

Abstract

A series of aryl piperazinyl ureas that act as covalent inhibitors of fatty acid amide hydrolase (FAAH) is described. A potent and selective (does not inhibit FAAH-2) member of this class, JNJ-40355003, was found to elevate the plasma levels of three fatty acid amides: anandamide, oleoyl ethanolamide, and palmitoyl ethanolamide, in the rat, dog, and cynomolgous monkey. The elevation of the levels of these lipids in the plasma of monkeys suggests that FAAH-2 may not play a significant role in regulating plasma levels of fatty acid ethanolamides in primates.
Keywords: FAAH, urea, ethanolamides, FAAH-2, enzyme, anandamide
Cannabis sativa has been used medicinally for the treatment of pain and other conditions for thousands of years,1 but it was not until the discovery and cloning of two cannabinoid receptors CB12 and CB23 in the early 1990s that a rationale for the analgesic effects of cannabis preparations became possible. The principal active ingredient of cannabis, Δ9-THC, is not an ideal drug due to numerous adverse side effects (hypothermia, catalepsy, hyperphagia, delayed gastric emptying, dizziness, and diminished motor coordination and memory) associated with its use.4 The later discovery of anandamide (AEA)5 (Figure 1), an agonist of CB1,6−8 and fatty acid amide hydrolase (FAAH),10 the enzyme that hydrolyzes AEA, opened the possibility of engaging the cannabinoid system without the use of direct exogenous cannabinoid agonists.
Figure 1.

Δ9-THC and substrates of FAAH.
Unfortunately, AEA is also a nonideal drug substance because it has poor physical properties and is rapidly (T1/2 on the order of a few minutes)9 metabolized by FAAH to give ethanolamine and arachidonic acid.10 FAAH also breaks down several other substances, including N-palmitoylethanolamide (PEA), N-oleoylethanolamide (OEA), oleamide (OA), 2-arachidonyl glyceryl ether (noladin), O-arachidonylethanolamine (virodamine), and N-arachidonyldopamine.11,12 PEA is known to have anti-inflammatory properties and exert analgesic effects through a noncannabinoid pathway.13,14 OEA appears to be involved in regulating satiety,15 whereas the similar OA is an important contributor to sleep induction.16 The pharmacology of the other FAAH substrates is less well understood.
FAAH appears to be the primary enzyme responsible for the metabolism of AEA, PEA, and OEA as the levels of these fatty acid amides (FAAs) are all greatly elevated in FAAH knockout mice.8 These knockout mice are healthy but are phenotypically less sensitive to pain, thus supporting the notion that pharmacologically induced FAAH inhibition may afford analgesia. More recently, another fatty acid amide metabolizing enzyme (FAAH-2) was discovered and cloned.17 FAAH-2 is not expressed in rats or dogs but is present in monkeys and humans. While the preferred substrates of FAAH-2 are primary amides, its expression in primates raises the question of whether or not a selective FAAH inhibitor would be able to elevate the levels of FAAs in primates significantly.
Numerous groups have reported the preparation and pharmacological testing of FAAH inhibitors (Figure 2). Boger's group described several series of highly potent α-keto heterocycles.18,19 Piomelli20 disclosed carbamate-based inhibitors as did Sanofi.21 Pfizer,22−24 Takeda,25 and Johnson & Johnson26 have disclosed phenyl and heteroaryl urea inhibitors of FAAH. All of these compounds are quite potent and form covalent bonds with Ser241 within the FAAH active site. OL-135 forms a reversible tetrahedral hemiketal intermediate derived from the attack of Ser241 onto the ketone.27,28 URB59729 and the carbamates reported by Sanofi carbamylate the active site Ser241 of the FAAH enzyme with the concomitant loss of a phenolic or alcoholic fragment, respectively.30 The ureas also form a carbamate with the FAAH enzyme but with the loss of an aniline or heteroaryl amine rather than an alcohol.31,32 OL-135,33 URB597,34 JNJ-1661010,26 and PF-0445784535 have been found to be efficacious in the treatment of pain in various animal models without the motor impairment associated with direct CB1 agonism. Encouragingly, at least two compounds have been evaluated in the clinic. Pfizer's PF-04457845 was evaluated as an analgesic (phase II) and Sanofi-Aventis' SSR411298 for major depression (phase II).36 While no data have been posted for SSR411298, the study was completed in 2010. PF-04457845, on the other hand, was found to be ineffective in people for the reduction of OA pain in the knee.37
Figure 2.

