

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease with poor outcomes with current therapies. Gemcitabine is the primary adjuvant drug used clinically, but its effectiveness is limited. In this study, our objective was to utilize a rationale-driven approach to identify novel biomarkers for outcome in patients with early-stage resected PDAC treated with adjuvant gemcitabine. Using a synthetic lethal screen in human PDAC cells, we identified 93 genes including 55 genes linked to DNA damage responses (DDR) that demonstrated gemcitabine sensitization when silenced, including CHD7 which functions in chromatin remodeling. CHD7 depletion sensitized PDAC cells to gemcitabine and delayed their growth in tumor xenografts. Moreover, CHD7 silencing impaired ATR-dependent phosphorylation of CHK1 and increased DNA damage induced by gemcitabine. CHD7 was dysregulated, ranking above the 90th percentile in differential expression in a panel of PDAC clinical specimens, highlighting its potential as a biomarker. Immunohistochemical analysis of specimens from 59 resected PDAC patients receiving adjuvant gemcitabine revealed that low CHD7 expression was associated with increased recurrence-free survival (RFS) and overall survival (OS), in univariate and multivariate analyses. Notably, CHD7 expression was not associated with RFS or OS for patients not receiving gemcitabine. Thus, low CHD7 expression was correlated selectively with gemcitabine sensitivity in this patient population. These results supported our rationale-driven strategy to exploit dysregulated DDR pathways in PDAC to identify genetic determinants of gemcitabine sensitivity, identifying CHD7 as a novel biomarker candidate to evaluate further for individualizing PDAC treatment.
Keywords: Pancreatic Cancer, DNA Damage Response, CHD7, Biomarker, Gemcitabine, Replication Stress, ATR, CHK1
Introduction
Pancreatic adenocarcinoma (PAC) has a poor prognosis with a five-year overall survival (OS) rate around 5% (1). Patients with early-stage PAC who undergo resection demonstrate the best prognosis, particularly when resection is followed by adjuvant chemotherapy with or without radiation therapy (2, 3). Still, recurrence is common and OS remains poor even for patients who undergo complete resection and adjuvant therapy. Recent developments have suggested that PAC is a genetically heterogeneous disease (4) and, as such, patients may benefit from the identification of predictive biomarkers for responsiveness to adjuvant therapy.
Gemcitabine is the primary chemotherapeutic agent used to treat patients with PAC in the adjuvant setting (2, 5). The cytotoxic effects of gemcitabine are mediated in part through incorporation into DNA as a terminal nucleoside analogue and in part through inhibition of ribonucleotide reductase, which depletes nucleotides required for DNA synthesis. However, the efficacy of gemcitabine for PAC is limited. A better understanding of which patients are likely to respond to gemcitabine treatment would facilitate personalization of therapy and optimize the clinical benefit to toxicity ratio associated with adjuvant therapy.
The DNA Damage Response (DDR) pathway is critical for the maintenance of genome integrity and serves as a cancer barrier by mobilizing DNA repair, cell cycle arrest, and/or apoptosis (6, 7). In human precancerous lesions, aberrant DNA replication induces DDR activation, which constrains tumor development. Thus the DDR acts as a barrier against genomic instability and cancer development. Tumor cells may in turn develop mutations or epigenetic silencing of protective DDR genes, leading to the proliferation of genetically unstable cells and ultimately resulting in cancer. Indeed, a large number of DDR genes are somatically mutated in PAC, including ATM, BRCA2, CDKN2A, FANCI, HELB, and RAD9 (8). These genetic changes in the DDR pathway can lead to PAC and can also weaken the ability of cancer cells to respond to treatment by decreasing activity in DNA repair pathways. Often the cancer cell will become reliant on backup pathways that can be targeted to cause cell death through the principal of synthetic lethality (inactivation of one gene or pathway is sublethal but inactivation of both causes cell death). As such, determining genetic alterations and cancer treatments that are synthetically lethal may lead to the identification of novel druggable targets as adjuncts to gemcitabine treatment or novel biomarkers to predict response to gemcitabine therapy. Utilizing this rationale, we sought to exploit dysregulated DDR pathways in PAC by identifying genetic determinants that are synthetically lethal with gemcitabine treatment and evaluating their clinical relevance as biomarkers for outcome in patients with early-stage resected PAC treated with adjuvant gemcitabine.
Chromodomain helicase DNA binding protein 7 (CHD7), is a member of a family of chromodomain enzymes that belong to the ATP-dependent chromatin remodeling protein SNF2 superfamily. Mutations in CHD7 lead to congenital CHARGE syndrome, named for its characteristic traits: coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness (9); and Kallman Syndrome, a genetic disorder marked by hypogonadotropic hypogonadism and anosmia (10). CHD7 is also dysregulated in 13-35% of cases of PAC, with aberrant expression, copy number variation, and somatic mutations (11-13) (see Supplemental Table S3). CHD7 helps to regulate neural crest gene expression (14), regulates ribosomal RNA biogenesis (15), and interacts with SOX2 to regulate gene expression (16). CHD7 is also a putative substrate of the ATM/ATR checkpoint kinases suggesting that it may play a role in the DDR (17, 18). The clinical significance of CHD7 expression in PAC has not previously been reported.
