

Abstract
A ligand-based method based on the SMARTCyp approach that predicts the sites of cytochrome P450 2D6-mediated metabolism of druglike molecules has been developed. The method uses only two descriptors besides the reactivity from SMARTCyp: the distance to a protonated nitrogen atom and the distance to the end of the molecule. Hence, the site of metabolism is predicted directly from the 2D structure of a molecule, without requiring calculation of electronic properties or generation of 3D structures. Testing on an independent test set gives an area under the curve value of 0.94, and a site of metabolism is found among the top two ranked atoms for 91% of the compounds.
Keywords: CYP2D6, cytochrome P450, drug metabolism
Cytochrome P450 (CYP) constitutes an ubiquitous family of enzymes, and from a drug perspective, its most important function is to metabolize drug compounds. In the development of drugs, it is important at an early stage to be able to identify the site of metabolism (SOM) of the lead compounds to be able to guide the development of compounds with a desirable pharmacokinetic profile.
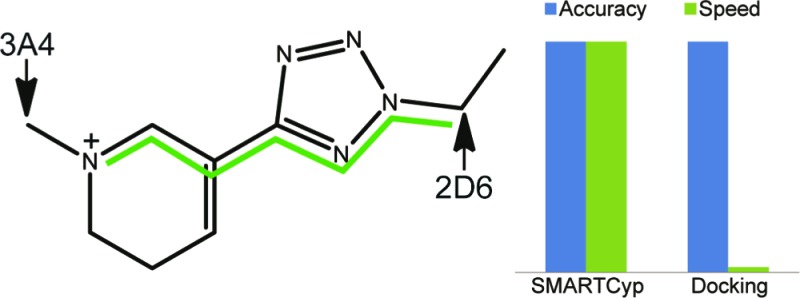
The CYP 3A4 isoform is the most important enzyme in the degradation of drug compounds and metabolize about 50% of them. CYP 3A4 is promiscuous and is capable of converting both small and large compounds, and crystal structures have shown that the volume of the active site can change dramatically upon binding of ligands.1−3 This is probably also why it is possible to use ligand-based models to predict how a druglike compound is metabolized for CYP 3A4, because binding in this flexible pocket is not too restrictive, and thus, the intrinsic reactivity plays a significant role. For example, it was possible with the SMARTCyp method,4,5 which primarily takes the intrinsic reactivity into account, to predict a SOM within the top two ranked atoms for 81% of the compounds within a set of 361 druglike compounds.5
The second most drug-metabolizing CYP enzyme, the 2D6 isoform, is more selective in its recognition of the substrates. Generally, many medium-sized amines are metabolized, however, not many of them by N-dealkylation of amines but rather further away from this functional group. The crystal structure of CYP 2D6 reveals that a there are two negatively charged amino acids in the upper part of the binding cavity of 2D6, that is, Glu216 and Asp301, which may facilitate the binding of the positively charged parts of the substrates.6 The fact that the enzyme induces the binding suggests that it is relevant to use the protein structures to predict how drug compounds are metabolized. This is probably why only few studies of pure ligand-based models on SOM prediction for CYP2D6 exist,7 and structural information of protein has been included for these purposes. de Groot and co-workers combined structural models of CYP2D6 and pharmacophore modeling with AM1 energies of intermediates and products to predict the SOMs.8,9 Later studies have shown the importance of including water molecules and using ensembles of protein structures to get accurate predictions.10−12 In addition to docking the substrates into an ensemble of 1000 structures for predicting SOMs for CYP 2D6, Moors et al. included reactivities from SMARTCyp and StarDrop,13 which improved the prediction rate significantly.12
The drawback of using models that explicitly make use of the protein structures is that they are significantly slower, especially when multiple structures are used for ensemble docking. However, because the binding plays such a large role, it is necessary to include implicitly this information in the ligand-based models to predict the SOMs. Here, we present a variant of the SMARTCyp method that, in addition to the intrinsic reactivity and accessibility, takes the presence of a positive charge in the compound into account and shows that this gives a model able to accurately predict CYP 2D6 metabolism.
The data sets from Moors et al.12 were, with some modifications, used to build and validate the SMARTCYP-based 2D6 models. Because we are building models based on 2D structures, first, we removed one of each duplicate R and S isomers. The pairs of stereoisomers always give rise to the same metabolites according to Moors et al. Second, we moved six compounds from the large data set to the small data set. This was done because, in the original data sets, there were no compounds that were metabolized through N-dealkylation in the small data set. The modified large data set was then used as a training set, and the modified small data set was used as a test set. Statistics on the two data sets are shown in Table 1.