Some known FAAH inhibitors.
Our internal HTS efforts identified a series of potent FAAH inhibitors from a purchased combinatorial library (Figure 3). Resynthesis of an example showed that FAAH inhibition came not from the presumed structure but from an unknown impurity. An analysis of the likely synthesis and side products suggested the possibility of a benzyl piperidine urea contaminant. A combination of synthesis and cheminformatics analyses of our corporate collection identified benzyl piperazine urea (1). Compound (1) was a potent FAAH inhibitor, and its modular structure suggested the possibility of rapidly optimizing this chemotype to yield more potent analogues.
Figure 3.

From an impure HTS hit to benzyl piperazine ureas.
We elected to first optimize the pendent benzyl substituent, keeping the phenyl urea constant. Treatment of 1-Boc piperazine with phenyl isocyanate and then removing the Boc group with 4.0 N HCl in dioxane gave (2), a common intermediate that allowed for rapid diversification of the benzyl group (Scheme 1).23 Treatment of (2) with a benzaldehyde, diisopropylethylamine, and resin supported triacetoxyborohydride [MP-BH(OAc)3] in THF gave the desired final products in reasonable yield. A selection of compounds prepared in this manner is shown in Table 1. Because these aryl ureas are known to be covalent inhibitors of FAAH, their activities are reported as apparent IC50 values after 1 h of incubation with the enzyme.22
Scheme 1. Preparation of Anilinyl Piperazinyl Ureas: Variation of Benzyl Substituents.
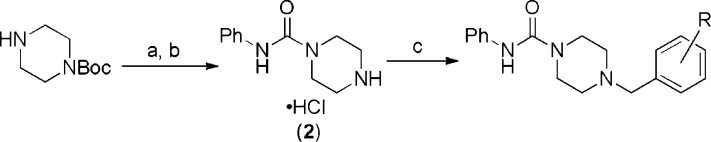
Reagents and conditions: (a) 1.1 mol equiv PhNCO, methylene chloride, 4 h, rt, 93%. (b) 4 N HCl in 1,4-dioxane, methylene chloride, 40 h, rt, 96%. (c) 1.5 mol equiv aldehyde (R defined in Table 1), 9 mol equiv diisopropylethylamine, 2.5 mol equiv MP-BH(OAc)3, THF, rt, 24 h, 17–66%.
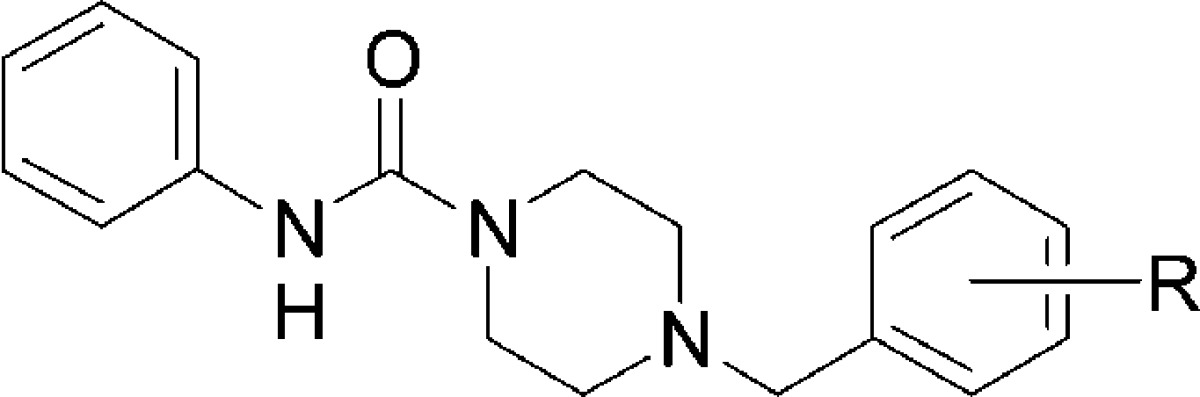
Table 1. SAR of the Pendant Benzyl Group.