The purpose of this analysis was to utilize a rationale-driven approach to identify novel biomarkers for outcome in patients with early-stage resected PAC treated with adjuvant gemcitabine (Fig 1A). We initially completed a synthetic lethal siRNA screen to identify genetic determinants of gemcitabine sensitivity in human pancreatic cancer cells and identified the top 15% of these genes for further analysis. Genes validated by a secondary screen and/or linked to the DDR were then analyzed for dysregulation and differential expression in PAC by mining published data sets to determine their potential as biomarkers. Finally, we correlated CHD7 gene expression characterized by immunohistochemistry (IHC) with clinical outcome in patients with early-stage resected PAC treated with adjuvant gemcitabine.
Fig 1.
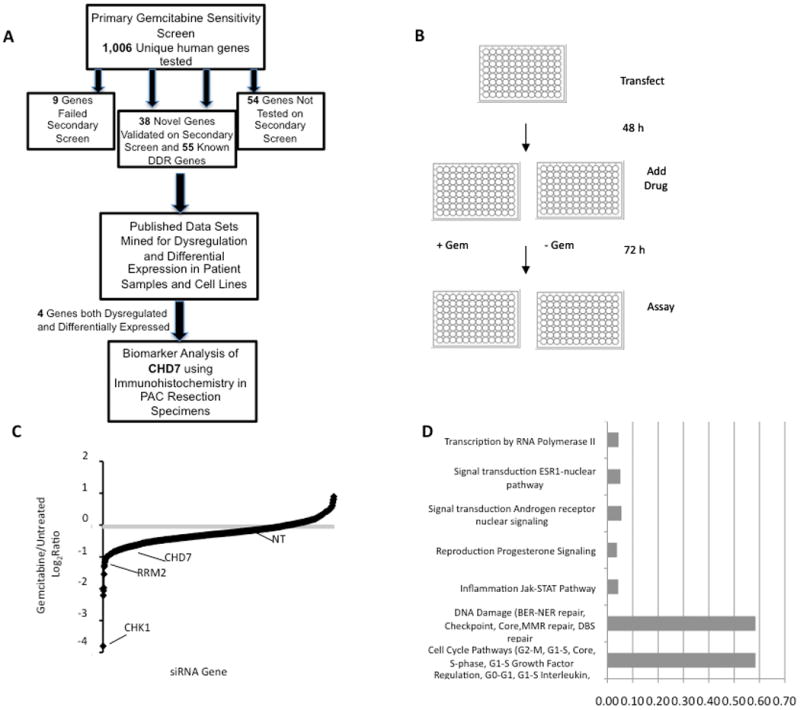
Primary gemcitabine sensitivity screen. (A) Flow diagram of approach for identifying novel biomarkers for outcome in early-stage resected PAC patients treated with adjuvant gemcitabine. (B) Diagram of primary screen as described in text. (C) Results of primary screen. The log2ratio of treated versus untreated cell viability relative to the non-targeting (NT) siRNA for each gene is shown. Mean from three replicas of primary screen is shown. (D) Proportion of top 15% of gemcitabine sensitivity genes with statistically significant involvement in known pathways. The top 18 pathways identified on network analysis via MetaCore ExPlain Process Network Analysis can be consolidated into the listed categories. 55% of all identified genes were involved in DDR pathways.
Materials and Methods
Cell culture, siRNA, and transfection
MIA PaCa-2 cells were grown in Dulbecco Modified Eagle Medium (DMEM, Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY) and 2.5% horse serum (HS, Gibco, Grand Island, NY). HPAC cells were grown in 1:1 DMEM:Hams F12 supplemented (Gibco, Grand Island, NY) with 40 ng/ml hydrocortisone, 10 ng/ml epidermal growth factor (EGF), and 5% FBS. BxPC-3 and AsPC-1 cells were grown in RPMI 1640 supplemented with 10% FBS, and CAPAN-1 cells were grown in Iscove's Modified Dulbecco's Medium supplemented with 20% FBS. Cell lines were grown in a humidified incubator at 37°C with 5% carbon dioxide.
