

Abstract

Natural and synthetic histone deacetylase (HDAC) inhibitors generally derive their strong binding affinity and high potency from a key functional group that binds to the Zn2+ ion within the enzyme active site. However, this feature is also thought to carry the potential liability of undesirable off-target interactions with other metalloenzymes. As a step toward mitigating this issue, here, we describe the design, synthesis, and structure–activity characterizations of cyclic α3β-tetrapeptide HDAC inhibitors that lack the presumed indispensable Zn2+-binding group. The lead compounds (e.g., 15 and 26) display good potency against class 1 HDACs and are active in tissue culture against various human cancer cell lines. Importantly, enzymological analysis of 26 indicates that the cyclic α3β-tetrapeptide is a fast-on/off competitive inhibitor of HDACs 1–3 with Ki values of 49, 33, and 37 nM, respectively. Our proof of principle study supports the idea that novel classes of HDAC inhibitors, which interact at the active-site opening, but not with the active site Zn2+, can have potential in drug design.
Keywords: HDAC inhibitors, zinc binding group, cancer therapy, tetrapeptide, apicidin
Histone deacetylases (HDACs) catalyze the removal of the acetyl group from N-Ac-lysine side chains of histone tails and other proteins.1,2 HDACs are among the key players in the complex epigenetic regulation of cellular processes and, consequently, are important targets for therapeutic approaches in several chronic diseases, including cystic fibrosis (CF),3 sickle cell anemia,4 and neurodegenerative disorders.5 HDAC inhibitors can also reactivate gene expression to induce cell cycle arrest and apoptosis.6 In that regard, there has been considerable interest in HDAC inhibitors as anticancer agents,7 and two drugs (1 and 2) have marketing approval for treating refractory cutaneous T-cell lymphoma.8,9 HDACs in classes 1, 2, and 4 contain a catalytic Zn2+ ion in the active site, which can be targeted by various compounds that possess metal binding functional groups (Figure 1). HDAC inhibitors commonly employ the following Zn2+-binding groups: hydroxamic acid (e.g., 1), thiol (e.g., 2), carboxylic acid (e.g., 3), ketone (e.g., 4), or 2-aminoanilide (e.g., 5) (Figure 1).10,11 However, these functional groups can also bind strongly with other important metalloenzymes, leading to dose-limiting toxicity that can impede the wider application of HDAC inhibitors in pharmacotherapy.12,13
Figure 1.
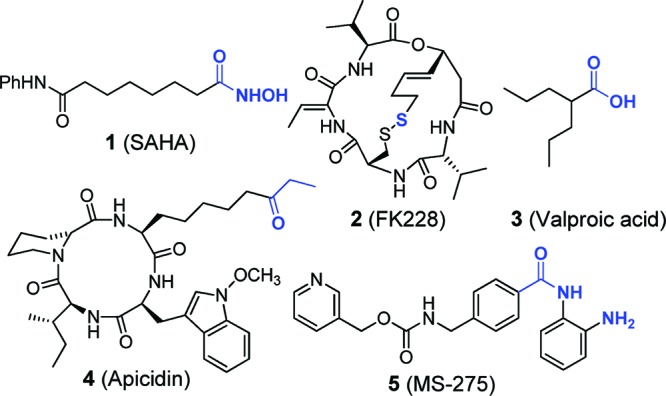
Common HDAC inhibitors with Zn2+-binding groups highlighted in blue.