| IC50 (nM)a |
|||
|---|---|---|---|
| entry | R | hFAAH | rFAAH |
| 3 | H | >10000 | >10000 |
| 4 | 2-Cl | 4670 ± 1100 | >10000 |
| 5 | 3-Cl | 840 ± 150 | 5600 ± 860 |
| 6 | 4-Cl | 128 ± 60 | 2670 ± 2000 |
| 7 | 3-F | 2470 ± 650 | >10000 |
| 8 | 4-F | 2770 ± 780 | >10000 |
| 9 | 3-CN | 6670 ± 2300 | 10000 ± 0 |
| 10 | 4-CN | 4670 ± 1800 | 7670 ± 1800 |
| 11 | 3-CF3 | 350 ± 90 | 830 ± 200 |
| 12 | 4-CF3 | 50 ± 19 | 260 ± 70 |
| 13 | 3-OMe | 1530 ± 220 | >10000 |
| 14 | 4-OMe | 490 ± 200 | >10000 |
| 15 | 3-OPh | 74 ± 14 | 6670 ± 800 |
| 16 | 4-OPh | 2830 ± 1400 | >10000 |
| 17 | 3-(4-chlorophenoxy) | 6 ± 4 | 300 ± 90 |
| 18 | 4-(4-chlorophenoxy) | 62 ± 28 | 4000 ± 1000 |
| 19 | 3-(4-methylphenoxy) | 11 ± 2.4 | 640 ± 120 |
| 20 | 3-(4-methoxy-phenoxy) | 12 ± 0.7 | 133 ± 11 |
| 21 | 3-(4-t-butyl-phenoxy) | 46 ± 12 | 75 ± 16 |
These are apparent IC50 values with values obtained after a 60 min preincubation with the enzyme; n ≥ 3 ± standard error.
From Table 1, it is apparent that substitution is required on the benzyl group for there to be some affinity for the FAAH enzyme. Generally, substituents in the 3- and 4-positions of the benzyl group were better accommodated than those in the 2-position. Polar substituents, such as nitriles, were less preferable than lipophilic groups, which is consistent with the lipophilic nature of the enzyme binding pocket. When the lipophilic groups are small (Cl, CF3, and OMe), there is a larger boost in potency when they are in the 4-position. However, as the lipophilic groups get larger, it becomes clear that the enzyme can better accommodate extra bulk in the 3-position of the benzyl group, thus giving rise to potent FAAH inhibitors (17 and 19–21).
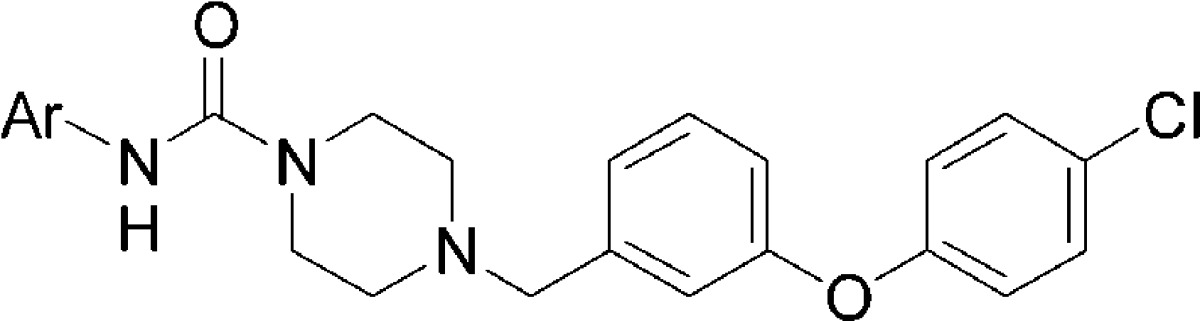
Recognizing that anilines have a propensity to be genotoxic38−40 and that a covalent FAAH inhibitor would release an equivalent of aniline upon inactivation of the enzyme, we began to examine heteroaromatic amines as possible substitutes. As 17 was both one of the most potent analogues made and had the least obvious metabolic liabilities, we elected to keep the 3-(4-chlorophenoxy)benzyl substituent constant while exploring aniline replacements. Heteroaryl ureas were not synthesized from isocyanates but rather from their corresponding phenyl carbamates, which can be prepared in a single step (Scheme 2). Consistent with previous reports,32 the 3-pyridyl analogue (23, JNJ-40355003) was the most potent inhibitor of both h- and rFAAH.