Table 1. Description of the Data Sets Used for Training and Testing the Algorithm.
| training set | test set | |
|---|---|---|
| no. of compounds | 86 | 45 |
| no. of metabolic sites | 95 | 56 |
| metabolic sites/compound | 1.1 | 1.2 |
| aliphatic hydroxylations | 15 (16%) | 8 (18%) |
| N-dealkylations | 19 (20%) | 7 (16%) |
| O-dealkylations | 40 (42%) | 14 (31%) |
| aromatic hydroxylations | 21 (22%) | 27 (60%) |
As mentioned previously, binding plays a larger role for CYP 2D6 than for CYP 3A4. Compounds with a positive charge would tend to bind to Glu216 and Asp301,14 resulting in an orientation that positions positively charged amine groups far away from the catalytic heme group. Thus, the likelihood that a basic amine will undergo N-dealkylation is much smaller in CYP2D6, despite the fact that these are very reactive sites. To capture this effect in the ligand-based 2D6 model, we introduce a descriptor that describes the distance (calculated as number of bonds) from the atom of interest to nitrogen atoms, which would interact with these amino acids (N+dist).
A second difference in the SOM preference of CYP2D6 as compared to CYP3A4, for which SMARTCyp has been validated previously, is that the SOMs in CYP2D6 are much more likely to be situated at the end of a molecule than in the center of a molecule. This is most likely due to less space for free rotation of molecules in the CYP2D6 active site, as well as the interactions with Glu216 and Asp301. To capture this effect, we have applied two different descriptors (shown in Figure 1). First, we have tried the relative span, which is defined as the maximum shortest bond path of the atom of interest divided by the maximum shortest bond path in the molecule (i.e., the relative distance of an atom to the center of the molecule). By definition, this descriptor will always have a value between 0.5 and 1.0, where 0.5 is assigned to atoms that are positioned in the 2D center of a molecule, and 1.0 is assigned to atoms that are positioned at the end of a molecule. This is also the descriptor used to describe accessibility in SMARTCyp. Second, we have defined a novel descriptor, Span2End, which describes the number of bonds between the atom of interest and the end of the molecule. Span2End is defined as the maximum shortest bond path in the molecule minus the maximum shortest bond path of the atom of interest. Hence, Span2End will always have integer values ranging from 0 to half the value of the maximum shortest bond path in the molecule. A value of 0 is assigned to an atom, which is positioned at the end of a molecule, and the highest value is assigned to an atom in the 2D center of the molecule.
Figure 1.
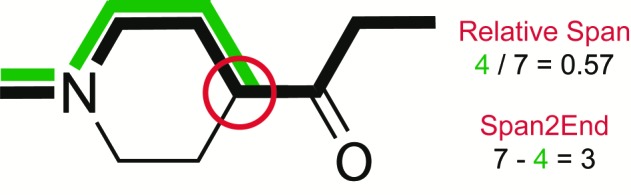
Description of the two different accessibility descriptors, with an example for the atom in the red circle.
Two optimization criteria were used when building models using these descriptors. First, the area under curve (AUC) of a receiver operator characteristic (ROC) curve calculated from the ranks of the atoms in all of the molecules in the data set (AUCrank) was used. The AUC value can range from 0.5 (random prediction) to 1.0 (perfect prediction). For evaluation of the final model, the AUC was also computed using the scores of all atoms in the data sets (AUCscore). We choose to optimize the model toward the AUCrank instead of AUCscore, because most of the time a user of a SOM prediction model is only interested in comparing atoms within a molecule and not atoms in different molecules (which is what is described by AUCscore). Second, we have used the top two prediction accuracy. This is defined as the percent of molecules in data set for which we find a SOM among the top two ranked atoms. The top two measure was applied because there are many different ways to achieve a similar AUCrank, but when a medicinal chemist applies methods of this type, they will most of the time only look at the top-ranked atoms. Hence, among the models built that gave a very similar AUCrank, we chose the model with highest top two value.
When optimizing the contributions of the two different accessibility descriptors (relative span and Span2End), we found that both could give a high numerical prediction accuracy by itself (AUCrank of 0.96), but they were not complementary. This is obviously because they are two different formulations of the same property, the number of bonds from the atom of interest to the end of the molecule, either as a value relative to the size of the molecule (relative span) or as an absolute value (Span2End). However, looking at the actual optimized correction values, we chose to use Span2End. This is because the correction using the relative span descriptor becomes very different when applied to molecules of varying sizes. When using the relative span, two neighboring atoms will not have the same difference in accessibility correction in a small molecule as in a large molecule. In addition, for small molecules, this difference becomes totally unphysical using the optimized constant of −108 kJ/mol (resulting in penalty differences for the relative span alone of up to ∼15 kJ/mol per bond in small molecules).