Transfections were done using HiPerfect transfection reagent (Qiagen, Netherlands) according to the manufacturer's protocol. Primary and secondary screen siRNAs were purchased from Thermo Scientific (Waltham, MA). siRNA sequences are listed below:
NT: (ATGAACGTGAATTGCTCAATT)
ATR: (CCUCCGUGAUGUUGCUUGA)
ATRIP: (GGTCCACAGATTATTAGA)
CHK1: (CTGAAGAAGCAGTCGCAGT)
CHD7-1: (UAACGUACCUAACCUAUUA)
CHD7-2: (CGACAAGGCUAGUUUGAAA)
CHD7-3: (GGGAAGCUAUUAUAUCUGA)
CHD7-4: (GUAGAUAACCAAGAACUAA)
TRC lentiviral shRNA was purchased from Thermo Scientific (Waltham, MA): shControl (RHS4080), shCHD7-1 (1RHS3979-201747986), shCHD7-2 (1RHS3979-201747990).
Gemcitabine sensitivity screen
MIA PaCa-2 cells were transfected in 96-well plates using HiPerfect reagent (Qiagen, Netherlands) with 25 nM siRNA from a custom siGenome siRNA library (Thermo Scientific, Waltham, MA) of 4,024 siRNAs corresponding to 1,006 unique human nuclear enzyme genes (pools of 4 siRNAs targeting a unique sequence of each gene) using a one gene per well format. Twenty-four hours later plates were split 1:4, and then treated following another 24 hours with or without 13nM gemcitabine (Hospira Inc., Lake Forest, IL) for 72 hours prior to assaying for cell proliferation using WST-1 reagent (Roche Diagnostics, Indianapolis, IN). Each plate contained two positive controls (ATR and CHK1) and several negative controls (NT), and plate-to-plate variability was controlled by normalizing the values on each plate to the average of the negative control values on that plate. A ratio of gemcitabine treated/untreated viability was calculated and normalized to that of non-targeting (NT) siRNA. Principal Components Analysis (PCA) was used to account for possible variability between the cell viability of the three replicates for each gene. These genes were then sorted by increasing average cell viability via PCA, and the top 15% of genes were categorized as possible ‘hits.’
Secondary validation screen
MIA Pa-Ca 2, BxPC3, or HPAC cells were transfected in 96-well plates with 25 nM siRNA, split 1:4 24 hours later, and then treated following another 24 hours with or without gemcitabine at inhibitory concentration (IC)5, IC25, or IC50 for 72 hours prior to assaying for cell proliferation using WST-1 reagent. A ratio of gemcitabine treated/untreated viability was calculated and normalized to that of non-targeting (NT) siRNA. MIA PaCa-2, HPAC, CAPAN-1, BxPC3, and AsPC-1 cells were treated with the indicated concentrations of gemcitabine for 72 hours prior to assaying for cell proliferation using WST-1 reagent.
Colony formation assay
Cells were transfected with 25nM siRNA. Following a 24 hour knockdown, 500 cells were seeded into 6-well plates in triplicate. Cells were allowed to culture overnight and were then treated for 24 hours with increasing concentrations of gemcitabine. Following the gemcitabine incubation, the plates were washed with PBS and fresh media was added for 8-12 days prior to staining colonies with a 0.5% crystal violet (AMPRESCO, Solon, OH) solution.
Western blot analysis
MIA PaCa-2 cells transfected with siRNA for 48 hours or MIA PaCa-2, HPAC, CAPAN-1, BxPC3, and AsPC-1 cells were harvested with NP40 buffer containing: 200 mM NaCl, 1% NP40, 50 mM Tris-HCl (pH 8.0), and supplemented with fresh protease inhibitors. Samples were loaded into a SDS-PAGE gel, transferred to a PVDF membrane, and subsequently probed with an anti-CHD7 antibody (NBP1-77393, Novus Biologicals, Littleton, CO) and anti-GAPDH antibody (GeneTex, GTX627408) followed by LI-COR IRDye secondary antibodies. Detection was performed using the Odyssey system (LI-COR Biosciences, Lincoln, NE).
To analyze phosphorylation of CHK1, MIA PaCa-2 cells were transfected with 25 nM siRNA for 48 hours and treated with 1mM gemcitabine for 6 hours. Cells then were harvested, washed with PBS and lyzed in cold RIPA buffer (25 mM Tris·HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) with protease and phosphatase inhibitors for 30 minutes. Lysates were clarified by centrifugation at 16 g for 10 minutes and 150 μg of protein for each sample were used for Western blot analysis. Primary antibody pCHK1 S317 (Cell Signaling, #2344) and CHK1 (Santa Cruz Technologies, sc-8408) were used for detection of phosphorylated and total CHK1, respectively.
In vivo tumor growth inhibition assay
Male nude mice were maintained in a pathogen-free environment, and all in vivo procedures were approved by the Emory University, Institutional Animal Care and Use Committee. Stable MIA PaCa-2 shCHD7-2 and MIA PaCa-2 shControl cells (1.5 × 106 / 0.1 mL of 20% Matrigel gel in serum free media) were injected subcutaneously into the flank of 5 week old mice. Mice bearing established tumors (100-125 mm3) were randomized into treatment groups of 4. Animals were treated on days 0, 7 and 14 via the tail vein with either vehicle or 100 mg/kg gemcitabine. Tumor growth inhibition was determined as described previously (19).