Scheme 2. Preparation of Heteroaryl Piperazinyl Ureas.

Reagents and conditions (note: HetAr as defined in Table 2 as a subset of Ar): (a) 0.9 mol equiv ClCO2Ph, 1.1 mol equiv pyridine, CH3CN, 0 °C–rt, 2 h, 80–81%. b) 1/3 mol equiv ClCO2Ph, CH3CN, rt, 31 h, 90%. (c) 1 mol equiv 1-Boc-piperazine or (40), 1–1.2 mol equiv heteroaryl phenyl carbamate, 2 mol equiv Et3N, 1,2-dichloroethane or DMSO, 18–24 h, rt, 34%-quant. (d) 2 M HCl, MeOH, 18 h, rt, 75% or methylene chloride/trifluoroacetic acid, quant. (e) 1.2–2 mol equiv 3-(4-chlorophenoxy)benzaldehyde, 2–4 mol equiv MP-BH(OAc)3, 6.0 mol equiv Et3N, THF, rt, 18–48 h, 36–82%.
Table 2. Heteroaryl Urea SAR.
| IC50 (nM)a |
|||
|---|---|---|---|
| entry | Ar | hFAAH | rFAAH |
| 17 | phenyl | 6 ± 4 | 300 ± 90 |
| 22 | 4-pyridyl | 15 ± 6.1 | 520 ± 80b |
| 23 | 3-pyridyl | 1.4 ± 0.41 | 33 ± 9 |
| 24 | 2-pyridyl | 12 ± 2.5 | 470 ± 180 |
| 25 | 4-pyrimidyl | 41 ± 17 | 1500 ± 620 |
These are apparent IC50 values with values obtained after a 60 min preincubation with the enzyme; n ≥ 3 ± standard error.
N = 2.
We modeled JNJ-40355003 in the crystal structure of humanized rFAAH31,41 with the carbonyl bound covalently to Ser241, forming a tetrahedral hemiketal-like intermediate (Figure 1 in the Supporting Information). The benzyl substituent is in an environment very similar to that of the reported crystal structure, making a key face-to-edge π-interaction with Phe192. Before collapse of the tetrahedral intermediate and expulsion of 3-aminopyridine, there is a possibility of hydrogen bond interactions between the urea N–H and the carbonyl of Met191 and the side chain of Thr236 (alternatively, rotation of the aryl group could expose the nitrogen to water molecules in this cavity).
We elected to profile JNJ-40355003 in greater detail. JNJ-40355003 has excellent physical properties42 and is readily formulated for both po and iv dosing. JNJ-40355003 has two basic nitrogens, and the bis-HCl salt 1.5 hydrate is soluble to the extent of >20 mg/mL in deionized water. Concentrated samples of JNJ-40355003 gave rise to highly acidic solutions, which, if intended for iv delivery, were titrated to pH 4.5 with aqueous NaOH.
Rats dosed orally with JNJ-40355003 showed a dose proportional increase in compound concentrations both in the plasma and in the brain with a brain to plasma ratio of 2.62 (Figure 3 in the Supporting Information).43 A time–course study of FAAH inhibition and FAA elevation was conducted (Figure 4 in the Supporting Information) using JNJ-40355003 at a dose of 3 mg/kg (po). Consistent with observations by others,20,22 an almost complete blockade of the FAAH enzyme is required to elevate AEA, with peak concentrations observed approximately 4 h (Cp of JNJ-40355003 ≈ 800 nM) postdosing. PEA and OEA concentrations peaked approximately 6 h (Cp of JNJ-40355003 ≈ 600 nM) postdosing. PEA and OEA increased more readily and to a much greater degree than AEA, which is consistent with AEA being a better substrate for the enzyme than the other two ethanolamides.44 By 12 h postdose (Cp of JNJ-40355003 ≈ 200 nM), levels of the FAAs are decreasing even though FAAH is still 90% inhibited. By 24 h (Cp of JNJ-40355003 ≈ 15 nM), AEA, PEA, and OEA levels had returned to their basal levels, and by 48 h, there was complete recovery of enzymatic activity.