We have been studying macrocyclic tetrapeptides and peptidomimetics related to apicidin (4) as HDAC inhibitors.14,15 In the course of our work, we sought to eliminate the zinc-binding group to ascertain how much residual potency could be retained. Thus, we synthesized α3β cyclic peptide scaffolds depicted in Chart 1, based on 4.14,15 The β-amino acid in the α3β scaffold facilitates peptide cyclization during synthesis and can also impart proteolytic stability.16 All peptides were prepared using standard solid-phase peptide synthesis (SPPS) followed by cyclization in solution as described previously.14,15 Our initial series of compounds varied only at the R1 position (Chart 1, red bracket, peptides 6–15), which usually contains the Zn2+-binding group. We replaced the Zn2+-binding side chain with various straight-chain or branched alkyl groups of different lengths, as shown. All of the compounds were screened for inhibitory activity at concentrations of 10 and 1 μM against recombinant HDAC3/NCoR2. Surprisingly, the compound containing a propyl side chain (15) was the most potent inhibitor from this series (Chart 1 and Figure S1 in the Supporting Information). It is intriguing that an alkyl chain that is shorter (6, 7) or longer (8, 14) than three carbon atoms resulted in substantially decreased activity (Chart 1 and Table S1 in the Supporting Information). We also found that incorporating unsaturation (12, 13) or branching (9–11) at position R1 did not improve activity. Crystal structures of several Zn2+-dependent HDACs show a narrow binding pocket (long enough to accommodate a chain of 5–6 atoms) leading to the active-site zinc atom,17 and a recent cocrystal structure of HDAC8 bound to a cyclic depsipeptide confirmed that a long linker is necessary for binding to the zinc.18 We next prepared two additional series of compounds that varied at side chain R2 (Chart 1, green bracket, compounds 16–25) or at side chain R3 (Chart 1, blue bracket, compounds 26–29) while keeping the propyl side chain at R1 constant. The β-homoleucine amino acid was not varied, because we have found it to be optimal in HDAC inhibitors of this type.14,15 Compounds varied at position R2 showed activity at 10 μM; however, at 1 μM, only the alanine and proline substitutions stood out. At position R3, where bicyclic aromatic side chain groups have been shown to facilitate inhibitor efficacy,14,1526 gave the best activity against HDAC3/NCoR2, showing 60% inhibition at 1 μM, an unprecedented value for a molecule without a Zn2+-binding group (Table S1 in the Supporting Information).10,11 Surprisingly, the 1-naphthylalanine-containing compound (27) showed little activity even at 10 μM, despite showing better activity than tryptophan in our previous report, where a carboxylic acid Zn2+-binding group was used.15
Chart 1. SAR Data for Compounds vs HDAC3/NCoR2a.

a Purified compounds 6–29 were synthesized and assayed against recombinant HDAC3/NCoR2 (Supporting Information). Percent inhibition data represent averages of at least two separate experiments performed in duplicate. Error bars indicate the standard deviation for each value. aaR1, aaR2, and aaR3 refer to the amino acid at position R1, R2, or R3 in the cyclic peptide.
We determined IC50 values for our two lead compounds, 15 and 26 (Figure 2), against a group of recombinant HDACs from classes 1 (HDACs 1–3) and 2b (HDAC6), which are the major deacetylase enzymes in the nucleus and cytoplasm, respectively (Table 1). Intriguingly, 26 had in vitro activity comparable to several state-of-the-art inhibitors, including being 40–200-fold more potent than valproic acid, an FDA-approved drug for epilepsy (Table 1). Compound 26 was also only 2–6-fold less active than MS-275 (Table 1), a compound in clinical trials for cancer therapy. Archetypal hydroxamic acid inhibitor 1 (SAHA) is only ∼10-fold more potent against recombinant HDACs than 26 (Table 1).
Figure 2.
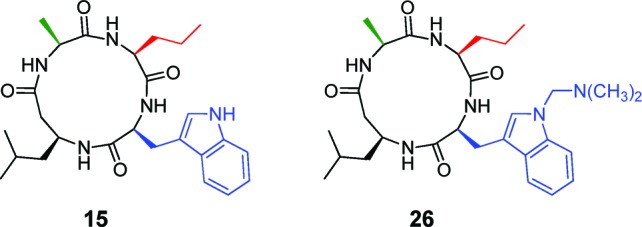
Lead non-Zn2+-binding HDAC inhibitors.