γH2AX DNA damage assay
MIA PaCa-2 cells were treated with or without 13 nM gemcitabine for 20 hours, washed, released for the indicated time points, and processed for γH2AX staining by indirect immunofluorescence. Cells were washed with 1×PBS, fixed with 2% paraformaldehyde for 10 minutes at room temperature, permeabilized with 0.5% Triton-X-100 (Fisher-Scientific), and blocked in a 5% bovine serum albumin solution (Sigma). Cells were then immunostained with anti-phospho-histone H2AX (Ser139) antibody (Millipore, 05-636) and anti-mouse secondary antibody with AlexaFluor 488 (Invitrogen, A21206). After incubation, cells were mounted onto slides with a mounting media containing 4′, 6-diamidino-2-phenylindole (DAPI) and dried. Analysis was performed using a Zeiss Observer Z1 microscope with Axiovision Rel 4.8 software using the 63× oil objective. Foci quantitation was conducted by counting 250 healthy cells and scoring cells with 10 or more foci as positive. Experiments were done in triplicate.
Cell Cycle Analysis
MIA PaCa-2 cells were transfected with 25 nM siRNA for 48 hours and treated with or without gemcitabine (13nM) for 24 hours. After fixing with ice-cold 70% ethanol cells were washed in PBS and propidium iodide (PI; 25 μg/mL; Sigma) and RNase A (10 μg/mL; Qiagen) were added to determine DNA content. Cells were analyzed on a FACS Canto II (BD Bioscience) and FlowJo software.
Biomarker selection
Gemcitabine sensitivity genes validated on secondary screen or known DDR genes were analyzed for evidence of dysregulation by identifying genes overexpressed in The Compendium of Potential Biomarkers of Pancreatic Cancer (20) or somatically mutated in the pancreatic Catalogue for Somatic Mutations in Cancer (COSMIC) database (11). For determination of differential expression, we extracted expression data from the two GEO submissions based on the Affymetrix U133 Plus 2.0 platform: 1) GSE12654; a 22 pancreatic cancer cell line study (19), and 2) GSE16515; 20 pancreatic patient tumors (21, 22). Within each study, after processing and normalization, we performed a genome-wide filter to identify genes with ‘large’ expression differences among tumors, and separately, among cell lines, using a variance approach. We define ‘differential expression’ as genes whose expression variability is ‘large’ relative to all other genes on the array, where ‘large’ is defined according to whether expression variability associated with a gene was greater than the 90th percentile from all genes. We then compared this list of genes to the lists in Supplemental Tables S2 and S4. CHD7 was chosen as a potential biomarker based on evidence of both dysregulation and differential expression.
Immunohistochemistry patient selection
Patients were selected for this analysis from a prospectively maintained database of patients who underwent resection for early-stage PAC between January 2000 and October 2008; data for these patients has been included in other cohorts previously reported (23-26). These 59 patients received adjuvant chemotherapy with or without adjuvant radiation. The gemcitabine patient population was composed of 42 of these patients who received gemcitabine as a component of the adjuvant chemotherapy regimen. An additional 17 patients received adjuvant chemotherapy with agents other than gemcitabine. Overall Survival (OS) was calculated from date of surgery to patient death. Recurrence-free survival (RFS) was measured based on surveillance imaging obtained at regular intervals after resection. Patient demographics, pathologic characteristics, and treatment characteristics were originally collected from pathologic record and chart review. Permission was obtained from Emory Institutional Review Board (IRB) 00048816, and patient confidentiality was maintained according to the Health Insurance and Patient Accessibility Act of 1996.
Immunohistochemical analysis
An experienced pathologist identified representative sections of tumor and normal tissue from formalin-fixed paraffin embedded slides. The tissue was stained using an anti-CHD7 mouse monoclonal antibody (NBP1-77393 Novus Biologicals, Littleton, CO) at a concentration of 1:200. Specificity of the anti-CHD7 antibody was validated by western blot analysis following siRNA silencing (Fig 2B). An expression score was calculated using a previously defined scoring system (23, 27). Overall score was dichotomized into low (<3.1) and high (>3.1) expression groups for this analysis (Supplemental Fig S1).
Fig 2.