We then conducted a dose–response (0.1–60 mg/kg) study of FAA elevation with JNJ-40355003. FAA levels were measured at the 4 h time point postdose (Figure 5 in the Supporting Information) in both the brain and the plasma. Maximum FAA elevations in the brain were observed at the 10 mg/kg dose (approximate fold increase: AEA, 1.75; PEA, 3.7; OEA, 3.25; FAA levels plateau at higher doses), whereas maximal plasma concentrations were achieved at 3 mg/kg (approximate fold increase: AEA, 3; PEA, 2.25; and OEA, 1.9).
The behavior of JNJ-40355003 in the dog was somewhat surprising. The PK properties of JNJ-40355003 in beagle dogs were reasonable (Cl = 2.3 L/h/kg, Vz = 1.4 L/kg, Vss = 1.24 L/kg, and F = 39%), but the degree to which the individual FAAs were elevated was quite unexpected (Figure 6 in the Supporting Information). Whereas in rats FAAs typically achieve a 2–4-fold increase over basal levels, the peak concentrations of all three FAAs measured in dogs were >30-fold over basal levels. AEA and OEA concentrations peaked 2–3 h postdosing, whereas PEA levels peaked in the 6–8 h time frame. These dramatic elevations of FAAs in dogs were not associated with any adverse behavioral findings.
Unlike humans and monkeys, neither the rat nor the dog expresses functional FAAH-2 enzyme. The degree to which enzymatically active FAAH-2 in primates would offset any benefits of inhibition of FAAH is, perhaps, an open question. PF-04457845 is reported to be completely selective for FAAH, at least among serine hydrolases,24 but no specific mention of selectivity over FAAH-2 was published. However, a closely related molecule, PF-3845, has been found to have an IC50 >10 μM against this FAAH isoform.32,45 As JNJ-40355003 also lacks FAAH-2 inhibitory activity (>10 μM), we were interested in determining the effect that this compound would have on FAA levels in a species expressing FAAH-2. To this end, we measured FAA levels in cynomolgous monkeys dosed with JNJ-40355003.
JNJ-40355003 had good PK properties in the monkey (Cl = 1.52 L/h/kg, Vz = 2.18 L/kg, Vss = 1.89 L/kg, and F = 62%), and more importantly, plasma levels for all three FAAs examined were elevated (300–400% for AEA, 150–200% OEA, and 150–175% PEA; Figure 7 in the Supporting Information). The degree of AEA elevation was greater than that observed in the rat but less than in the dog. The peak levels of all three FAAs were reached 4 h after iv dosing. As PF-04457845 raised plasma levels of the FAAs in human subjects, it may be reasonable to postulate that dual inhibition of FAAH and FAAH-2 is not necessary to achieve elevated levels of FAAH's primary substrates in primates. Because it is not known what FAA levels could be achieved if both FAAH and FAAH-2 were inhibited,10,46 we cannot rigorously conclude that inhibition of FAAH-2 would have no impact on the efficacy of a FAAH inhibitor. However, it is reassuring that FAA levels can be influenced by the selective inhibition of FAAH.
In conclusion, we have described a series of substituted phenyl and heteroaryl urea FAAH inhibitors. The highly potent FAAH inhibitor JNJ-40355003 was found to have good physical and PK properties and elevated the plasma and brain concentrations of AEA, PEA, and OEA in rats after oral dosing. Similarly, plasma levels of these three lipids were found to be elevated in dogs and monkeys orally dosed with JNJ-40355003. The elevation of FAAs in the monkey strongly suggests that FAAH-2 is not contributing greatly to the breakdown of fatty acid ethanolamides in the plasma; thus, its inhibition is unlikely to be required for there to be a pharmacological benefit from blockade of the FAAH enzyme in humans. Additional studies involving urea inhibitors of FAAH will be reported in due course.