Table 1. IC50 Values for Lead HDAC Inhibitorsa.
| HDAC enzyme |
||||
|---|---|---|---|---|
| inhibitors | 1 | 2 | 3b | 6 |
| 15c,d | 0.98 | 1.5 | 1.4 | >20c |
| 26c,d | 0.68 | 1.4 | 0.80 | >20c |
| SAHAe | 0.069 | 0.18 | 0.043 | 0.018 |
| FK228f | 0.2 nM | 1.0 nM | 3.0 nM | 0.2 |
| VPAg | 39 | 62 | 161 | >2000 |
| apicidinh | 0.022 | 0.035 | 0.029 | >10 |
| MS-275i | 0.12 | 0.25 | 0.4 | >10 |
All values are in μM unless otherwise specified.
HDAC3/NCoR2.
Compounds 15 and 26 have 37 and 46% inhibition, respectively, against HDAC6 at 20 μM.
Values are averages of at least two separate experiments performed in duplicate.
From ref (19).
From ref (20).
From ref (21).
From ref (15).
From ref (22).
Human tissue culture cytotoxicity assays were carried out to determine if our lead compounds could penetrate cells and act against intracellular HDAC complexes, which can have different properties and selectivity profiles as compared to the recombinant proteins. We screened 15, 26, apicidin (4), and SAHA (1) against several cancer cell lines, including Jurkat (T-cell leukemia), HL-60 (acute promyelocytic leukemia), HeLa (cervical cancer), MCF-7 (breast cancer), and HEK293 (human embryonic kidney) cells (Table 2). We found the best efficacy against the leukemia cell lines, where 15 caused 50% growth inhibition (GI50) at 12 and 8.2 μM against Jurkat and HL-60 cells, respectively. Compound 26 had GI50 values of 8.1 and 2.8 μM against Jurkat and HL-60 cells, respectively (Table 2).
Table 2. Cytotoxicity Data for Selected Compoundsa.
| cell type | 15 | 26 | apicidin | SAHA |
|---|---|---|---|---|
| HL-60 | 8.2 | 2.8 | 0.28 | 0.93 |
| Jurkat | 12 | 8.1 | 0.83 | 1.1 |
| HeLa | 81 | 62 | 1.4 | 2.5 |
| MCF-7 | 40 | 47 | 6.8 | 8.6 |
| HEK293 | >100b | 100 | 11 | 13 |
Data represent growth inhibition (GI50) values in units of μM. HL-60 (acute promyelocytic leukemia), Jurkat (T-cell leukemia), HeLa (cervical cancer), MCF-7 (breast cancer), and HEK293 (human embryonic kidney). Values are an average of two independent experiments performed in duplicate.
33% growth inhibition at 100 μM.
We further carried out Western blots for acetylated and total histone H3 to confirm that 15 and 26 act as HDAC inhibitors in cells. Indeed, we observed a dose-dependent increase in acetylated H3 in the presence of 15 and 26 (Figure S1 in the Supporting Information).
Enzyme kinetic analyses can provide valuable insights into the likely mechanism by which an inhibitor deactivates the enzyme. Accordingly, we measured progression curves for HDACs 1, 2, and 3 in the presence of varying concentrations of 26 to determine if it operated via a fast-on/fast-off or a slow-binding mechanism.23,24 The optimized procedure for the real-time enzyme-coupled assay used in these studies can be found in the Supporting Information.
Briefly, if the HDAC displays a constant rate of substrate turnover, there will be a constant rate of fluorescence increase (substrate deacetylation) over time, and the graph (over that time period) will be linear indicating a fast-on/fast-off mechanism of inhibition.23,24 An example of this type of inhibition is seen with 1 (SAHA)11,23 and, presumably, other hydroxamic acid-bearing HDAC inhibitors. However, if the rate of substrate conversion decreases over time (before the substrate has been significantly depleted), then the inhibitor can be described as having a slow-binding mechanism of action.23,24 From the data plotted in Figure 3a–c, it appears that 26 binds HDACs 1–3 with a fast-on/fast-off mechanism, thus rendering less likely the involvement of protein conformational changes or an allosteric mode of inhibition. Moreover, to establish whether substrate and inhibitor 26 binding to the enzyme were mutually exclusive events, we measured product formation under conditions of varying substrate and inhibitor concentration for HDACs 1 and 3, followed by a Lineweaver–Burk analysis (Figure S2 in the Supporting Information). We found that 26 fits the model for a competitive inhibitor for both HDACs. Thus, 26 appears to bind to the enzyme active site pocket in a similar manner as its progenitor, despite lacking the Zn2+-binding side chain. Lastly, we determined the inhibition constant (Ki) for 26 with HDACs 1, 2, and 3. This was accomplished by using eq 1 for a fast-on/fast-off inhibitor, where vp is the velocity in the presence of the inhibitor, va is the velocity in the absence of the inhibitor, [I] is the concentration of the inhibitor, Km is the Michaelis–Menten constant for the substrate against each HDAC (10 μM), and [S] is the concentration of the substrate.23
| 1 |
Figure 3.

