

Abstract

Inhibition of MK2 has been shown to offer advantages over that of p38 MAPK in the development of cures for inflammatory diseases such as arthritis. P38 MAPK knockout in mice was lethal, whereas MK2-null mice demonstrated strong inhibition of disease progression in collagen-induced arthritis and appeared normal and viable. However, it is challenging to develop ATP-competitive MK2 inhibitors due to high ATP binding affinity to the kinase. Non-ATP-competitive MK2 inhibitors interact and bind to the kinase in a mode independent of ATP concentration, which could provide better selectivity and cellular potency. Therefore, it is desirable to identify non-ATP-competitive MK2 inhibitors. Through structure optimization of lead compound 1, a novel series of dihydrooxadiazoles was discovered. Additional structure–activity relationship (SAR) study of this series led to the identification of compound 38 as a non-ATP-competitive MK2 inhibitor with potent enzymatic activity and good cellular potency. The SAR, synthesis, and biological data of dihydrooxadiazole series are discussed.
Keywords: mitogen-activated protein kinase-activated protein kinase 2, non-ATP-competitive inhibitors
Activation of the p38/mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2 or MK2) pathway has been associated with promoting inflammatory diseases, such as rheumatoid arthritis, due to overproduction of pro-inflammatory cytokines including tumor necrosis factor α (TNFα) and interleukin 6 (IL6).1−3 The role that p38 MAPK kinase plays in proinflammatory cytokine production has been well documented, and it is known to regulate many downstream cellular processes. However, inhibition of p38 MAPK has been shown to cause unwanted side effects. Therefore, despite numerous efforts to identify p38 MAPK inhibitors, no clinical compounds have progressed beyond phase II trials due to related toxicity and limited efficacy.4−6 MK2 is the downstream substrate kinase activated by p38 in response to inflammatory stimuli (e.g., lipopolysaccharides, LPS) and environmental stress.7−9 MK2 knockout mice show no LPS-induced TNFα release and appear normal and viable, whereas knockout of p38 in mice is lethal.9,10 Furthermore, MK2-null mice demonstrated strong inhibition of disease progression in collagen-induced arthritis.10
Several small molecule MK2 inhibitors from different research laboratories have been reported.11−25 Adenosine-5′-triphosphate (ATP)-competitive MK2 inhibitors have demonstrated efficacy in cellular and animal assays, such as Pfizer's compound PF-3644022.14 However, it is challenging to develop ATP-competitive MK2 inhibitors due to high ATP binding affinity to the kinase (ATP Km = 2 μM), resulting in difficulty in obtaining sufficient selectivity and cellular potency. Non-ATP-competitive MK2 inhibitors interact and bind to the kinase in a mode independent of ATP concentration, which can offer an advantage over the issues associated with ATP-competitive inhibitors.26 In a program to identify non-ATP-competitive MK2 inhibitors in our laboratory, compound 1 (Scheme 1), a furan-2-carboxamide-based scaffold, was discovered through high-throughput screening and initial lead optimization.27 It showed good enzymatic and cellular activity as a non-ATP-competitive inhibitor. Inspired by this amide motif, we decided to modify the core linkage of 1 by forming a heterocyclic ring to fix the conformation of the rotameric amide bond. We hypothesized that this modification could potentially improve potency by offering a rigidified conformation with limited bond rotation, although structure modeling needs to be performed to provide future guidance. On the basis of this hypothesis, we designed and synthesized a series of compounds with different heterocyclic core structures (Table 1). The SAR data showed that the ring size and geometry affect the potency significantly. Compound 2 with a 3,4-substituted isoxazole core was 30-fold less active than 1. The isomeric isoxazole analogue 3 with different substitution geometry was inactive in the assay. The related oxadiazolone 4 also displayed reduced activity. However, analogue 5 that contained a dihydrooxadiazole core demonstrated potency 2-fold better than compound 1. Relocation of the stereocenter in 5 led to analogue 6, which offered much weaker activity than 5. A six-membered dihydrooxadiazine compound 7 possessed similar potency to compound 1. In contrast, analogue 8, which had a seven-membered tetrahydrooxadiazepine core, lost 10-fold activity as compared with 1. From this initial amide replacement and structure–activity relationship (SAR) study, the novel dihydrooxadiazole core structure was taken as a lead for further optimization.
Scheme 1. Design of Novel Heterocyclic Non-ATP-Competitive Inhibitors.

Table 1. In Vitro Potency of Compounds 2–8 in Enzyme and Cell Assay.