Supporting Information Available
Detailed experimental procedures, annotated 1H NMR, and MS data for the described compounds as well as graphical representations of in vivo data for JNJ-40355003 can be found in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Calixto J. B.; Beirith A.; Ferreira J.; Santos A. R.; Filho V. C.; Yunes R. A. Naturally occurring antinociceptive substances from plants. Phytother. Res. 2000, 14, 401–418. [DOI] [PubMed] [Google Scholar]
- Matsuda L. A.; Lolait S. J.; Brownstein M. J.; Young A. C.; Bonner T. I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature (London, U.K.) 1990, 346, 561–564. [DOI] [PubMed] [Google Scholar]
- Munro S.; Thomas K. L.; Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature (London, U.K.) 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- Grotenhermen F. The toxicology of cannabis and cannabis prohibition. Chem. Biodiversity 2007, 4, 1744–1769. [DOI] [PubMed] [Google Scholar]
- Devane W. A.; Hanus L.; Breuer A.; Pertwee R. G.; Stevenson L. A.; Griffin G.; Gibson D.; Mandelbaum A.; Etinger A.; Mechoulam R. Isolation and structure of a brain constituent that bind to the cannabinoid receptor. Science 1992, 258, 1946–1949. [DOI] [PubMed] [Google Scholar]
- Lichtman A. H.; Hawkins E. G.; Griffin G.; Cravatt B. F. Pharmacological Activity of Fatty Acid Amides Is Regulated, but Not Mediated, by Fatty Acid Amide Hydrolase in Vivo. JPET 2002, 302, 73–79. [DOI] [PubMed] [Google Scholar]
- Steffens M.; Zentner J.; Honegger J.; Feuerstein T. J. Binding affinity and agonist activity of putative endogenous cannabinoids at the human neocortical CB1 receptor. Biochem. Pharmacol. 2005, 69, 169–178. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Giang D. K.; Mayfield S. P.; Boger D. L. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature (London, U.K.) 1996, 384, 83–87. [DOI] [PubMed] [Google Scholar]
- Willoughby K. A.; Moore S. F.; Martin B. R.; Ellis E. F. The biodistribution and metabolism of anandamide in mice. J. Pharmacol. Exp. Ther. 1997, 282, 243–247. [PubMed] [Google Scholar]
- Cravatt B. F.; Demarest K.; Patricelli M. P.; Bracey M. H.; Giang D. K.; Martin B. R.; Lichtman A. H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 9371–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Cravatt B. F. Fatty acid amide hydrolase competitively degrades bioactive amides and esters through a nonconventional catalytic mechanism. Biochemistry 1999, 38, 14125–14130. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Fecik R. A.; Patterson J. E.; Miyauchi H.; Patricelli M. P.; Cravatt B. F. Fatty acid amide hydrolase substrate specificity. Bioorg. Med. Chem. Lett. 2000, 10, 2613–2616. [DOI] [PubMed] [Google Scholar]
- Lambert D. M.; Vandevoorde S.; Jonsson K. O.; Fowler C. J. The palmitoylethanolamide family: A new class of anti-inflammitory agents. Curr. Med. Chem. 2002, 9, 663–674. [DOI] [PubMed] [Google Scholar]
- Lo Verme J.; Fu J.; Astarita G.; La Rana G.; Russo R.; Calignano A.; Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammitory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [DOI] [PubMed] [Google Scholar]
- Thabuis C.; Destaillats F.; Tissot-Favre D.; Martin J.-C. Oleoyl-Ethanolamide (OEA): A bioactive lipid derived from oleic acid and phosphatidylethanol-amine. Lipid Technol. 2007, 19, 225. [Google Scholar]
- Boger D. L.; Henriksen S. J.; Cravatt B. F. Oleamide: An endogenous sleep-inducing lipid and prototypical member of a new class of biological signaling molecules. Curr. Pharm. Des. 1998, 4, 303–314. [PubMed] [Google Scholar]
- Wei B. Q.; Mikkelsen T. S.; McKinney M. K.; Lander E. S.; Cravatt B. F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006, 281, 36569–36578. [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Miyauchi H.; Du W.; Hardouin C.; Fecik R. A.; Cheng H.; Hwang I.; Hedrick M. P.; Leung D.; Acevedo O.; Guimaraes C. R.; Jorgensen W. L.; Cravatt B. F. Discovery of a potent, selective, and efficacious class of reversible α-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J. Med. Chem. 2005, 48, 1849–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Du W.; Hwang I.; Rayl T. J.; Kimball F. S.; Leung D.; Hoover H. S.; Apodaca R. L.; Breitenbucher J. G.; Cravatt B. F.; Boger D. L. Potent and selective α-ketoheterocycle inhibitors of the anandamide and oleamide catabolizing enzyme, fatty acid amide hydrolase. J. Med. Chem. 2007, 50, 1058–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D.; Gaetani S.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. [DOI] [PubMed] [Google Scholar]
- Abouabdellah A.; Burnier P.; Hoornaert C.; Jeunesse J.; Puech F.. Piperidinyl- and piperazinyl-alkylcarbamates, their use as fatty acid amido hydrolase (FAAH) inhibitors for treating FAAH-related pathologies. PCT Int. Appl. 2004099176, 2004.