Progress curves for 26 shown as an average of two separate experiments performed each in duplicate. Panels a–c depict substrate turnover by increasing RFU (relative fluorescent units) over time with different inhibitor concentrations, [I] (labeled next to each curve), for HDACs 1, 2, and 3, respectively. Panels d–f are plots of reaction velocity in the presence of inhibitor (vp) over the absence of inhibitor (va) vs [I] used to determine the Ki for 26 against HDACs 1–3.
The measured vp/va values (Figure 3a–c and Table S2 in the Supporting Information) were fit to eq 1 using GraphPad Prism to give the calculated Ki values for 26 against HDACs 1–3 of 49, 33, and 37 nM, respectively.
Although HDAC inhibitors are promising therapeutic agents for many diseases, acute dose-limiting toxicity and a narrow therapeutic index limit their widespread clinical utility for treating chronic diseases. We have developed inhibitors that lack the commonly used Zn2+-binding group as a step toward mitigating these problems. We reported a macrocyclic peptide scaffold that does not require a Zn2+-binding group for activity against HDACs 1, 2, and 3. Lead compounds 15 and 26 have translatable potency from in vitro experiments with recombinant enzymes to efficacy in cell-based assays. Our results provide the impetus for further research in advancing metal ion-binding independent classes of HDAC inhibitors and assessing their potential in drug design.
Acknowledgments
We thank our colleagues Jeff Kelly and Benjamin Cravatt for providing the cell lines and Bruce Maryanoff, Joel M. Gottesfeld, C. James Chou, and Dennis W. Wolan for invaluable discussions.
Supporting Information Available
Experimental procedures for compound synthesis, HDAC inhibition assays, cell growth assays, and kinetic assays as well as characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Technical University of Denmark, Kemitorvet 207, DK-2800, Kgs. Lyngby, Denmark.
We gratefully acknowledge financial support from the Skaggs Institute for Chemical Biology and NSF (predoctoral fellowship to C.J.V.).
The authors declare no competing financial interest.