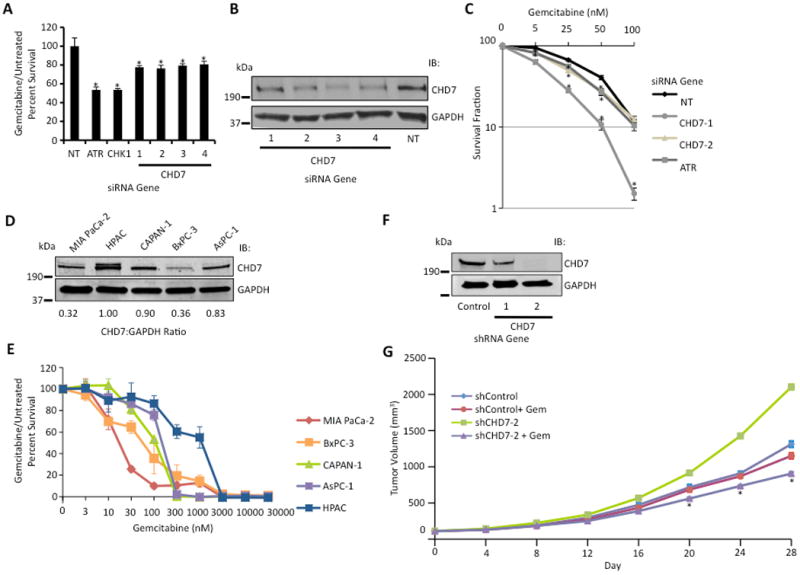
CHD7 knockdown causes gemcitabine sensitization. (A) Four siRNAs targeting CHD7 caused gemcitabine sensitization in MIA PaCa-2 cells. Treated versus untreated percent viability was calculated and the mean and standard deviation from three replicas is shown. * indicates p < 0.05. (B) Western blot analysis demonstrating efficiency of CHD7 knockdown with indicated siRNAs. (C) Clonogenic assay demonstrating gemcitabine sensitization with CHD7 silencing. MIA PaCa-2 cells transfected with siRNA against CHD7, ATR, or NT were seeded for colony formation, treated with indicated concentrations of gemcitabine for 24 hours, and assayed for surviving colonies 8-12 days later. Percent survival of colonies from treated versus untreated cells is indicated. Mean and standard deviation from three replicas are shown. * indicates p < 0.05. (D) Western blot analysis of cell lysate from MIA PaCa-2, HPAC, CAPAN-1, BxPC-3, and AsPC-1 cells with the indicated antibodies. The CHD7:GAPDH ratio of representative blot from three independent experiments is shown. (E) Gemcitabine sensitivity of MIA PaCa-2, HPAC, CAPAN-1, BxPC-3, and AsPC-1 cells following treatment with indicated concentrations of gemcitabine for 72 hours is shown. (F) Western blot analysis demonstrating efficiency of CHD7 knockdown with indicated shRNAs in MIA PaCa-2 cells. (G) Athymic nude mice with shCHD7 and shControl MIA PaCa-2 tumor xenografts were treated with or without gemcitabine (100 mg/kg) on days 0, 7, and 14, and tumor growth was measured every 4 days. Mean and standard error of mean from 6 tumors are shown. * indicates p < 0.05.
Statistical analysis
Descriptive statistics were generated for patient characteristics, tumor characteristics, and treatment characteristics. Similar statistical analyses were performed for patients receiving adjuvant therapy, patients receiving gemcitabine-based therapy, and patients receiving non-gemcitabine based therapy. Kaplan-Meier log-rank survival analysis was performed to determine prognostic factors for RFS and OS. Univariate (UV) and multivariate (MV) Cox regression analyses were performed for all patients to examine the correlation of CHD7 expression level on both RFS and OS. Factors examined on UV analysis included age, sex, ethnicity, receipt of adjuvant and neoadjuvant therapy, tumor size, margin status, grade, nodal status, perineural invasion, lymphovascular invasion, receipt of radiation therapy, CA19-9 levels, and type of adjuvant chemotherapy. Clinically relevant covariates significant to a level of p<0.2 on univariate analysis for either RFS or OS were included in the multivariate model; these included tumor size, margin status, nodal status, perineural invasion, lymphovascular invasion, and tumor grade. Data was analyzed using the Statistical Package for the Social Sciences 19.0 software for Windows (IBM, Armonk, NY).
Results
Gemcitabine sensitivity screen
To identify genetic determinants of gemcitabine sensitivity, we completed a siRNA screen to identify genes that when silenced, cause either sensitization or resistance to a low dose of gemcitabine in human pancreatic cancer cells. Since gemcitabine induces DNA damage and replication stress, we reasoned that gemcitabine sensitivity genes would likely be involved in the DDR. We therefore optimized a high-throughput assay using ATR and CHK1 siRNA as positive controls and a non-targeting (NT) siRNA as a negative control with cell proliferation as a read-out (Fig 1B). The primary screen was completed in MIA PaCa-2 cells, which consistently gave the highest signal to noise ratio among several tested cell types (Supplemental Fig S3A and data not shown). Briefly, cells were transfected with pools of 4 siRNAs targeting a unique sequence of each gene arrayed in a one gene/one well format in 96-well plates. Forty-eight hours after transfection, cells were treated with or without 13 nM gemcitabine (equivalent to the inhibitory concentration (IC) 25 under these conditions, see Fig 2E) for 72 hours prior to assaying for cell proliferation using WST-1 reagent. Each plate contained two positive controls (ATR and CHK1) and several negative controls (NT), and plate-to-plate variability was controlled by normalizing the values on each plate to the average of the negative control values on that plate. We completed three replicas of the primary screen using a library of 4,024 siRNAs, corresponding to 4 unique siRNA duplexes, targeting each of 1,006 unique human genes (Fig 1C). The library consisted predominantly of nuclear enzymes, which we reasoned were more likely to function directly in the DDR and be targetable. Results of the primary screen were ranked by PCA score (Supplemental Table S1). The top 15% of these genes (156 genes) included 55 genes linked to the DDR (Fig 1D, Supplementary Fig S2, and Supplemental Table S2) including well-characterized ATR signaling pathway genes ATR, CHK1, RAD9, RAD1, and HUS1 and nucleotide metabolism genes RRM1 and RRM2, known to regulate gemcitabine sensitivity (28), demonstrating that our screen can yield DDR genes that determine gemcitabine sensitivity.
