITMN-191.11
We have disclosed a molecular modeling-derived strategy that led us to design HCV NS3/4a protease inhibitors that contain the P2 to P4 macrocyclic constraint.12 This design arose from an analysis of the crystal structure of full-length NS3/4A with and without inhibitors docked in the active site.13 Our strategy coupled with a modular synthetic approach, which relies on a key ring-closing metathesis (RCM) reaction,14 allowed for the rapid exploration of these molecules and the identification of clinical candidates, vaniprevir (1)15,16 and MK-1220 (2).17 Herein, we describe the discovery of a clinical candidate with broad activity across genotypes (gt) and resistant HCV variants.18,19
With the development of vaniprevir progressing, we set a goal for the ongoing discovery program to be the identification of a second generation NS3/4a protease inhibitor. We wanted to maintain or improve the PK profile seen with our previous compounds and make significant improvements in activity against the gt 3a enzyme as well as key clinically relevant gt 1 mutant enzymes. Looking back at the gt 3a enzyme potency in previous series led us to the following observation: a consistent trend for increasing gt 3a activity was observed with the addition of a large substituent to the P2 heterocycle. For example, the gt 3a activity of compound 3 (Ki = 200 nM, Table 1) was improved by approximately 4-fold with the addition of a phenyl group to give closely related compound 4 (Ki = 45 nM). On the basis of this observation, we undertook a systematic investigation of substituents at this position of the P2 quinoline, which will be published elsewhere. As part of this investigation, we also made a small number of fused ring analogues exemplified by tricylic compound 5, which exhibited greatly improved gt 3a enzyme activity (Ki = 12 nM). This represented a 16-fold increase in gt 3a potency over bicyclic compound 3 and showed that a fused ring could give results similar to those seen with quinoline substituents. This increase in potency was accompanied by an increase in rat liver exposure to 96 μM at the 4 h time point versus 21 μM for compound 3.
Table 1. In Vitro Activity.
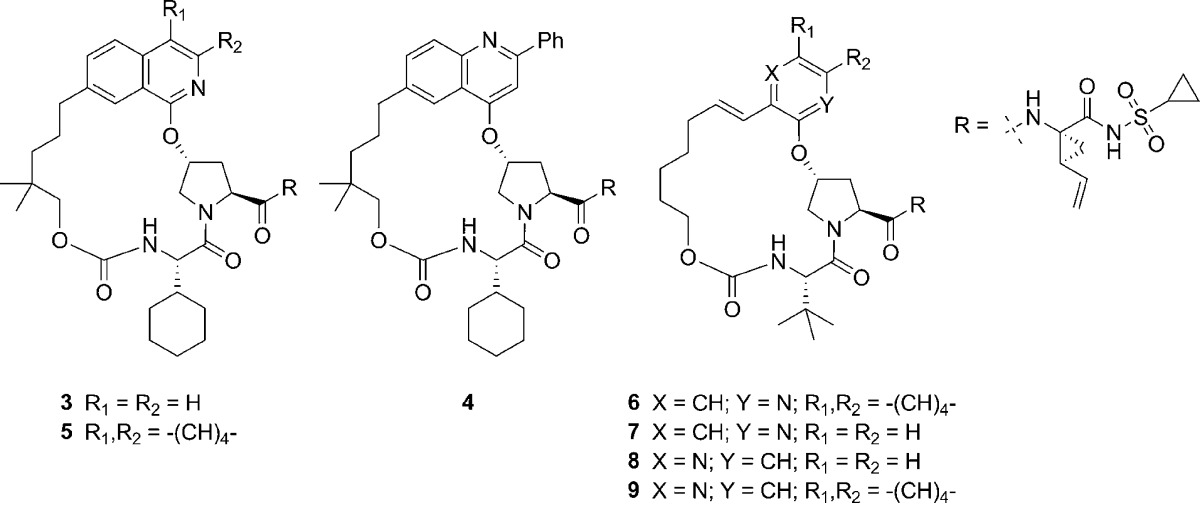
|
Ki (nM)a |
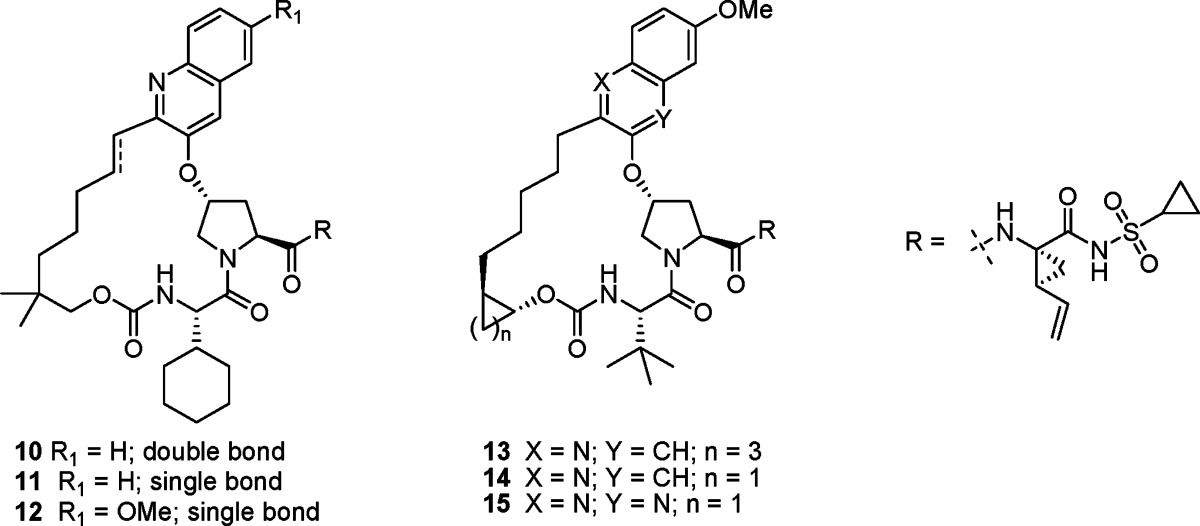
||||