Supplementary Material
References
- Kouzarides T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Margueron R.; Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt D. M.; Olsen C. A.; Vickers C. J.; Herman D.; Chalfant M.; Montero A.; Leman L. J.; Burkle R.; Maryanoff B. E.; Balch W. E.; Ghadiri M. R. Potential Agents for Treating Cystic Fibrosis: Cyclic Tetrapeptides that Restore Trafficking and Activity of ΔF508-CFTR. ACS Med. Chem. Lett. 2011, 2, 703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J. E.; Mak R.; Tanguturi S. K.; Mazitschek R.; Haggarty S. J.; Ross K.; Chang C. Y.; Bosco J.; West N.; Morse E.; Lin K.; Shen J. P.; Kwiatkowski N. P.; Gheldof N.; Dekker J.; DeAngelo D. J.; Carr S. A.; Schreiber S. L.; Golub T. R.; Ebert B. L. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 12617–12622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A. G.; Thompson L. M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discovery 2008, 7, 854–868. [DOI] [PubMed] [Google Scholar]
- Xu W. S.; Parmigiani R. B.; Marks P. A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [DOI] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discovery 2006, 5, 769–784. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- Piekarz R. L.; Frye R.; Prince H. M.; Kirschbaum M. H.; Zain J.; Allen S. L.; Jaffe E. S.; Ling A.; Turner M.; Peer C. J.; Figg W. D.; Steinberg S. M.; Smith S.; Joske D.; Lewis I.; Hutchins L.; Craig M.; Fojo A. T.; Wright J. J.; Bates S. E. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 2011, 117, 5827–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Balado C.; Nebbioso A.; Rodriguez-Graña P.; Minichiello A.; Miceli M.; Altucci L.; de Lera A. R. Bispyridinium dienes: histone deacetylase inhibitors with selective activities. J. Med. Chem. 2007, 50, 2497–2505. [DOI] [PubMed] [Google Scholar]
- Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D. 4th; Liu H.; Lowe J. T.; Marie J. C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B. C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y. L.; Fass D. M.; Reis S. A.; Zhao W. N.; Haggarty S. J.; Palmer M.; Foley M. A. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: Discovery of macrocyclic histone deacetylase inhibitors. J. Am. Chem. Soc. 2010, 132, 16962–16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen F. E.; Lewis J. A.; Cohen S. M. A new role for old ligands: discerning chelators for zinc metalloproteinases. J. Am. Chem. Soc. 2006, 128, 3156–3157. [DOI] [PubMed] [Google Scholar]
- Flipo M.; Charton J.; Hocine A.; Dassonneville S.; Deprez B.; Deprez-Poulain R. Hydroxamates: Relationships between structure and plasma stability. J. Med. Chem. 2009, 52, 6790–6802. [DOI] [PubMed] [Google Scholar]
- Montero A.; Beierle J. M.; Olsen C. A.; Ghadiri M. R. Design, synthesis, biological evaluation, and structural characterization of potent histone deacetylase inhibitors based on cyclic alpha/beta-tetrapeptide architectures. J. Am. Chem. Soc. 2009, 131, 3033–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C. A.; Ghadiri M. R. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic alpha3beta-tetrapeptides. J. Med. Chem. 2009, 52, 7836–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frackenpohl J.; Arvidsson P. I.; Schreiber J. V.; Seebach D. The outstanding biological stability of beta- and gamma-peptides toward proteolytic enzymes: An in vitro investigation with fifteen peptidases. ChemBioChem 2001, 2, 445–455. [DOI] [PubMed] [Google Scholar]
- Finnin M. S.; Donigian J. R.; Cohen A.; Richon V. M.; Rifkind R. A.; Marks P. A.; Breslow R.; Pavletich N. P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Cole K. E.; Dowling D. P.; Boone M. A.; Phillips A. J.; Christianson D. W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IC50 values are from BPS Bioscience, San Diego, CA.
- Bowers A. A.; Greshock T. J.; West N.; Estiu G.; Schreiber S. L.; Wiest O.; Williams R. M.; Bradner J. E. Synthesis and conformation-activity relationships of the peptide isosteres of FK228 and largazole. J. Am. Chem. Soc. 2009, 131, 2900–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass D. M.; Shah R.; Ghosh B.; Hennig K.; Norton S.; Zhao W.-N.; Reis S. A.; Klein P. S.; Mazitschek R.; Maglathlin R. L.; Lewis T. A.; Haggarty S. J. Short-Chain HDAC Inhibitors Differentially Affect Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2011, 2, 39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.; Altamura S.; De Francesco R.; Paz O. G.; Kinzel O.; Mesiti G.; Monteagudo E.; Pescatore G.; Rowley M.; Verdirame M.; Steinkühler C. A novel series of potent and selective ketone histone deacetylase inhibitors with antitumor activity in vivo. J. Med. Chem. 2008, 51, 23502353. [DOI] [PubMed] [Google Scholar]
- Chou C. J.; Herman D.; Gottesfeld J. M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggleby R. G.; Attwood P. V.; Wallace J. C.; Keech D. B. Avidin is a slow-binding inhibitor of pyruvate carboxylase. Biochemistry 1982, 21, 3364–3370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.