To create the correction using N+dist and Span2End, a linear correction with as simple function as possible was used, with only three variables; a cutoff for each penalty (beyond which the penalties should be constant) and one common constant for both corrections. The N+dist correction has its maximum value at the position of the protonated nitrogen atom and decreases linearly until the cutoff, beyond which the correction is zero. The Span2End correction is zero at the end of the molecule and increases linearly until the Span2End value reaches the cutoff; beyond this, the value is constant. The mathematical formulation of the two corrections is shown below. While separate constants for the two descriptors might seem more appropriate, extensive tests of a large number of different cutoffs and constants did not result in better accuracy than using the two cutoffs shown below and a common constant.
The final prediction algorithm can be described as follows:
Here, reactivity is the activation energy assigned by SMARTCyp, and N+dist_correction and Span2End_correction are assigned by the following equations:
The “0.01 × Span2End” in the last equation is used to separate atoms that otherwise would get exactly the same score. Because the optimized cutoff for Span2End is 4, small molecules are not affected by this cutoff, and then, the two corrections become linear (see examples in Figure 2).
Figure 2.

Two examples of how the N+dist correction and the Span2End correction affect the atom scores. Atoms 4 and 5 in the top molecule have the same Span2End correction because they are both three bonds away from the end of the molecule.
The final algorithm is relatively robust. Modifications of cutoffs or the constant by ±1 give changes to AUCrank of <0.01.
The final N+dist cutoff of eight bonds agrees with the pharmacophore model derived by de Groot, in which the distance between the SOM and the basic nitrogen atom is 5 or 10 Å for a few of the substrates and 7 Å for the most substrates.8
For the two example molecules in Figure 2, the sum of the corrections is between 54–68 kJ/mol; at the same time, the activation energies for the atoms within these two molecules vary from 40 to 90 kJ/mol. Hence, in our CYP2D6 model, the sum of the orientational descriptors and the reactivity have similar weight. This is different from the original SMARTCyp CYP 3A4 model, in which the accessibility descriptor has a weight that is less than 10% of the reactivity, indicating that, as expected, the reactivity is not as important for CYP 2D6 metabolism.
The prediction accuracy for both the training and the test sets are shown in Table 2 and show that the SMARTCyp 2D6 model gives higher accuracy as compared to both the docking-based model of Moors et al.12 and the ligand-based model in the StarDrop software,13 especially with regard to finding a SOM in the top-ranked position.
Table 2. Results for the Training and Test Sets and Comparison to Results from StarDrop and Moors et al.12.
| SMARTCyp 2D6 | Moors | StarDrop | |
|---|---|---|---|
| training set | |||
| top 1 (%)a | 70 | 65 | 63 |
| top 2 (%)a | 88 | 88 | 80 |
| top 3 (%)a | 92 | N/A | 93 |
| AUCrank | 0.96 | N/A | N/A |
| AUCscore | 0.93 | 0.93b | N/A |
| test set | |||
| top 1 (%)a | 78 | 62 | 60 |
| top 2 (%)a | 91 | 83 | 80 |
| top 3 (%)a | 91 | N/A | 91 |
| AUCrank | 0.93 | N/A | N/A |
| AUCscore | 0.94 | 0.92b | N/A |
Percent molecules in the data set that are found to have at least one metabolic site among the top-ranked atoms.
Computed on the original, unmodified data sets from Moors et al.12
Analyzing the predictions on the test set, we can conclude that the structurally based model is superior to the SMARTCyp-2D6 model for some compounds where the binding contribution to the metabolism is not easily described by only the distance to a protonated amine (ondansetron, propafenone, and tamoxifen; see Figure 3). On the other hand, our model more accurately select the correct atom when reactivity is the property that determines which atom in a specific group is oxidized (amitriptyline, cinnarizine, flunarizine, and nortriptyline; see Figure 3). Hence, a more carefully implemented reactivity correction might improve the predictions of structurally based prediction methods.
Figure 3.