|---|---|---|---|---|
| compd | 1b | 3a | 1b replicon IC50 50% NHS (nM)b | rat [liver] at 4 h (μM)c |
| 3 | 0.02 | 200 | 10 | 21 |
| 4 | 0.05 | 45 | 19 | 7.8 |
| 5 | 0.05 | 12 | 19 | 96 |
| 6 | 0.06 | 130 | 21 | 25 |
| 7 | 1.1 | 1200 | ND | ND |
| 8 | 1.9 | 520 | ND | ND |
| 9 | 0.08 | 26 | 44 | 0.6 |
NS3/4a protease time-resolved fluorescence assay, mean of n ≥ 3 measurements.
Cell-based replicon assay, mean of n ≥ 3.
Liver levels from PK experiments described in the Supporting Information.
Compounds 3–5 were docked in both the gt 1 and the gt 3a NS3/4a active site via molecular modeling in an effort to explain this observed increase in gt 3a activity. Figure 1 shows the three compounds docked in the gt 1b active site, and the major differences between the two genotypes are highlighted in cyan. All three compounds adopt similar conformations as expected, but it is clear that both compounds 4 and 5 extend further into the S2 pocket of the active site than compound 3. Furthermore, this pocket is a conserved region between gt 1 and gt 3a, leading us to hypothesize that increasing contacts with the conserved regions of the active sites of the two genotypes should lead to increases in activity at gt 3a without causing a loss in potency versus gt 1. It is also clear from the modeling that the P2 to P4 linker falls into a region of the active site that is not conserved between genotypes and is also the region where two important gt 3a polymorphisms occur. The docked poses of 4 and 5 suggest that the addition of a larger P2 substituent may also bias the macrocyclic ring to shift toward these residues, possibly improving complementarity to the gt 3a site, especially with respect to the R123T polymorph, which presumably leads to a significantly different active site surface in this region. In addition to the D168Q difference between gt 1b and gt 3a, D168 and R155 mutations are observed in the gt 1b enzyme in clinical studies, and these mutations cause potency shifts against both vaniprevir and MK-1220 (1 and 2, see Table 2). This second observation led us to hypothesize that removing steric bulk from the P2 region of the molecule as well as increasing the flexibility of our P2 to P4 macrocylic linker may allow for further improvements in gt 3a activity and mutant profile.