- Ahn K.; Johnson D. S.; Fitzgerald L. R.; Liimatta M.; Arendse A.; Stevenson T.; Lund E.. T.; Nugent R. A.; Nomanbhoy T. K.; Alexander J. P.; Cravatt B. F. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 2007, 46, 13019–13030. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Ahn K.; Kesten S.; Lazerwith S. E.; Song Y.; Morris M.; Fay L.; Gregory T.; Stiff C.; Dunbar J. B.; Liimatta M.; Beidler D.; Smith S.; Nomanbhoy T. K.; Cravatt B. F. Benzothiophene piperazine and piperadine urea inhibitors of fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 2009, 19, 2865–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. S.; Stiff C.; Lazerwith S. E.; Kesten S. R.; Fay L. K.; Morris M.; Beidler D.; Liimatta M. B.; Smith S. E.; Dudley D. T.; Sadagopan N.; Bhattachar S. N.; Kesten S. J.; Nomanbhoy T. K.; Cravatt B. F.; Ahn K. Discovery of PF-04457845: A highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med. Chem. Lett. 2011, 2, 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T.; Kori M.; Miyazaki J.; Kiyota Y.. Preparation of piperidinecarboxamides and piperazinecarboxamides as fatty acid amide hydrolase (FAAH) inhibitors. PCT Int. Appl. 2006054652, 2006.
- Keith J. M.; Apodaca R.; Xiao W.; Seierstad M.; Pattabiraman K.; Wu J.; Webb M.; Karbarz M. J.; Brown S.; Wilson S.; Scott B.; Tham C.-S.; Luo L.; Palmer J.; Wennerholm M.; Chaplan S.; Breitenbucher J. G. Thiadiazolopiperazine ureas as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2008, 18, 4838–4843. [DOI] [PubMed] [Google Scholar]
- Mileni M.; Garfunkle J.; DeMartino J. K.; Cravatt B. F.; Boger D. L.; Stevens R. C. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by α-ketoheterocycle inhibitors revealed from cocrystal structures. J. Am. Chem. Soc. 2009, 131, 10497–10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes C. R.; Boger D. L.; Jorgensen W. L. Elucidation of fatty acid amide hydrolase inhibition by potent α-ketoheterocycle derivatives from Monte Carlo simulations. J. Am. Chem. Soc. 2005, 127, 17377–17384. [DOI] [PubMed] [Google Scholar]
- Mileni M.; Kamtekar S.; Wood D. C.; Benson T. E.; Cravatt B. F.; Stevens R. C. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: Discovery of a deacylating water molecule and insight into enzyme inactivation. J. Mol. Biol. 2010, 400, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander J. P.; Cravatt B. F. Mechanism of carbamate inactivation of FAAH: Implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 2005, 12, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PF-750:Mileni M.; Johnson D. S.; Wang Z.; Everdeen D. S.; Liimatta M.; Pabst B.; Bhattacharya K.; Nugent R. A.; Kamtekar S.; Cravatt B. F.; Ahn K.; Stevens R. C. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12820–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PF-3845:Ahn K.; Johnson D. S.; Mileni M.; Beidler D.; Long J. Z.; McKinney M. K.; Weerapana E.; Sadagopan N.; Liimatta M.; Smith S. E.; Lazerwith S.; Stiff C.; Kamtekar S.; Bhattacharya K.; Zhang Y.; Swaney S.; Van Becelaere K.; Stevens R. C.; Cravatt B. F. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem. Biol. 2009, 16, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D.; Gaetani S.; Duranti A.; Tontini A.; Mor M.; Tarzia G.; Piomelli D. Characterization of the fatty-acid amide hydrolase inhibitor URB597: effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. [DOI] [PubMed] [Google Scholar]
