

Abstract
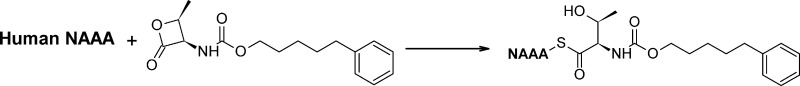
The cysteine amidase N-acylethanolamine acid amidase (NAAA) is a member of the N-terminal nucleophile class of enzymes and a potential target for anti-inflammatory drugs. We investigated the mechanism of inhibition of human NAAA by substituted β-lactones. We characterized pharmacologically a representative member of this class, ARN077, and showed, using high-resolution liquid chromatography–tandem mass spectrometry, that this compound forms a thioester bond with the N-terminal catalytic cysteine in human NAAA.
Keywords: NAAA, cysteine amidase, covalent inhibitors, high resolution mass spectrometry, proteomics
The fatty acid ethanolamides (FAEs) are a family of bioactive lipid molecules that have attracted considerable interest due to their potential role in the control of pain, inflammation, and energy metabolism. Two structurally and functionally distinct classes of FAEs have been described. Polyunsaturated FAEs, such as arachidonoylethanolamide (anandamide) and eicosapentaenoylethanolamide, are endogenous agonists of G protein-coupled cannabinoid receptors.1,2 These compounds are thought to act as retrograde messengers at synapses of the central nervous system and as local mediators in peripheral tissues. In the brain, anandamide-mediated signaling at CB1 type cannabinoid receptors has been implicated in the regulation of anxiety,3 depression,4,5 stress-induced analgesia,6 and nausea.7 Outside the brain, anandamide released at sites of tissue injury is thought to control the initiation of emerging pain signals,8,9 presumably by reducing the local release of proalgesic and proinflammatory factors from nociceptive nerve terminals.10 The biological actions of anandamide are interrupted by a two-step deactivation process consisting of carrier-mediated transport into cells11,12 followed by intracellular hydrolysis, which is catalyzed by the membrane-bound serine hydrolase, fatty acid amide hydrolase (FAAH). Inhibitors of this enzyme protect anandamide from deactivation and selectively magnify its intrinsic actions, producing an array of pharmacological effects that include reduced anxiety, depression, and pain.13 Saturated and monounsaturated FAEs, which include oleoylethanolamide (OEA) and palmitoylethanolamide (PEA), do not productively interact with cannabinoid receptors. These compounds are, however, potent or moderately potent agonists of nuclear peroxisome proliferator-activated receptor-α (PPAR-α), which is responsible for most of their analgesic and anti-inflammatory properties.14−16 OEA and PEA are produced in many mammalian tissues, including neurons17 and innate immune cells,18 where a selective phospholipase D releases them by cleaving the membrane precursor, N-acyl-phosphatidylethanolamine.19 They are deactivated either by FAAH, which prefers anandamide and OEA over PEA, or by NAAA, which conversely prefers PEA and OEA over anandamide.20 Despite its functional similarity with FAAH, NAAA shares no homology with this or other enzymes of the same “amidase signature” family. Rather, NAAA is an N-terminal nucleophile (Ntn) hydrolase and belongs to the choloylglycine hydrolase family of hydrolases, which are characterized by the ability to cleave nonpeptide amide bonds. Like other Ntn enzymes, NAAA is converted by proteolysis into a shorter active form upon incubation at acidic pH.20 Considering its predominant localization in lysosomes,21 it is conceivable that NAAA finds within these organelles the acidic pH (≈5.0) necessary for autocatalytic proteolysis and activation. Processing of NAAA renders a cysteine residue (Cys131 in mice and rats and Cys126 in humans) the N-terminal amino acid and catalytic nucleophile.18,21,22 Pharmacological blockade of intracellular NAAA activity results in a normalization of OEA and PEA levels, which are markedly reduced in inflammatory states,18,23,24 and exerts profound anti-inflammatory effects in animal models.18 These findings suggest that NAAA inhibition might represent a novel mechanistic approach to control inflammation. A few groups of NAAA inhibitors have been developed to test this hypothesis.18,25,26 Among them is a class of substituted β-lactones, which shows promising potency and target selectivity.18,26 The prototype of this class, (S)-N-(2-oxo-3-oxetanyl)-3-phenylpropanamide [(S)-OOPP, Figure 1], is a potent noncompetitive NAAA inhibitor that is highly effective at preventing PEA and OEA hydrolysis in activated inflammatory cells and at reducing tissue reactions to various proinflammatory stimuli.18
Figure 1.