Figure 1.
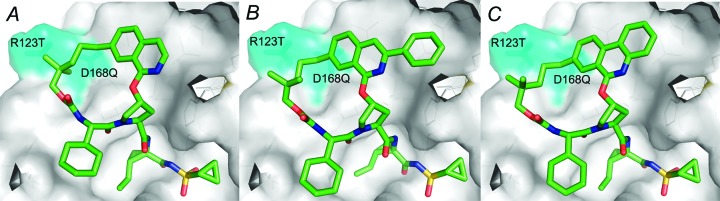
Compounds 3 (A), 4 (B), and 5 (C) docked in the gt 1b NS3/4a active site. Cyan = areas of diversity between the gt 1b and gt 3a enzymes. White = conserved areas.
Table 2. In Vitro Activity.
|
Ki (nM)a |
||||||||
|---|---|---|---|---|---|---|---|---|
| compd | 1b | 3a | 1b R155K | 1b A156T | 1b A156V | 1b D168V | 1b replicon IC50 50% NHS (nM)b | rat [liver] at 4 h (μM)c |
| 1 | 0.05 | 54 | 19 | 22 | 88 | 77 | 19 | 9.9 |
| 2 | 0.09 | 70 | 15 | 540 | 790 | 27 | 11 | 23 |
| 10 | 0.02 | 2.8 | 0.32 | 18 | 79 | ND | 37 | 1.5 |
| 11 | 0.02 | 4.0 | 0.20 | 27 | 80 | ND | 18 | 63 |
| 12 | 0.02 | 1.9 | 0.16 | 12 | 64 | ND | 28 | 9.8 |
| 13 | 0.02 | 0.39 | 0.07 | 2.4 | 11 | 0.06 | 16 | 6.3 |
| 14 | 0.02 | 0.31 | 0.02 | 2.9 | 5.7 | 0.05 | 13 | 20 |
| 15 | 0.02 | 0.70 | 0.07 | 5.3 | 12 | 0.14 | 7.4 | 23 |
NS3/4a protease time-resolved fluorescence assay, mean of n ≥ 3 measurements.
Cell-based replicon assay, mean of n ≥ 3.
Liver levels from PK experiments described in the Supporting Information.