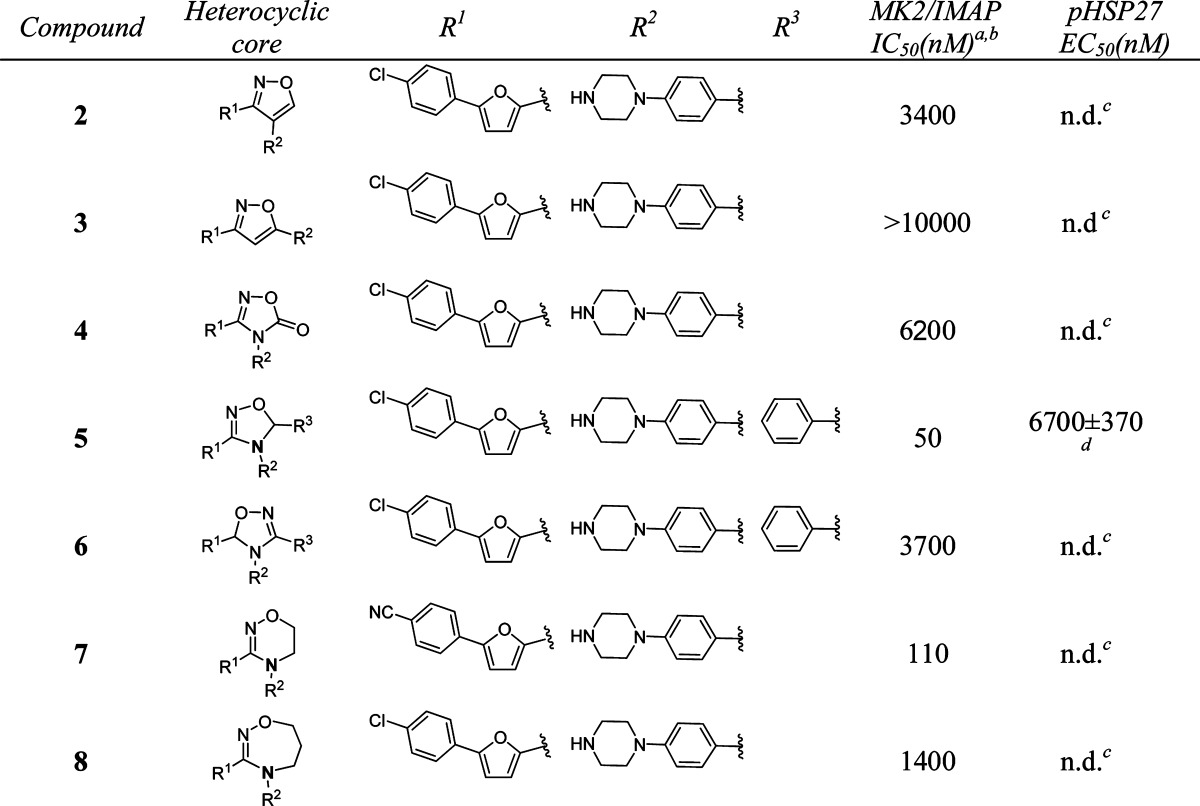
Data represent an average of multiple determinations (n ≥ 2).
Assays were conducted in the presence of 100 μM ATP.
n.d., not determined.
An average of multiple determinations ± standard deviations (n ≥ 2).
We then turned our attention to modifying the right-hand side of the dihydrooxadiazole. A series of compounds with different aromatic groups were prepared to explore the potential in this region (Table 2). Pyridyl (9–12) and pyrimidyl (13–14) groups were well tolerated and maintained good potency in enzymatic assay. Compounds with fluoro-substituted phenyl rings (15–17) were slightly less active than 5. Imidazoyl derivative (18) also showed good activity. It was noted that the 5-(2-phenyl)pyrimidyl group of 19 and the 4-(2-pyrimidyl)phenyl group of 20 were detrimental to the potency, whereas 4-(5-pyrimidyl)phenyl analogue 21 retained similar activity to that of pyrimidyl compound 14. Relocation of the pyrimidyl substitution on the phenyl ring from para- (21) to meta- (22) caused a loss of 4-fold in potency. However, ortho-(5-pyrimidyl)phenyl analogue 23 was twice as active as 21. Additional SAR showed that similar ortho-(3-pyridyl)phenyl and ortho-biphenyl analogues 24 and 25 were inferior to compound 23.
Table 2. In Vitro Potency of Compounds 9–25 in Enzyme and Cell Assay.