CHD7 knockdown causes gemcitabine sensitization
Sixty-eight of our hits were identified in previously published DNA damage sensitivity screens (17, 20, 29-36) and 27 are putative ATM/ATR substrates (17) (Supplemental Table S3). We utilized these criteria to validate 47 of the 99 hits not characterized in the DDR in a secondary screen using deconvoluted individual siRNAs to confirm their gemcitabine sensitivity and eliminate false positives due to off-target effects, and 38 of these genes induced gemcitabine sensitivity in at least two out of four siRNAs tested, including CHD7 (Supplemental Table S4). Four of four siRNAs targeting CHD7 caused gemcitabine sensitization (Fig 2A). Western blot analysis confirmed decreased levels of CHD7 following siRNA knockdown as well as specificity of the anti-CHD7 antibody used for IHC analysis (Fig 2B). A similar gemcitabine sensitization after CHD7 silencing was observed using a range of gemcitabine concentrations and in BxPC-3 and HPAC pancreatic cancer cells, suggesting that the phenotype is not cell-type specific (Supplemental Fig S3A-C). CHD7 silencing in the absence of gemcitabine treatment reduced cell viability (Supplemental Fig S3D). We also determined the gemcitabine sensitivity of CHD7 depleted cells using a colony formation assay. MIA PaCa-2 cells silenced for CHD7 demonstrated a significantly reduced percentage of surviving colonies following a 24 hour pulse of gemcitabine in a dose-dependent manner compared to a NT control (Fig 2C), confirming the gemcitabine sensitization of CHD7 depleted cells observed with WST-1 reagent. Consistent with these findings, MIA PaCa-2 and BxPC3 pancreatic cancer cells, which express lower levels of CHD7 than HPAC, CAPAN-1, and AsPC-1 pancreatic cancer cells, demonstrated increased gemcitabine sensitivity (Fig 2D-E), suggesting that CHD7 expression may predict response to gemcitabine in PAC cells. To determine if CHD7 silencing causes gemcitabine sensitization of pancreatic cancer tumors in vivo, we generated a xenograft model using MIA PaCa-2 cells stably expressing shCHD7 or shControl (Fig 2F). CHD7 silencing significantly delayed tumor growth in mice treated with gemcitabine compared with a control treated with gemcitabine (Fig 2G), suggesting that CHD7 silencing also causes gemcitabine sensitization in vivo. No significant difference in body weight was observed in mice bearing tumors with shCHD7 compared with shControl and treated with or without gemcitabine (Supplemental Fig S4).
CHD7 is a DNA damage response protein
The gemcitabine hypersensitivity of CHD7 depleted cells suggests that CHD7 may function in the DDR. CHD7 silencing significantly increased the percentage of cells staining with γH2AX, a marker for DNA damage, following treatment with gemcitabine (Fig 3A), suggesting that CHD7 silencing potentiates gemcitabine-induced DNA damage. However, no significant difference in repair kinetics was observed between cells silenced with CHD7 compared with a NT siRNA (Fig 3A). CHD7 silenced cells showed a decreased percentage of cells in S-phase and an increased percentage of cells in G2/M in the absence of gemcitabine treatment (Supplemental Fig S5A); however, no significant difference in cell cycle profile was observed between CHD7 depleted compared with NT control cells following gemcitabine treatment (Supplemental Fig S5B). There was also no significant difference in protein levels of CHD7 in response to gemcitabine treatment (Supplemental Fig S6). To determine if CHD7 functions in ATR-dependent signaling in response to gemcitabine treatment, we examined cells for the phosphorylation of CHK1 Ser317. CHD7 silencing significantly reduced CHK1 Ser317 phosphorylation but not total CHK1 protein levels in response to gemcitabine treatment (Fig 3B), suggesting that CHD7 functions in controlling ATR dependent phosphorylation of CHK1 in response to gemcitabine treatment.
Fig 3.