To test these hypotheses, compound 6, a bicyclic analogue of 5, was targeted. This compound was designed by removing two of the carbons of the tricylic system, effectively lengthening the P2 to P4 linker by two carbons and maintaining the interaction with the conserved pocket, which seems to enhance gt 3a activity. Compound 6 maintained excellent gt 1 activity (Ki = 0.06 nM) and liver exposure (25 μM) and showed good gt 3a (Ki = 130 nM) activity considering that a large structural change had been made. This result with compound 6 led us to reconsider results with some compounds made early in the program. Compounds 7 and 8 have simple 2-pyridyl and 3-pyridyl substituents in the P2 position and do not have the gt 1 potency seen in advanced NS3/4a inhibitors. However, when just the relative potency of these compounds is considered, compound 8 seems to have an advantage with regard to gt 3a potency. Moving the nitrogen from the 2-pyridyl position (7) to the 3-pyridyl position (8) leads to a 2-fold reduction in potency vs gt 1b (Ki = 1.1 vs 1.9 nM) but also leads to a 2-fold increase in potency against the gt 3a enzyme (Ki = 1200 vs 520 nM). On the basis of modeling, this result is difficult to explain, as there does not seem to be a specific hydrogen-bonding interaction associated with the pyridyl nitrogen in either position. This led us to hypothesize a more favorable electrostatic interaction with R155 in the case of compound 8 or a change in hydrogen bonding to active site water molecules. On the basis of the results with 7 and 8, we performed this same nitrogen switch on bicyclic 2-quinoline 6, giving 3-quinoline 9, which leads to a 5-fold increase in gt 3a potency (Ki = 26 nM). Although liver exposure was markedly reduced (4 h liver exposure: 9 = 0.6 μM vs 6 = 26 μM), compound 9 emerged as a key lead for the second generation program, and the focus became improving liver exposure as well as evaluation of potency vs gt 1b mutant enzymes observed in clinical studies with vaniprevir.20
We previously observed that increasing lipophilicity in the P2 to P4 macrocylic series can lead to increases in liver exposure.15,17 Adding a dimethyl linker substituent to compound 9 gave 10 and led to only a slight increase in liver exposure (Table 2). This compound, however, had an improved Ki of 2.8 nM against the gt 3a enzyme, representing an almost 20-fold improvement over both of our previous clinical candidates, 1 (gt 3a Ki = 54 nM) and 2 (gt 3a Ki = 70 nM). Further profiling of 10 showed that activity against the key R155K mutant (Ki = 0.32 nM) had been improved by almost 50-fold in comparison to both 1 (gt 1b R155K Ki = 19 nM) and 2 (gt 1b R155K Ki = 15 nM). The activity of 10 vs the A156 mutants was improved by 10–30-fold in relation to compound 2 but was comparable to that of compound 1. Saturation of the linker olefin of 10 gave 11, which had a similar potency profile but showed a 40-fold increase in rat liver exposure to 63 μM after a 5 mg/kg oral dose at 4 h. These dramatic changes in rat liver exposure have been observed throughout the program and highlight the need for a rat liver screening strategy that we have adopted.15,17 Substituent effects were studied on the quinoline scaffold, and a 7-methoxy substituent (12) was optimal for activity, giving slight increases in potency against gt 3a and 1b mutants R155K, A156T and A156V.
In general, increasing potency against the A156 mutants was very difficult, because as seen in Figure 2, the P2–P4 macrocyles sit almost directly on top of this residue. The activity against R155 and D168 mutants was somewhat more addressable because the P2 substituent in the quinoline series mostly avoids this region of the active site. One observed change that led to improvement in A156 activity was the installation of a fused ring linker. This effect was seen in a variety of compound subclasses throughout the program, and details will be published elsewhere. In this case, fusion of a cyclopentyl ring gave compound 13 and resulted in a broad activity profile against genotypes and key mutant enzymes (Table 2). Adjusting to a fused cyclopropyl further improved the activity and gave a compound (14) with excellent liver exposure. Modeling results with compounds 12 and 14 give some insight into the reasons for the 4–10-fold improvement against A156 mutant enzymes (Figure 2). The cyclopropyl constraint of 14 seems to shift the macrocycle away from the A156 residue as compared to compound 12. Because the A156 residue is positioned close to the middle of the macrocyclic ring, this slight shift may allow more space for the macrocycle of 14 to accommodate the A156T or A156V mutation. Another potential explanation could be that the constraint restricts bond rotation leading to a decreased entropic penalty upon binding and gives an overall increase in potency. Compound 14 met our potency and PK criteria and was evaluated in a battery of studies as a prelude to its selection as a development candidate.
Figure 2.