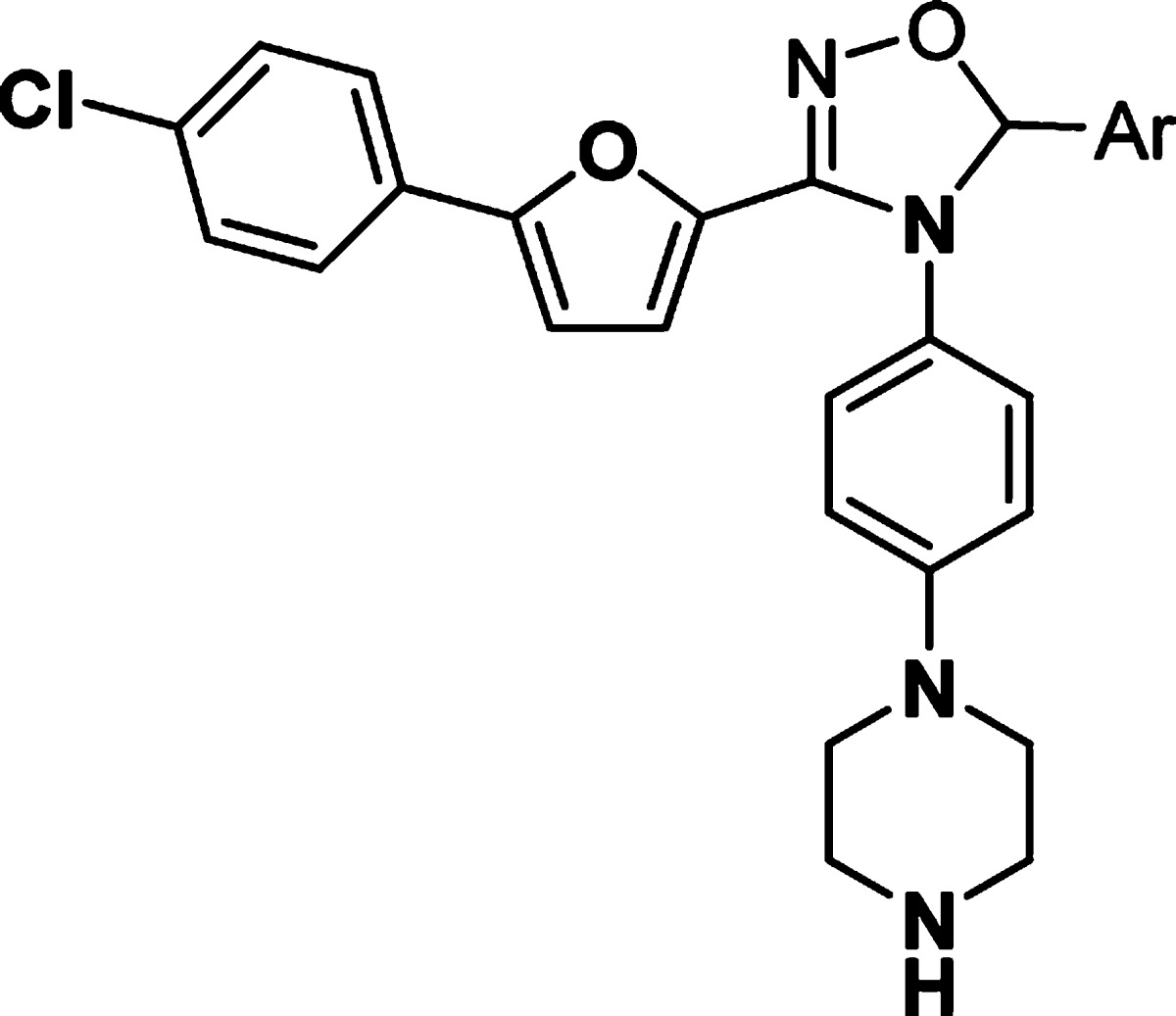
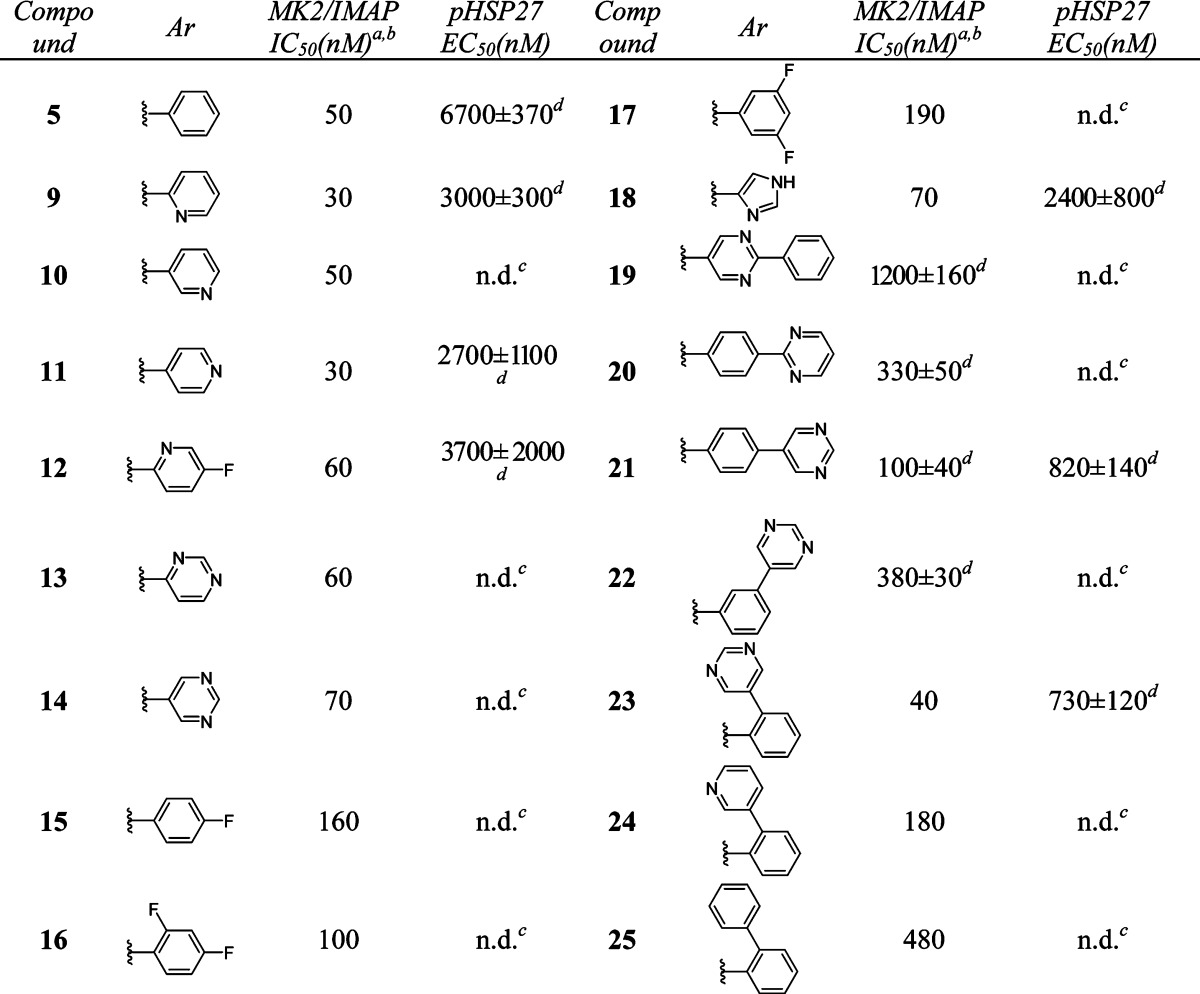
Data represent an average of multiple determinations (n ≥ 2).
Assays were conducted in the presence of 100 μM ATP.
n.d., not determined.
An average of multiple determinations ± standard deviations (n ≥ 2).
Further optimization of the left-hand side aromatic group showed very tight SARs (Table 3). Changing the position of chloro substitution on the phenyl ring from para to meta was not tolerated (23 vs 26). However, 4-fluorophenyl analogue 27 retained similar activity to 23. Compounds with dihalogen-substituted phenyl (28, 29), pyridyl (30), or 4-methoxyphenyl (31) groups showed much weaker activity. 