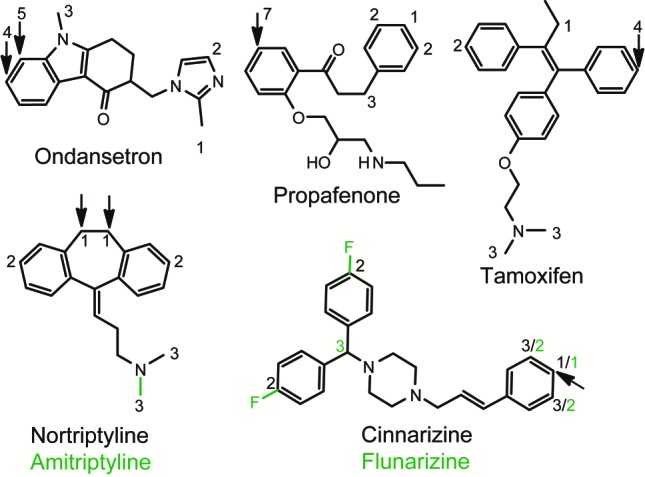
Examples of molecules in the test set for which the SMARTCyp 2D6 model outperforms structurally based models (nortriptyline/amitriptyline and cinnarizine/flunarizine) and examples where it fails (ondansetron, tamoxifen, and propafenone). The sites of metabolisms are labeled with arrows, and the numbers represent the SMARTCyp 2D6 rankings. The two pairs of molecules with dual names have the same structure except for the green parts, which only exist in the molecules with green names. Where the SMARTCyp 2D6 rankings for these molecules vary, they are shown in green for the corresponding molecules.
The model was further validated on the training set used by Sheridan et al. to construct a ligand-based model for CYP 2D6.7 For this data set consisting of 124 compounds, our model predicts a SOM within the top two ranked atoms for 84% of the compounds (as compared to 72% for Sheridan et al.). While the overlap with our training set is significant (only 46 compounds were not within our training set; for these, our model gets 80% top two rate), we do not have access to the results for individual compounds from Sheridan et al. and cannot compare the performance for only the nonoverlapping compounds. For the test set from Sheridan et al., which only contains 10 compounds, our model gets 80% top two rate, as compared to 70% for Sheridan et al.
The fact that this CYP2D6 model based on SMARTCyp works quite well shows that there is no need for tens or hundreds of descriptors to approach this problem. Using only a few chemically sensible descriptors can result in quite accurate prediction models, which are at least as good as much more time-consuming models based upon docking into ensembles of many structures. It is encouraging to see that the model is good at identifying compounds with a metabolic position ranked highest, in particular because it is a pure 2D method that gives an extremely fast prediction.
In summary, we have developed a method based on SMARTCyp for predicting the SOM for drug metabolism mediated by CYP2D6. The method uses only the 2D structure of a drug, from which the reactivity of a site is deduced by fragment matching, the binding into the CYP2D6 enzyme is described by two additional descriptors, the distance from the end of the molecule, and the largest distance to a protonated nitrogen atom.
Computational Methods
The reactivity values were taken from the SMARTCyp program (version 1.5.3).4,5 Matching of SMARTS patterns, computation of the topological bond path distances, and rendering of 2D structures were performed using the CDK and JChemPaint java libraries.15,16
The N+dist descriptor was computed by using the topological bond path distances already available through SMARTCyp and identifying the nitrogen atoms of interest by fragment matching using SMARTS. The following two SMARTS patterns were used to identify these nitrogen atoms:
N+dist is then calculated as the number of bonds between the atom of interest and an atom matching the SMARTS patterns above. If there are multiple atoms matching the SMARTS patterns, then the one resulting in the largest N+dist value is used. AUC values were computed from the ROC curves using the trapezoid formula to compute the values.
For comparison, the training and test sets were also computed using StarDrop, version 5.0.13 The StarDrop software is a ligand-based model that combines steric accessibility descriptors with on-the-fly reactivity calculations (using the semiempirical AM1 method). The new model has been implemented in SMARTCyp version 2.0 and is available from http://www.farma.ku.dk/smartcyp/.
Acknowledgments
The authors thank David E. Gloriam for assistance with the programming of the new method in SMARTCyp.
Glossary
Abbreviations
- CYP
cytochrome P450
- SOM
site of metabolism
- AUC
area under curve
- ROC
receiver operator characteristics
Supporting Information Available
Training and test sets in sdf format. This material is available free of charge via the Internet at http://pubs.acs.org.
The work was supported by a grant from Lhasa Ltd.
The authors declare no competing financial interest.