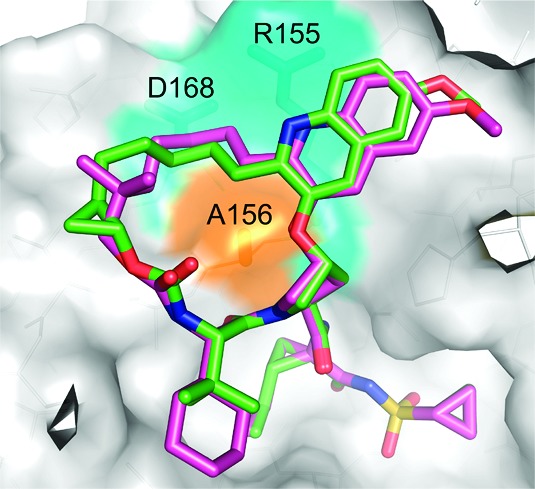
Comparison of the energy-minimized conformations of compounds 12 (magenta) and 14 (green) docked in the gt 1b NS3/4a active site.
During this candidate workup, an important pharmacokinetics-related issue regarding the behavior of pharmaceutical salts of 14 emerged and represented a major development hurdle. While the crystalline potassium salt of 14 has good aqueous solubility (9.7 mg/mL), it readily disproportionates to a crystalline zwitterionic form that has a greatly reduced aqueous solubility of <0.009 mg/mL. This behavior is driven by the moderate basicity of the quinoline P2 group of 14 (calculated pKa = 4.47)21 along with moderate acidity of the acylsulfonamide (calculated pKa = 3.67). In vivo, this process is promoted in the stomach where the native pH of the dosing solution is attenuated, giving rise to nonreproducible pharmacokinetics.22 Poor absorption of the crystalline zwitterionic form of 14 that is formed in vivo significantly capped the achievable plasma and liver exposures of 14 in preclinical species. Given the excellent overall profile of compound 14, we examined alternative P2 heterocycles with less basic functionality that would potentially maintain a similar potency and PK profile. From this work, P2 quinoxalines emerged as the most promising subclass, with lower pKa reducing the risk of zwitterion formation (calculated pKa ∼ 1.2). Incorporating a quinoxaline into our framework gave compound 15. Overall, compound 15 maintained the excellent potency against the gt 3a enzyme as well as a broad panel of mutant enzymes, has excellent potency in the replicon system [gt 1b IC50 (50% NHS) = 7.4 nM; gt 1a IC50 (40% NHS) = 7 nM], and showed excellent rat liver exposure. The potassium salt of 15 showed aqueous solubility of 1.8 mg/mL, somewhat lower than 14; however, no disproportionation was observed, making this compound a viable development candidate.
All of the P2–P4 macrocyclic compounds shown were prepared using a generally similar synthetic sequence.15,17 Specifically for compound 15, the synthesis starts with 4-methoxy-phenylenediamine hydrochloride, which was heated with diethyloxalate to give 17 in 69% yield (Scheme 1). Chemoselective chlorination with thionyl chloride followed by addition to activated cis-hydroxyproline derivative 18(23) and deprotection then led to intermediate 19 in 69% yield. Straightforward amide coupling with 20, prepared in enantiopure form in six steps, led to chloroquinoxaline 21. Next, while vinylation proceeded smoothly, the key RCM using the Zhan 1b catalyst24 was problematic, and the desired macrocycle was isolated in 25% yield. Reduction of the resulting olefin gave macrocycle 22, which was readily converted to 15 (MK-5172) in 89% yield by hydrolysis and coupling to the known cyclopropylamino acylsulfonamide 23.25
Scheme 1. Synthesis of Compound 15 (MK-5172).

Reagents and conditions: (a) Diethyloxalate, TEA, 150 °C. (b) Thionyl chloride, DMF, 110 °C. (c) Compound 18,23 Cs2CO3, NMP. (d) HCl, dioxane. (e) Compound 20, HATU, DIPEA, DMF. (f) Potassium vinyltrifluoroborate, TEA, dichloro[1,1-bis(diphenylphosphino)ferrocene]palladium(II) chloride, EtOH. (g) Zhan 1B catalyst,24 DCE. (h) H2, 10% Pd/C, MeOH, dioxane. (i) LiOH, THF, water. (j) Compound 23,25 TBTU, DIPEA, DMAP, DMF.