4-(N-Methylformyl)phenyl substitution of 32 was not tolerated. However, a breakthrough was achieved when a 4-cyanophenyl group was introduced in compound 33. This analogue showed excellent activity [MK2/immobilized metal ion affinity-based fluorescence polarization (IMAP) IC50 = 8 nM]. On the other hand, the 3-cyanophenyl group of 34 was detrimental to the activity.
Table 3. In Vitro Potency of Compounds 26–34 in Enzyme and Cell Assay.
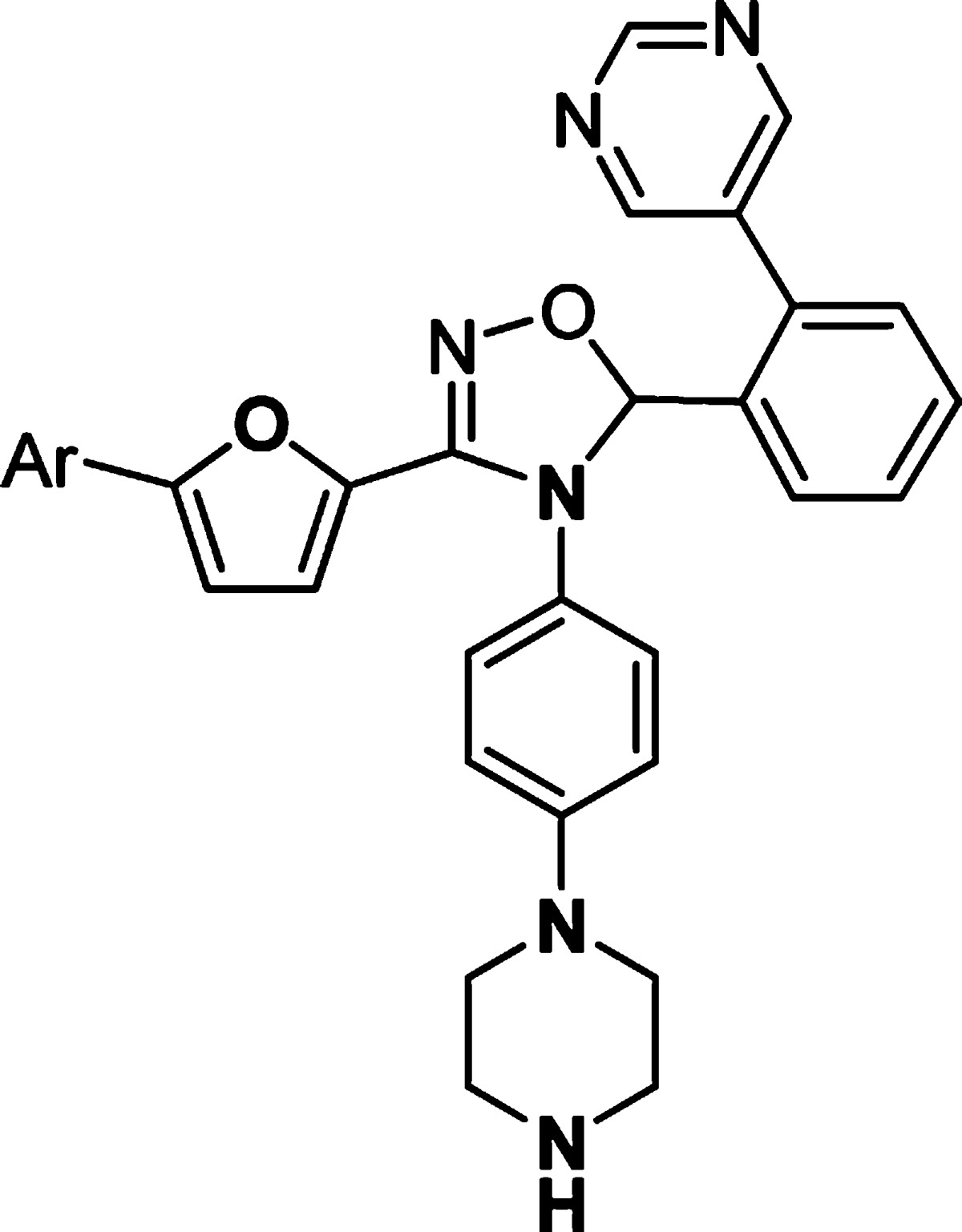
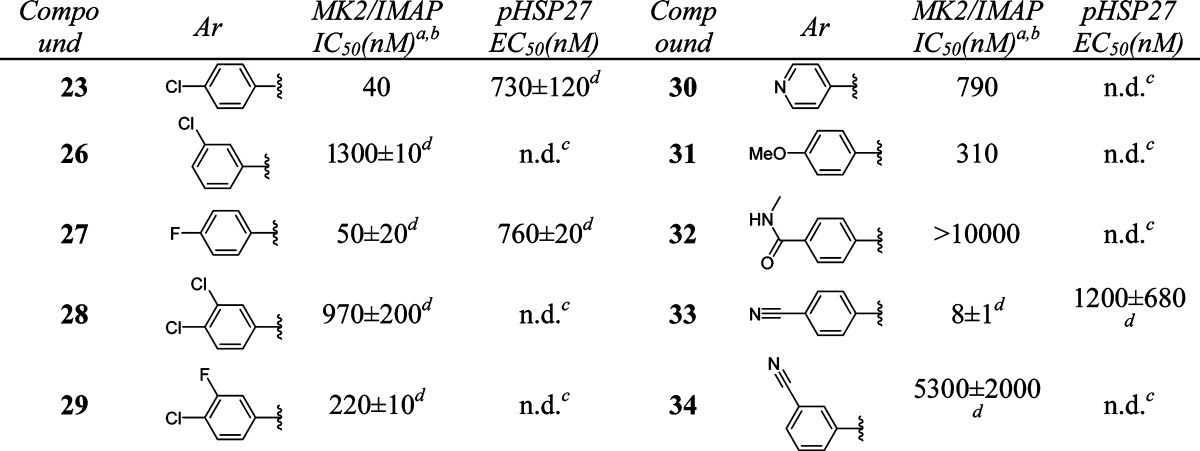
Data represent an average of multiple determinations (n ≥ 2).
Assays were conducted in the presence of 100 μM ATP.
n.d., not determined.
An average of multiple determinations ± standard deviations (n ≥ 2).
Having explored SAR in the right- and left-hand sides, we continued the optimization efforts at the bottom part of the structure (Table 4). It was clear that 4-(piperazin-1-yl)phenyl group was the optimal substitution. Replacement of either one of nitrogen atom in the piperazine ring caused the loss of activity (35–36 vs 11). Compound 37 with 4-(piperazin-1-yl)benzyl substitution was inactive, suggesting that the length of the substitution at this position was critical for the potency.
Table 4. In Vitro Potency of Compounds 35–37 in Enzyme and Cell Assay.