Chemical structures of β-lactone NAAA inhibitors, (S)-N-(2-oxo-3-oxetanyl)-3-phenylpropanamide [(S)-OOPP], N-[(2S,3R)-2-methyl-4-oxo-oxetan-3-yl] carbamate (ARN077), and N-[(2S,3R)-2-methyl-4-oxo-oxetan-3-yl]-7-phenyl-heptanamide (ARN768).
We have recently used the structure of (S)-OOPP as a starting point to discover β-lactone-based NAAA inhibitors with improved potency. This investigation lead us to identify several new compounds that selectively inhibit NAAA at low nanomolar concentrations, including 5-phenylpentyl N-[(2S,3R)-2-methyl-4-oxo-oxetan-3-yl] carbamate (ARN077, Figure 1). The structure–activity relationship properties of ARN077 will be described elsewhere (Ponzano, S.; et al. Submitted for publication). In the present study, we used this molecule as a probe to characterize the mechanism through which substituted β-lactones inhibit NAAA. Our results indicate that the N-terminal cysteine of human recombinant NAAA (h-NAAA) is acylated by ARN077 and that the new bond formed is a thioester between the cysteine thiol and the carbonyl group of the β-lactone.
ARN077 (Figure 1) inhibited the activity of h-NAAA with a median effective concentration (IC50) of 7.3 ± 1.6 nM (n = 3) (Figure 2A) and a Hill coefficient of −0.8. By contrast, at the concentrations tested (up to 30 μM), the compound had no significant effect on the activity of FAAH or acid ceramidase, a cysteine amidase that exhibits 33–35% amino acid identity with NAAA27 (Supporting Information, Table 1). Kinetic and time–course experiments showed that the inhibitory effect of ARN077 was noncompetitive (Figure 2B and Supporting Information, Table 1) and reached half-maximal inhibition in 2.4 min and maximum inhibition within 10 min of incubation with the enzyme (Figure 2C).
Figure 2.

Inhibition of human NAAA by ARN077. (A) Concentration–response curve for inhibition of recombinant hNAAA by ARN077 after 30 min of incubation. (B) Michaelis–Menten analysis of ARN077 (vehicle, ●; 10 nM ARN077, ■; and 30 nM ARN077, ▲). (C) Preincbation time course of ARN077 (100 nM). (D) Reversibility study of ARN077 (vehicle, open bars; ARN077, black bars). *p < 0.05 ARN077 postdialysis vs vehicle postdialysis; **p < 0.01 ARN077 postdialysis vs ARN077 predialysis; and ***p < 0.001 ARN077 predialysis vs vehicle predialysis.
Following centrifugation through a dialyzing membrane, the inhibition of h-NAAA activity produced by ARN077 (100 nM) was markedly reduced but not completely reversed (Figure 2D). The results indicate that ARN077 is a potent h-NAAA inhibitor that acts through a noncompetitive and partially reversible mechanism. Compounds that contain a β-lactone ring are known to interact covalently with biological nucleophiles, and some have been shown to inhibit Ntn enzymes such as NAAA.26 This led us to hypothesize that ARN077 might interact with h-NAAA through a covalent binding to the enzyme's catalytic cysteine. h-NAAA is translated as a 331 amino acid protein, which undergoes autoproteolytic cleavage at acidic pH 4.5 forming a new N-terminus, Cys126, which serves as the catalytic nucleophile.28 We examined, therefore, whether ARN077 might form a covalent adduct with the N-terminal cysteine of the active form of h-NAAA. If present, this modification should be carried by the corresponding N terminus peptide produced by trypsin digestion (C126TSIVAQDSR; MW 1078.51). The position of this peptide within the primary sequence of the expressed h-NAAA is underlined below, along with the position of the trypsin cleavage sites (in bold):
MRTADREARPGLPSLLLLLLAGAGLSAASPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFC126TSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNLTIYTTVMSAGSPDKYMTRIRNPSRK
A control incubation of h-NAAA at pH 4.5 for 90 min yielded a peptide of mass-to-charge ratio (m/z) 540.26 (Figure 3A), which was unambiguously identified as the product of autoproteolytic cleavage of NAAA (Figure 3B), because the corresponding MS/MS spectrum shows a complete match of the y fragment ions series.29 We next searched, in incubations of intact h-NAAA with ARN077 (both at 5 μM), for adducts that might be generated by the nucleophilic attack of the thiol group of Cys126 on ARN077. As illustrated in Scheme 1, three such adducts are expected to be produced: the thioether a, formed through S-alkylation of the carbon in the 2-position of the oxooxetan ring of ARN077; the thioester b, formed through S-acylation of the oxooxetan carbonyl carbon of ARN077; and the thioesters c and c′, formed by the attack of Cys 126 on the carbamate group of ARN077.