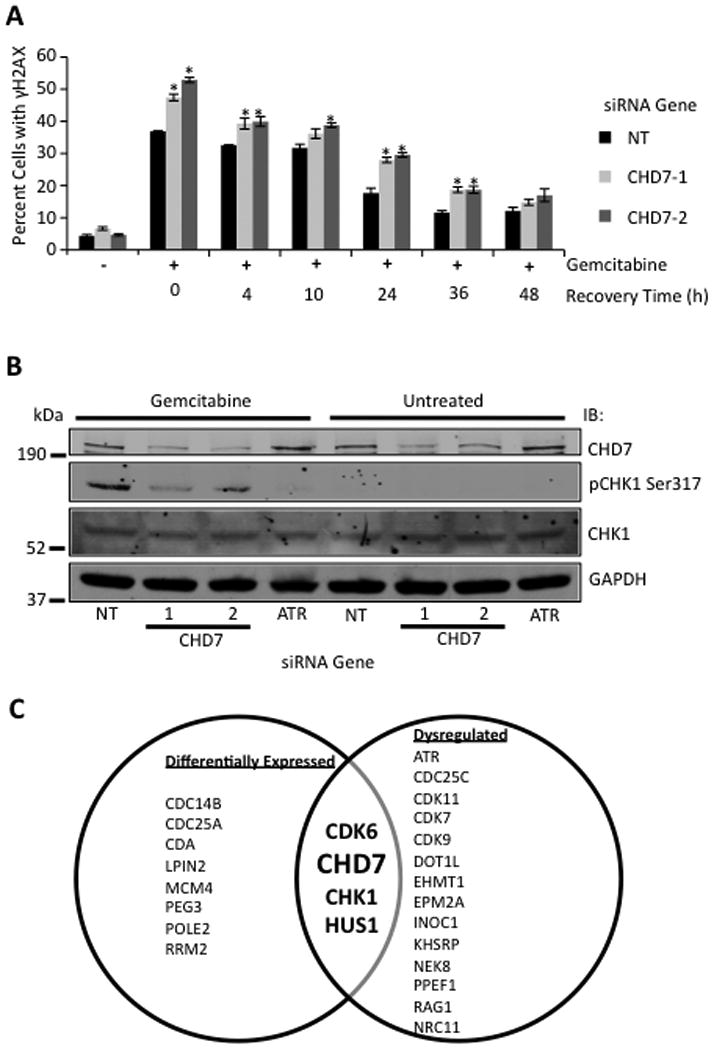
CHD7 is a DNA damage response protein. (A) MIA PaCa-2 cells were treated with or without gemcitabine for 20 hours, washed, released for the indicated time points, and processed for γH2AX staining by indirect immunofluorescence. The percentage (mean and standard deviation) of γH2AX positive cells from three replicas is shown. * indicates p < 0.05 (B) Western blot analysis of cell lysate from MIA PaCa-2 cells treated with or without gemcitabine for 6 hours and probed with anti-CHD7, pCHK-1 Ser317, CHK1, and GAPDH antibodies. (C) Venn diagram showing gemcitabine sensitivity genes dysregulated and/or above the 90th percentile in differential expression amongst a panel of pancreatic cancer cell lines and tissue samples.
CHD7 is dysregulated and differentially expressed in PAC
Genes validated by our secondary screen or linked to the DDR were then analyzed for dysregulation and differential expression in PAC by mining of published data sets to determine their potential as biomarkers. Of these, eight genes demonstrate aberrant expression or somatic mutations in PAC (Fig 3C and Supplemental Table S3) as reported in The Compendium of Potential Biomarkers (20) and the Catalogue for Somatic Mutations in Cancer (COSMIC) database (11). Twelve of the genes are above the 90th percentile in differential expression amongst a panel of 22 PAC cell lines or 20 PAC tissue samples (Fig 3C and Supplemental Table S3) (21, 22). Four of the genes exhibit both dyregulation and differential expression, including CHD7, which was selected for further analysis as a biomarker.
Survival analyses
Patient demographics, pathologic and treatment characteristics can be seen in Table 1. CHD7 expression was low in 84.7% of patients. Median tumor size was 3.4 cm (range 1 – 6 cm), and 60% of patients were node positive. In addition to CHD7 expression, significant covariates on UV analysis included tumor size, margin status, lymph node status, PNI, LVI, and grade (p<0.2). On Kaplan-Meier analysis for patients receiving gemcitabine as a component of adjuvant therapy (n=42), low CHD7 expression was associated with increased RFS (15 months vs 7 months; p=.025; Fig 4A) and increased OS (KM: 18 months vs 10 months; p=.015; Fig 4B). On MV analysis (Table 2), low CHD7 expression remained associated with increased RFS (HR 0.12 [95% CI 0.04-0.42]; p=.001) and increased OS (HR 0.09 [95% CI 0.03-0.29); p<.0001). In the subset of patients receiving adjuvant therapy with agents other than gemcitabine (most commonly 5-FU), CHD7 was not associated with RFS (p=0.1, data not shown) or OS (p=0.4, data not shown). On Kaplan-Meier analysis for all patients (n=59), low CHD7 expression via IHC scoring was associated with increased RFS (15 months vs 7 months; p=.015; Fig 4C) and increased OS (KM: 19.5 months vs. 9 months; p=.001; Fig 4D). These results remained significant on MV analysis (Table 3). To ensure stability of the MV model given the small number of events, the three least significant factors on UV analysis were removed from the model (Grade, PNI and LVI) and the significance of CHD7 expression remained unchanged.