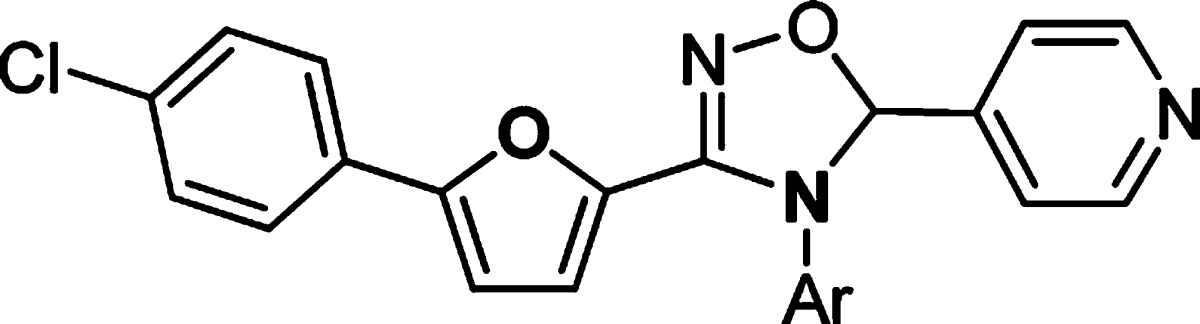
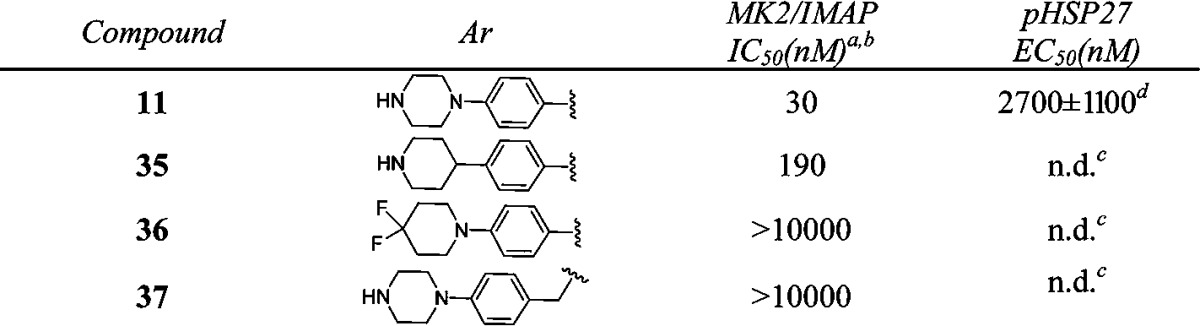
Data represent an average of multiple determinations (n ≥ 2).
Assays were conducted in the presence of 100 μM ATP.
n.d., not determined.
An average of multiple determinations ± standard deviations (n ≥ 2).
Compound 33 was resolved by chiral separation to provide two enantiomers 38 and 39.28 Enantiomer 38 retained excellent enzymatic activity and good cellular potency [MK2/IMAP IC50 = 6 ± 1 nM, phospho heat-shock protein 27 (pHSP27) EC50 = 170 ± 20 nM], whereas isomer 39 was much less active (MK2/IMAP IC50 = 340 nM). From the data presented above, it can be seen that this series of compounds demonstrated poor correlations between enzymatic and cellular potency in general. Solubility and plasma protein binding could be two of the most common factors affecting shifts in cell data as compared to isolated enzyme potencies, although we did not perform routine evaluation of plasma protein binding and solubility for these compounds (compound 38 solubility = 20 μM in 10 mM sodium phosphate buffer/2% DMSO solution, pH = 7.4; plasma protein binding = 96.5% human, 96.9% rat). The binding of compound 38 to MK2 was determined in house to be non-ATP competitive (Figure 1). As is illustrated in the figure, as the ATP concentration increases above the Km for ATP (MK2's Km for ATP ∼ 2 μM), the IC50 value of the inhibitor 38 does not change, indicating that the molecule is not affected by the binding of ATP or may not occupy the same binding pocket. Compound 38 showed a poor pharmacokinetic (PK) profile in rat (rat po 10 mg/kg AUC0–6h = 0 nM·h). We hypothesized that this could be due to low/zero bioavailability and/or high in vivo clearance and other undetermined reasons, although we do not have these data in hand to support at the moment (compound 38 rat hepatocyte clearance = 35 μL/min/M cell, Caco 2 permeability absorption = moderate). With these preliminary data in hand, additional structure optimization is needed to improve the PK profile of this series.
Figure 1.

Characterization of non-ATP-competitiveness for compound 38.
The synthesis of compounds 2–8 is summarized in the Supporting Information. Analogues 9–39 were prepared by a similar method to that described for 5.
In summary, we have explored several series of heterocyclic scaffolds as MK2 inhibitors. Among these series, the dihydrooxadiazoles were identified as a promising new lead series that led to discovery of compound 38 as a potent non-ATP-competitive inhibitor. Additional structure optimization is needed to further improve the PK profile of this series. The results will be the subject of a future publication.
Acknowledgments
We thank Drs. Chad Bennett and Jared Cumming for proofreading and comments on the preparation of manuscript.