Figure 3.

(A) Extracted ion chromatograms of the native (540.26 m/z) and acylated (685.43 m/z) peptide (for clarity purposes, peaks have not been normalized). (B and C) MS/MS spectra of m/z 540.26 and m/z 685.43. In both spectra, the y fragment ions series perfectly match a CTSIVAQDSR sequence (bearing a side chain adduct on the cysteine in the second case).
Scheme 1. Possible Nucleophylic Attacks of Cysteine on ARN077.
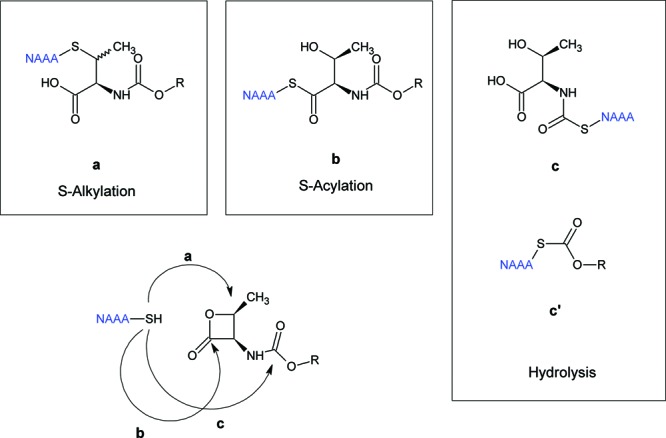
Path a, thioether adduct; path b, thioester adduct; and path c, hydrolysis of the carbamate, with two possible adducts (c and c′). R = (CH2)5-Ph.
The adducts a and b are isomers of formula C58H95N15O21S and are expected each to generate a doubly charged ion of m/z 685.43. A peptide with this m/z value was eluted approximately 20 min after the native, unmodified peptide (Figure 3A). MS/MS analysis of this component (Figure 3C) produced a y fragment ion series that matched that of the native peptide (Figure 3B), clearly indicating that ARN077 covalently links to the N-terminal cysteine of NAAA. The presence in the MS/MS spectrum of an ion of m/z 292.16, together with the corresponding loss of water (m/z 274.14), indicate a fragmentation of the acylated cysteine side chain (C16H22NO4). This is confirmed by the presence of the singly charged peptide ion at m/z 1079.52. However, this fragmentation did not allow us to conclusively determine whether the broken bond was a thioester or a thioether. Diagnostic ions that would permit to discriminate between S-alkylation and S-acylation are expected to be formed upon C–C cleavage of the thioether or α-cleavage of the thioester (Supporting Information, Figure 1). The former, which should produce an ion of m/z 264.12 (C14H18NO4), was not detected in our analyses, while the latter, which should produce an ion of m/z 264.16 (C16H24NO2), is rather weak and was covered by the second isotopic peak (m/z 264.16) of the very intense y2 ion of the backbone fragmentation at m/z 262.15. These results tentatively suggest that ARN077 might form a thioester bond with the N-terminal cysteine of h-NAAA, but additional evidence is needed to draw such conclusion. To fill this gap, we tested the ability of the compound ARN768 (Figure 1), an amide analogue of ARN077 that is a relatively potent h-NAAA inhibitor (IC50 314 ± 52 nM ; n = 3), to interact covalently with h-NAAA. We found that ARN0768 reacts with h-NAAA forming, as does ARN077, a peptide adduct whose MS/MS spectrum shows a side chain fragment ion at m/z 290.18 (C17H24NO3), the corresponding loss of water at m/z 272.17, and a clearly detectable ion at m/z 262.18 (Figure 4).
Figure 4.

Zoomed MS/MS spectra of peptide CTSIVAQDSR acylated by ARN077 and ARN768. Cysteine side chain fragment ions are indicated along with the y2 fragment (262.15 m/z). S-Acylation diagnostic fragment ion at 262.18 m/z is reported in the inset.
The latter corresponds to the cleavage of a thioester (C15H22NO3) rather than a thioether (C15H20NO3), whose corresponding ion at m/z 262.14 was not detected. Notably, attack on the carbamate group (mechanism c in Scheme 1) is expected to produce the modified peptides c (C47H81N15O21S) and c′ (C54H88N14O19S), with the alkyl-phenyl chain or the threonine residue as respective leaving groups. These peptides were not detected in our experiments, and the corresponding mechanism was therefore tentatively excluded. Furthermore, no evidence of adduct formation was found when the h-NAAA holoenzyme was incubated with ARN077, indicating that only the active form of the enzyme is subjected to S-acylation by this compound. Together, these findings suggest that substituted β-lactones, such as ARN077 and ARN768, react with Cys126 in h-NAAA to form a thioester bond. Moreover, we found no evidence of other peptide adducts with either inhibitor, suggesting that β-lactones selectively interact with the N-terminus cysteine.