- Piomelli D.; Tarzia G.; Duranti A.; Tontini A.; Mor M.; Compton T. R.; Dasse O.; Monaghan E. P.; Parrott J. A.; Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unfortunately, many compounds that inhibit FAAH-2 are not very selective esterase inhibitors. For examples, seeKarbarz M. J.; Luo L.; Chang L.; Tham C.-S.; Palmer J. A.; Wilson S. J.; Wennerholm M. L.; Brown S. M.; Scott B. P.; Apodaca R. L.; Keith J. M.; Wu J.; Breitenbucher J. G.; Chaplan S. R. Biochemical and Biological Properties of 4-(3-phenyl-[1,2,4]thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a Mechanism-Based Inhibitor of Fatty Acid Amide Hydrolase. Anesth. Analg. (Hagerstown, MD, U.S.) 2009, 108, 316–329. [DOI] [PubMed] [Google Scholar]
- Data from clinicaltrials.gov as of 6/21/11.
- Huggins J. P.; Smart T. S.; Langman S.; Taylor L.; Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 2012, 153, 1837–1846. [DOI] [PubMed] [Google Scholar]
- Basak S. C.; Gute B. D.; Grunwald G. D. Assessment of the mutagenicity of aromatic amines from theoretical structural parameters: A hierarchal approach. SAR QSAR Environ. Res. 1999, 10, 117–129. [DOI] [PubMed] [Google Scholar]
- Benigni R.; Passerini L.; Gallo G.; Giorgi F.; Cotta-Ramusino M. QSAR models for discriminating between mutagenic and nonmutagenic aromatic and heteroaromatic amines. Environ. Mol. Mutagen. 1998, 32, 75–83. [DOI] [PubMed] [Google Scholar]
- Aeschbacher H. U.; Turesky R. J. Mammalian cell mutagenicity and metabolism of heterocyclic aromatic amines. Mutat. Res. 1991, 259, 235–250. [DOI] [PubMed] [Google Scholar]
- PDB ID: 2wap.
- Physical properties: pKa1 (piperazine) = 6.08; pKa2 (pyridine) = 4.62; solubility of bis-HCl salt in deionized water = 24.5 mg/mL (resultant pH = 1.5); mp of bis-HCl salt 1.5 hydrate = 219 °C (onset); suitable formulations 30% SBE β-CDEX (83 mg/mL, resultant pH = 1.94), 20% SBE β-CDEX (62.6 mg/mL, resultant pH = 1.67.)
- Other rat PK parameters: Cl = 0.69 L/h/kg, Vz = 8.3 L/kg, Vss = 3.2 L/kg, and F = 53–69%.
- Ueda N.; Kurahashi Y.; Yamamoto K.; Yamamoto S.; Tokunaga T. Enzymes for anandamide biosynthesis and metabolism. J. Lipid Mediat. Cell Signal. 1996, 14, 57–61. [DOI] [PubMed] [Google Scholar]
- Li G. L.; Winter H.; Arends R.; Jay G. W.; Le V.; Young T.; Huggins J. P. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br. J. Clin. Pharmacol. 2012, 73, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; Smith S. E.; Liimatta M. B.; Beidler D.; Sadagopan N.; Dudley D. T.; Young T.; Wren P.; Zhang Y.; Swaney S.; Van Becelaere K.; Blankman J. L.; Nomura D. K.; Bhattachar S. N.; Stiff C.; Nomanbhoy T. K.; Weerapana E.; Johnson D. S.; Cravatt B. F. Mechanistic and pharmacological characterization of PF-04457845: A highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J. Pharmacol. Exp. Ther. 2011, 338, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.