Glossary
Abbreviations
- MAPKAPK2 or MK2
mitogen-activated protein kinase-activated protein kinase 2
- TNFα
tumor necrosis factor α
- IL6
interleukin 6
- ATP
adenosine-5′-triphosphate
- LPS
lipopolysaccharides
- IMAP
immobilized metal ion affinity-based fluorescence polarization
- pHSP27
phospho heat-shock protein 27
- PK
pharmacokinetic
Supporting Information Available
Experimental procedures for assay protocols and synthesis and characterization of compounds 2–37. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kumar S.; Boehm J.; Lee J. C. p38 MAP Kinases: Key signaling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discovery 2003, 2, 717. [DOI] [PubMed] [Google Scholar]
- Schindler J. F.; Monahan J. B.; Smith W. G. p38 Pathway kinases as anti-inflammatory drug targets. J. Dent. Res. 2007, 86, 800. [DOI] [PubMed] [Google Scholar]
- Schett G.; Zwerina J.; Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alten R. E.; Zerbini C.; Jeka S.; Irazoque F.; Khatib F.; Emery P.; Bertasso A.; Rabbia M.; Caulfield J. P. Efficacy and safety of pamapimod in patients with active rheumatoid arthritis receiving stable methotrexate therapy. Ann. Rheum. Dis. 2010, 69, 364. [DOI] [PubMed] [Google Scholar]
- Dambach D. M. Potential adverse effects associated with inhibition of p38α/β MAP kinases. Curr. Top. Med. Chem. 2005, 5, 929. [DOI] [PubMed] [Google Scholar]
- Gaestel M; Mengel A.; Bothe U.; Asadullah K. Protein kinases as small molecule inhibitor targets in inflammation. Curr. Med. Chem. 2007, 14, 2214. [DOI] [PubMed] [Google Scholar]
- Duraisamy S.; Bajpai M.; Bughani U.; Dastidar S. G.; Ray A.; Chopra P. MK2: A novel molecular target for anti-inflammatory therapy. Expert Opin. Ther. Targets 2008, 12, 921. [DOI] [PubMed] [Google Scholar]
- Haar E. T.; Prabhakar P.; Liu X.; Lepre C. Crystal structure of the p38α-MAPKAP kinase 2 heterodimer. J. Biol. Chem. 2007, 282, 14684. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A.; Neininger A.; Schubert C.; Eckert R.; Birchmeier C.; Volk H.-D.; Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat. Cell Biol. 1999, 1, 94. [DOI] [PubMed] [Google Scholar]
- Hegen M.; Gaestel M.; Nickerson-Nutter C. L.; Lin L.-L.; Telliez J.-B. MAPKAP Kinase 2-deficient mice are resistant to collagen induced arthritis. J. Immunol. 2006, 177, 1913. [DOI] [PubMed] [Google Scholar]
- Velcicky J.; Feifel R.; Hawtin S.; Heng R.; Huppertz C.; Koch G.; Kroemer M.; Moebitz H.; Revesz L.; Scheufler C.; Schlapbach A. Novel 3-aminopyrazole inhibitors of MK-2 discovered by scaffold hopping strategy. Bioorg. Med. Chem. Lett. 2010, 20, 1293. [DOI] [PubMed] [Google Scholar]
- Argiriadi M. A.; Ericsson A. M.; Harris C. M.; Banach D. L.; Borhani D. W.; Calderwood D. J.; Demers M. D.; DiMauro J.; Dixon R. W.; Hardman J.; Kwak S.; Li B.; Mankovich J. A.; Marcotte D.; Mullen K. D.; Ni B.; Pietras M.; Sadhukhan R.; Sousa S.; Tomlinson M. J.; Wang L.; Xiang T.; Talanian R. V. 2,4-Diaminopyrimidine MK2 inhibitors. Part I: Observation of an unexpected inhibitor binding mode. Bioorg. Med. Chem. Lett. 2010, 20, 330. [DOI] [PubMed] [Google Scholar]
- Harris C. M.; Ericsson A. M.; Argiriadi M. A.; Barberis C.; Borhani D. W.; Burchat A.; Calderwood D. J.; Cunha G. A.; Dixon R. W.; Frank K. E.; Johnson E. F.; Kamens J.; Kwak S.; Li B.; Mullen K. D.; Perron D. C.; Wang L.; Wishart N.; Wu X.; Zhang X.; Zmetra T. R.; Talanian R. V. 2,4-Diaminopyrimidine MK2 inhibitors. Part II: Structure-based inhibitor optimization. Bioorg. Med. Chem. Lett. 2010, 20, 334. [DOI] [PubMed] [Google Scholar]
- Mourey R. J.; Burnette B. L.; Brustkern S. J.; Daniels J. S.; Hirsch J. L.; Hood W. F.; Meyers M. J.; Mnich S. J.; Pierce B. S.; Saabye M. J.; Schindler J. F.; South S. A.; Webb E. G.; Zhang J.; Anderson D. R. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor α production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 2010, 333, 797. [DOI] [PubMed] [Google Scholar]