This paper was published on the Web on November 7, 2011, after which a small error in one of the python scripts used for analysis of the SMARTCyp results was located. This error does not affect the conclusions in any way but only resulted in an error of the top 1 accuracies of the model by 1%. The correct top 1 accuracies are 1% lower and higher for the training and test sets, respectively, as compared to the originally published numbers (70 vs 71% and 78 vs 77%). Table 2 was corrected as a result. The corrected version was reposted on December 21, 2011.
Supplementary Material
References
- Ekroos M.; Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J. K.; Wester M. R.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-angstrom resolution. J. Biol. Chem. 2004, 279, 38091–38094. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Vinkovi-ç D. M.; Ward A.; Angove H. C.; Day P. J.; Vonrhein C.; Tickle I. J.; Jhoti H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science 2004, 305, 683–686. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Gloriam D. E.; Zaretzki J.; Breneman C.; Olsen L. SMARTCyp: A 2D Method for Prediction of Cytochrome P450-Mediated Drug Metabolism. ACS Med. Chem. Lett. 2010, 1, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg P.; Gloriam D. E.; Olsen L. The SMARTCyp cytochrome P450 metabolism prediction server. Bioinformatics 2010, 26, 2988–2989. [DOI] [PubMed] [Google Scholar]
- Rowland P.; Blaney F. E.; Smyth M. G.; Jones J. J.; Leydon V. R.; Oxbrow A. K.; Lewis C. J.; Tennant M. G.; Modi S.; Eggleston D. S.; Chenery R. J.; Bridges A. M. Crystal structure of human cytochrome P450 2D6. J. Biol. Chem. 2006, 281, 7614–7622. [DOI] [PubMed] [Google Scholar]
- Sheridan R. P.; Korzekwa K. R.; Torres R. A.; Walker M. J. Empirical regioselectivity models for human cytochromes p450 3A4, 2D6, and 2C9. J. Med. Chem. 2007, 50, 3173–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot M. J.; Ackland M. J.; Horne V. A.; Alex A. A.; Jones B. C. Novel Approach To Predicting P450-Mediated Drug Metabolism:GÇë Development of a Combined Protein and Pharmacophore Model for CYP2D6. J. Med. Chem. 1999, 42, 1515–1524. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Ackland M. J.; Horne V. A.; Alex A. A.; Jones B. C. A Novel Approach to Predicting P450 Mediated Drug Metabolism. CYP2D6 Catalyzed N-Dealkylation Reactions and Qualitative Metabolite Predictions Using a Combined Protein and Pharmacophore Model for CYP2D6. J. Med. Chem. 1999, 42, 4062–4070. [DOI] [PubMed] [Google Scholar]
- de Graaf C.; Oostenbrink C.; Keizers P. H. J.; van der Wijst T.; Jongejan A.; Vermeulen N. P. E. Catalytic site prediction and virtual screening of cytochrome P450 2D6 substrates by consideration of water and rescoring in automated docking. J. Med. Chem. 2006, 49, 2417–2430. [DOI] [PubMed] [Google Scholar]
- Keizers P. H. J.; de Graaf C.; de Kanter F. J. J.; Oostenbrink C.; Feenstra K. A.; Commandeur J. N. M.; Vermeulen N. P. E. Metabolic regio- and stereoselectivity of cytochrome P450 2D6 towards 3,4-methylenedioxy-N-alkylamphetamines: in silico predictions and experimental validation. J. Med. Chem. 2005, 48, 6117–6127. [DOI] [PubMed] [Google Scholar]
- Moors S. L. C.; Vos A. M.; Cummings M. D.; Van Vlijmen H.; Ceulemans A. Structure-Based Site of Metabolism Prediction for Cytochrome P450 2D6. J. Med. Chem. 2011, 54, 6098–6105. [DOI] [PubMed] [Google Scholar]
- StarDrop, version 5.0; Optibrium Ltd.: 2011. [Google Scholar]
- Paine M. J. I.; McLaughlin L. A.; Flanagan J. U.; Kemp C. A.; Sutcliffe M. J.; Roberts G. C. K.; Wolf C. R. Residues glutamate 216 and aspartate 301 are key determinants of substrate specificity and product regioselectivity in cytochrome p450 2D6. J. Biol. Chem. 2003, 278, 4021–4027. [DOI] [PubMed] [Google Scholar]
- Steinbeck C.; Hoppe C.; Kuhn S.; Floris M.; Guha R.; Willighagen E. L. Recent developments of the Chemistry Development Kit (CDK) - An open-source Java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [DOI] [PubMed] [Google Scholar]
- Steinbeck C.; Han Y. Q.; Kuhn S.; Horlacher O.; Luttmann E.; Willighagen E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.