The high-resolution mass spectrometry studies reported here indicate that the substituted β-lactones, ARN077 and ARN768, inhibit h-NAAA activity through a mechanism that requires S-acylation of the catalytically active N-terminal cysteine residue of this enzyme. These findings are consistent with the pharmacological properties of ARN077, which blocks h-NAAA activity in a noncompetitive and partially reversible manner (as expected from the covalent yet hydrolyzable nature of a thioester bond). The results underscore the value of bottom-up proteomics as an essential tool for the direct validation of reaction mechanisms that are important in drug discovery. In the present case, the high-resolution tandem mass capabilities of the hybrid quadrupole-TOF instrument allowed us to unequivocally assign the structure of side chain fragment ions, which resulted in an unambiguous assignment of the chemical structure of the modified peptide. Furthermore, our results provide a solid mechanistic basis for the identification of a second generation of β-lactone-based NAAA inhibitors with improved potency, selectivity, and metabolic stability.
Acknowledgments
We thank Sine Mandrup Bertozzi for reverse phase and chiral HPLC purifications, Luca Goldoni for NMR technical support, and Silvia Venzano for compounds handling.
Supporting Information Available
Detailed description of the experimental procedures, MS/MS fragmentation analysis, and pharmacological data of ARN077. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
AA, GG, FB designed the experiments and analyzed the data. SP synthesized β-lactones. GT, MD and LM performed pharmacological profiling of ARN077. ER and CK produced the recombinant h-NAAA. DP oversaw the project with assistance from TB, AR, GT, MM. AA and DP wrote the manuscript.
Funding for this research was provided by the Istituto Italiano di Tecnologia.
The authors declare the following competing financial interest(s): A patent protecting ARN077 and related compounds was filed by the following authors: Piomelli, D.; Bandiera, T.; Mor, M.; Tarzia, G.; Bertozzi, F.; Ponzano, S.
Supplementary Material
References
- Devane W. A.; Hanus L.; Breuer A.; Pertwee R. G.; Stevenson L. A.; Griffin G.; Gibson D.; Mandelbaum A.; Etinger A.; Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [DOI] [PubMed] [Google Scholar]
- Kathuria S.; Gaetani S.; Fegley D.; Valino F.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; La Rana G.; Calignano A.; Giustino A.; Tattoli M.; Palmery M.; Cuomo V.; Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [DOI] [PubMed] [Google Scholar]
- Gobbi G.; Bambico F. R.; Mangieri R.; Bortolato M.; Campolongo P.; Solinas M.; Cassano T.; Morgese M. G.; Debonnel G.; Duranti A.; Tontini A.; Tarzia G.; Mor M.; Trezza V.; Goldberg S. R.; Cuomo V.; Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18620–18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M.; Mangieri R. A.; Fu J.; Kim J. H.; Arguello O.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry 2007, 62, 1103–1110. [DOI] [PubMed] [Google Scholar]
- Hohmann A. G.; Suplita R. L.; Bolton N. M.; Neely M. H.; Fegley D.; Mangieri R.; Krey J. F.; Walker J. M.; Holmes P. V.; Crystal J. D.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature 2005, 435, 1108–1112. [DOI] [PubMed] [Google Scholar]
- Parker L. A.; Limebeer C. L.; Rock E. M.; Litt D. L.; Kwiatkowska M.; Piomelli D. The FAAH inhibitor URB-597 interferes with cisplatin- and nicotine-induced vomiting in the Suncus murinus (house musk shrew). Physiol. Behav. 2009, 97, 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A.; La Rana G.; Giuffrida A.; Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature 1998, 394, 277–281. [DOI] [PubMed] [Google Scholar]
- Clapper J. R.; Moreno-Sanz G.; Russo R.; Guijarro A.; Vacondio F.; Duranti A.; Tontini A.; Sanchini S.; Sciolino N. R.; Spradley J. M.; Hohmann A. G.; Calignano A.; Mor M.; Tarzia G.; Piomelli D. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 2010, 13, 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J. D.; Aanonsen L.; Hargreaves K. M. Antihyperalgesic effects of spinal cannabinoids. Eur. J. Pharmacol. 1998, 345, 145–153. [DOI] [PubMed] [Google Scholar]