- Basabe P.; Martín M.; Bodero O.; Blanco A.; Marcos I. S.; Díez D.; Urones J. G. Synthesis of (+)-makassaric acid, a protein kinase MK2 inhibitor. Tetrahedron 2010, 66, 6008. [Google Scholar]
- Anderson D. R.; Meyers M. J.; Kurumbail R. G.; Caspers N.; Poda G. I.; Long S. A.; Pierce B. S.; Mahoney M. W.; Mourey R. J. Benzothiophene inhibitors of MK2. Part 1: Structure–activity relationships, assessments of selectivity and cellular potency. Bioorg. Med. Chem. Lett. 2009, 19, 4878. [DOI] [PubMed] [Google Scholar]
- Anderson D. R.; Meyers M. J.; Kurumbail R. G.; Caspers N.; Poda G. I.; Long S. A.; Pierce B. S.; Mahoney M. W.; Mourey R. J.; Parikh M. D. Benzothiophene inhibitors of MK2. Part 2: Improvements in kinase selectivity and cell potency. Bioorg. Med. Chem. Lett. 2009, 19, 4882. [DOI] [PubMed] [Google Scholar]
- Lin S.; Lombardo M.; Malkani S.; Hale J. J.; Mills S. G.; Chapman K.; Thompson J. E.; Zhang W. X.; Wang R.; Cubbon R. M.; O’Neill E. A.; Luell S.; Carballo-Jane E.; Yang L. Novel 1-(2-aminopyrazin-3-yl)methyl-2-thioureas as potent inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2009, 19, 3238. [DOI] [PubMed] [Google Scholar]
- Schlapbach A.; Feifel R.; Hawtin S.; Heng R.; Koch G.; Moebitz H.; Revesz L.; Scheufler C.; Velcicky J.; Waelchli R.; Huppertz C. Pyrrolo-pyrimidones: a novel class of MK2 inhibitors with potent cellular activity. Bioorg. Med. Chem. Lett. 2008, 18, 6142. [DOI] [PubMed] [Google Scholar]
- Xiong Z.; Gao D. A.; Cogan D. A.; Goldberg D. R.; Hao M.-H.; Moss N.; Pack E.; Pargellis C.; Skow D.; Trieselmann T.; Werneburg B.; White A. Synthesis and SAR studies of indole-based MK2 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1994. [DOI] [PubMed] [Google Scholar]
- Goldberg D. R.; Choi Y.; Cogan D.; Corson M.; DeLeon R.; Gao A.; Gruenbaum L.; Hao M. H.; Joseph D.; Kashem M. A.; Miller C.; Moss N.; Netherton M. R.; Pargellis C. P.; Pelletier J.; Sellati R.; Skow D.; Torcellini C.; Tseng Y.-C.; Wang J.; Wasti R.; Werneburg B.; Wu J. P.; Xiong Z. Pyrazinoindolone inhibitors of MAPKAP-K2. Bioorg. Med. Chem. Lett. 2008, 18, 938. [DOI] [PubMed] [Google Scholar]
- Trujillo J. I.; Meyers M. J.; Anderson D. R.; Hegde S.; Mahoney M. W.; Vernier W. F.; Buchler I. P.; Wu K. K.; Yang S.; Hartmann S. J.; Reitz D. B. Novel tetrahydro-b-carboline-1-carboxylic acids as inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2007, 17, 4657. [DOI] [PubMed] [Google Scholar]
- Anderson D. R.; Meyers M. J.; Vernier W. F.; Mahoney M. W.; Kurumbail R. G.; Caspers N.; Poda G. I.; Schindler J. F.; Reitz D. B.; Mourey R. J. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J. Med. Chem. 2007, 50, 2647. [DOI] [PubMed] [Google Scholar]
- Wu J.-P.; Wang J.; Abeywardane A.; Andersen D.; Emmanuel M.; Gautschi E.; Goldberg D. R.; Kashem M. A.; Lukas S.; Mao W.; Martin L.; Morwick T.; Moss N.; Pargellis C.; Patel U. R.; Patnaude L.; Peet G. W.; Skow D.; Snow R. J.; Ward Y.; Werneburg B.; White A. The discovery of carboline analogs as potent MAPKAP-K2 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4664. [DOI] [PubMed] [Google Scholar]
- Anderson D. R.; Hegde S.; Reinhard E.; Gomez L.; Vernier W. F.; Lee L.; Liu S.; Sambandam A.; Snider P. A.; Masih L. Aminocyanopyridine inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2005, 15, 1587. [DOI] [PubMed] [Google Scholar]
- Olsson H.; SjÖ P.; Ersoy O.; Kristoffersson A.; Larsson J.; Nordén B. 4-Anilino-6-phenyl-quinoline inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK2). Bioorg. Med. Chem. Lett. 2010, 20, 4738. [DOI] [PubMed] [Google Scholar]
- Huang X.; Shipps G. W.; Cheng C. C.; Spacciapoli P.; Zhang X.; McCoy M. A.; Wyss D. F.; Yang X.; Achab A.; Soucy K.; Montavon D. K.; Murphy D. M.; Whitehurst C. E. Discovery and hit-to-lead optimization of non-ATP competitive MK2 (MAPKAPK2) inhibitors. ACS Med. Chem. Lett. 2011, 2, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The absolute configurations of 38 and 39 were not determined. See the Supporting Information for HPLC separation conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.