The pharmacokinetic properties of the potassium salt of compound 15 were evaluated in multiple species (Table 3). In rat, 15 showed a plasma clearance of 28 mL/min/kg and a plasma half-life of 1.4 h. When dosed orally at 5 mg/kg, the plasma exposure of 15 was good with an AUC of 0.7 μM h. The liver exposure of the compound is quite good (23 μM at 4 h), and 15 remains in liver 24 h after a single 5 mg/kg oral dose. At 24 h, the liver concentration of 15 is 0.2 μM, which is >25-fold higher than the IC50 in the replicon assay with 50% NHS.
Table 3. Pharmacokinetic Parameters for the Potassium Salt of 15a.
| po |
||||||
|---|---|---|---|---|---|---|
| species | Clb | Vd (L/kg) | T1/2 (h) | AUC (μM h) | [liver] 4 h (μM) | [liver] 24 h (μM) |
| rat | 28 | 3.1 | 1.4 | 0.7 | 23 | 0.2 |
| dog | 5 | 0.7 | 3.0 | 0.4 | ND | 1.4 |
Rat iv (2 mg/kg, n = 3, DMSO), dog iv (0.5 mg/kg, n = 3, DMSO), rat po (5 mg/kg, n = 3, PEG400), and dog po (1 mg/kg, n = 3, PEG400).
mL/min/kg.
When dosed to dogs, compound 15 shows low clearance of 5 mL/min/kg and a 3 h half-life after iv dosing and has good plasma exposure (AUC = 0.4 μM h) after a 1 mg/kg oral dose. Dog liver biopsy studies showed that the liver concentration of 15 after the 1 mg/kg oral dose is 1.4 μM at the 24 h time point. Similar to its behavior in rats, 15 demonstrates effective partitioning into liver tissue and maintains high liver concentration, relative to potency, 24 h after oral dosing in dogs.
In summary, initial screening for gt 3a activity along with molecular modeling has led to the discovery of a series of P2 quinoline macrocycles with excellent broad activity vs NS3/4a genoytpes and clinically observed gt 1b mutant enzymes. This series was optimized for enzyme activity and liver exposure in preclinical species. Compound 15 emerged from this series via the introduction of a mildly basic quinoxaline P2 heterocycle to deal with disproportionation issues with the more basic quinoline P2 heterocycle. We believe the combination of good PK, and broad enzyme potency gives compound 15 the potential to be an important second generation NS3/4a protease inhibitor that could be a cornerstone of an all-oral treatment for hepatitis C. Further studies of 15 (MK-5172), including clinical investigations of the pharmacokinetic and efficacy profile, are ongoing.18
Acknowledgments
We thank Dr. Charles W. Ross, III, and Vince Van Nostrand for determination of high-resolution mass spectra.