- Beltramo M.; Stella N.; Calignano A.; Lin S. Y.; Makriyannis A.; Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 1997, 277, 1094–1097. [DOI] [PubMed] [Google Scholar]
- Fu J.; Bottegoni G.; Sasso O.; Bertorelli R.; Rocchia W.; Masetti M.; Guijarro A.; Lodola A.; Armirotti A.; Garau G.; Bandiera T.; Reggiani A.; Mor M.; Cavalli A.; Piomelli D. A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat. Neurosci. 2012, 15, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D.; Tarzia G.; Duranti A.; Tontini A.; Mor M.; Compton T. R.; Dasse O.; Monaghan E. P.; Parrott J. A.; Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Gaetani S.; Oveisi F.; Lo Verme J.; Serrano A.; Rodriguez De Fonseca F.; Rosengarth A.; Luecke H.; Di Giacomo B.; Tarzia G.; Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 2003, 425, 90–93. [DOI] [PubMed] [Google Scholar]
- Russo R.; Loverme J.; La Rana G.; Compton T. R.; Parrott J.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Calignano A.; Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J. Pharmacol. Exp. Ther. 2007, 322, 236–242. [DOI] [PubMed] [Google Scholar]
- LoVerme J.; La Rana G.; Russo R.; Calignano A.; Piomelli D. The search for the palmitoylethanolamide receptor. Life Sci. 2005, 77, 1685–1698. [DOI] [PubMed] [Google Scholar]
- Cadas H.; Gaillet S.; Beltramo M.; Venance L.; Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J. Neurosci. 1996, 16, 3934–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solorzano C.; Zhu C.; Battista N.; Astarita G.; Lodola A.; Rivara S.; Mor M.; Russo R.; Maccarrone M.; Antonietti F.; Duranti A.; Tontini A.; Cuzzocrea S.; Tarzia G.; Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 20966–20971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda N.; Tsuboi K.; Uyama T. Enzymological studies on the biosynthesis of N-acylethanolamines. Biochim. Biophys. Acta 2010, 1801, 1274–1285. [DOI] [PubMed] [Google Scholar]
- Ueda N.; Tsuboi K.; Uyama T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog. Lipid Res. 2010, 49, 299–315. [DOI] [PubMed] [Google Scholar]
- West J. M.; Zvonok N.; Whitten K. M.; Wood J. T.; Makriyannis A. Mass Spectrometric Characterization of Human N-Acylethanolamine-hydrolyzing Acid Amidase. J. Proteome Res. 2012, 11, 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhao L. Y.; Uyama T.; Tsuboi K.; Tonai T.; Ueda N. Amino acid residues crucial in pH regulation and proteolytic activation of N-acylethanolamine-hydrolyzing acid amidase. Biochim. Biophys. Acta 2008, 1781, 710–717. [DOI] [PubMed] [Google Scholar]
- Richardson D.; Pearson R. G.; Kurian N.; Latif M. L.; Garle M. J.; Barrett D. A.; Kendall D. A.; Scammell B. E.; Reeve A. J.; Chapman V. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Solorzano C.; Sahar S.; Realini N.; Fung E.; Sassone-Corsi P.; Piomelli D. Proinflammatory stimuli control N-acylphosphatidylethanolamine-specific phospholipase D expression in macrophages. Mol. Pharmacol. 2011, 79, 786–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K.; Hilligsmann C.; Vandevoorde S.; Lambert D. M.; Ueda N. N-cyclohexanecarbonylpentadecylamine: a selective inhibitor of the acid amidase hydrolysing N-acylethanolamines, as a tool to distinguish acid amidase from fatty acid amide hydrolase. Biochem. J. 2004, 379, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solorzano C.; Antonietti F.; Duranti A.; Tontini A.; Rivara S.; Lodola A.; Vacondio F.; Tarzia G.; Piomelli D.; Mor M. Synthesis and structure-activity relationships of N-(2-oxo-3-oxetanyl)amides as N-acylethanolamine-hydrolyzing acid amidase inhibitors. J. Med. Chem. 2010, 53, 5770–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K.; Sun Y. X.; Okamoto Y.; Araki N.; Tonai T.; Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J. Biol. Chem. 2005, 280, 11082–11092. [DOI] [PubMed] [Google Scholar]
- Tsuboi K.; Takezaki N.; Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA). Chem. Biodiversity 2007, 4, 1914–1925. [DOI] [PubMed] [Google Scholar]
- Biemann K. Appendix 5. Nomenclature for peptide fragment ions (positive ions). Methods Enzymol. 1990, 193, 886–887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.