Supporting Information Available
Assay and synthetic procedures and data for selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Hepatitis C—Global prevalence (update). Wkly. Epidemiol. Rec. 1999, 74, 425–427. [PubMed] [Google Scholar]
- Liang T. J.; Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 2004, 127, S62–S71. [DOI] [PubMed] [Google Scholar]
- Hadziyannis S. J.; Sette H.; Morgan T. R.; et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [DOI] [PubMed] [Google Scholar]
- Fried M. W. Side effects of therapy of hepatitis C and their management. Hepatology 2002, 36, S237–S244. [DOI] [PubMed] [Google Scholar]
- Gordon C. P.; Keller P. A. Control of hepatitis C: A medicinal chemistry perspective. J. Med. Chem. 2005, 48, 1–20. [DOI] [PubMed] [Google Scholar]
- Chen K. X.; Njoroge F. G. A review of HCV protease inhibitors. Curr. Opin. Invest. Drugs 2009, 10, 821–837. [PubMed] [Google Scholar]
- Lamarre D.; Anderson P. C.; Bailey M.; et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [DOI] [PubMed] [Google Scholar]
- Perni R. B.; Almquist S. J.; Byrn R. A.; et al. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3–4A serine protease. Antimicrob. Agents Chemother. 2006, 50, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm B. A.; Liu R.; Lahser F.; et al. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob. Agents Chemother. 2006, 50, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.-I.; Lenz O.; Fanning G.; et al. In Vitro Activity and Preclinical Profile of TMC435350, a Potent Hepatitis C Virus Protease Inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert S. D.; Andrews S. W.; Jiang Y.; et al. Preclinical Characteristics of the Hepatitis C virus NS3/4A Protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 2008, 52, 4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liverton N. J.; Holloway M. K.; McCauley J. A.; et al. Molecular modeling based approach to potent P2-P4 macrocyclic inhibitors of hepatitis C NS3/4a protease. J. Am. Chem. Soc. 2008, 130, 4607–4609. [DOI] [PubMed] [Google Scholar]
- Yao N.; Reichert P.; Taremi S. S.; et al. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 1999, 7, 1353–1363. [DOI] [PubMed] [Google Scholar]
- Grubbs R. H. Olefin metathesis catalysts for the preparation of molecules and materials (Nobel Lecture). Angew. Chem., Int. Ed. 2006, 45, 3760–3765. [DOI] [PubMed] [Google Scholar]
- McCauley J. A.; McIntyre C. J.; Rudd M. T.; et al. Discovery of Vaniprevir (MK-7009), a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. J. Med. Chem. 2010, 53, 2443–2463. [DOI] [PubMed] [Google Scholar]
- Liverton N. J.; Carroll S. S.; DiMuzio J. M.; et al. MK-7009: A Potent and Selective Inhibitor of Hepatitis C Virus NS3/4a Protease. Antimicrob. Agents Chemother. 2010, 54, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd M. T.; McCauley J. A.; Butcher J. W.; et al. Discovery of MK-1220: A Macrocyclic Inhibitor of Hepatitis C Virus NS3/4a Protease with Improved Preclinical Plasma Exposure. ACS Med. Chem. Lett. 2011, 2, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa V.; Ludmerer S. W.; McCauley J. A.;et al. MK-5172, a selective inhibitor of Hepatitis C Virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob. Agents Chemother. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.; Summa V.; Liverton N. J.; McCauley J. A.. Macrocyclic Quinoxaline Compounds as HCV NS3 Protease Inhibitors. U.S. Patent 7,973,040, 2011.
- Manns M. P.; Gane E. J.; Rodriguez-Torres M.; et al. Sustained viral response (SVR) rates in genotype 1 treatment-naive patients with chronic Hepatitis C (CHC) infection treated with Vaniprevir (MK-7009), a NS3/4A protease inhibitor, in combination with pegylated Interferon alfa-2A and Ribavirin for 28 days. Hepatology 2010, 524 Suppl.361A. [Google Scholar]
- MarvinSketch, publisher. ChemAxon Ltd., version 3.5.2; 2004; http://www.chemaxon.com/products/marvin/marvinsketch.
- Monteagudo E.; Fonsi M.; Chu X.; et al. The metabolism and disposition of a potent inhibitor of hepatitis C virus NS3/4A protease. Xenobiotica 2010, 40, 826–839. [DOI] [PubMed] [Google Scholar]
- Arasappan A.; Chen K. X.; Njoroge F. G.; et al. Novel Dipeptide Macrocycles from 4-Oxo, -Thio, and -Amino-Substituted Proline Derivatives. J. Org. Chem. 2002, 67, 3923–3926. [DOI] [PubMed] [Google Scholar]
- CAS# 918870-76-5.
- Li J.; Smith D.; Wong H. S.; et al. A Facile Synthesis of 1-Substituted Cyclopropylsulfonamides. Synlett 2006, 725–728. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.