Table 1. Patient demographics, tumor characteristics and treatment characteristics for all patients (N=59).
| Median (Range) | N= | Percent | |
|---|---|---|---|
| Patient Demographics | |||
| Male Sex | 31 | 52.5 | |
| Ethnicity | |||
| Asian | 2 | 3.4 | |
| Black | 12 | 20.3 | |
| White | 42 | 71.2 | |
| Age (years) | 60.0 (37-84) | ||
| Overall Survival (months) | 17.3 (4.8-114.6) | ||
| Recurrence Free Survival (months) | 14.5 (0.6-109.8) | ||
|
| |||
| Tumor Characteristics | |||
| Positive Margins | 20 | 24.5 | |
| Grade: | |||
| Well differentiated | 5 | 6.3 | |
| Moderately differentiated | 46 | 57.5 | |
| Poorly differentiated | 28 | 35.0 | |
| Positive Nodes | 48 | 60.0 | |
| Perineural Invasion | 70 | 87.5 | |
| Lymphovascular Invasion | 38 | 47.5 | |
| Low CHD7 Expression | 50 | 84.7 | |
| Tumor Size (cm) | 3.4 (1-6) | ||
|
| |||
| Treatment Characteristics | |||
| Neoadjuvant Therapy | 2 | 3.4 | |
| Radiation Therapy | 39 | 66.1 | |
| Received Gemcitabine | 42 | 69.5 | |
Fig 4.
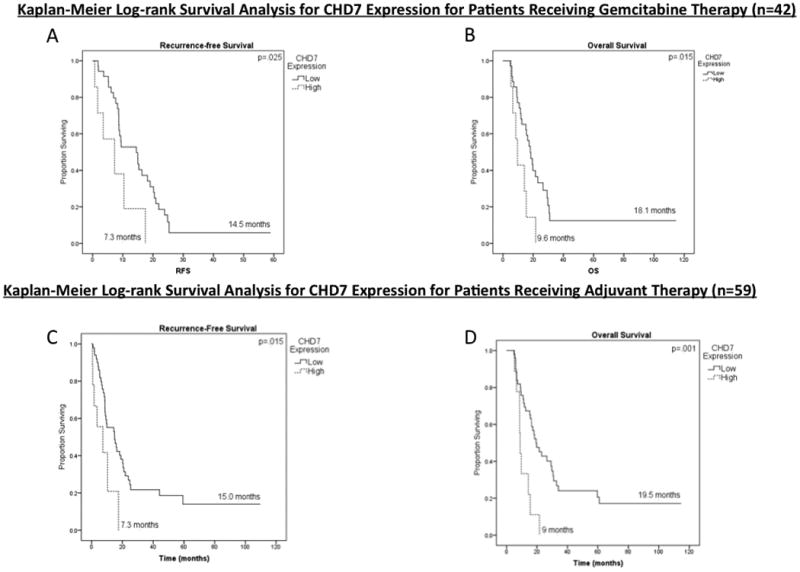
Kaplan-Meier log-rank survival analysis for CHD7 expression in patients receiving adjuvant therapy (n=59) and in patients receiving gemcitabine therapy (n=42). (A) Effect of CHD7 expression on recurrence-free survival (RFS) in patients receiving gemcitabine therapy (n=42). (B) Effect of CHD7 expression on overall survival (OS) in patients receiving gemcitabine therapy (n=42). (C) Effect of CHD7 expression on RFS in patients receiving adjuvant therapy (n=59). (D) Effect of CHD7 expression on OS in patients receiving adjuvant therapy (n=59).
Table 2. Multivariateα Cox Regression Analyses for patients receiving gemcitabine therapy (N=42).
| Outcome | ||||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| RFS | OS | |||||
|
|
|
|||||
| HR | 95% CI | P | HR | 95% CI | P | |
| Tumor Size | 1.708 | 1.223-2.387 | .002 μ | 1.685 | 1.171-2.425 | .005 |
| Positive Margins | .252 | .083-.767 | .015 | .187 | .059-.590 | .004 |
| Higher Grade | 1.126 | .566-2.242 | .735 | 1.064 | .555-2.040 | .851 |
| Positive Nodes | 1.177 | .5402.564 | .682 | .965 | .420-2.216 | .933 |
| PNI | .574 | .210-1.567 | .279 | 4.133 | 1.044-16.358 | .043 |
| LVI | 1.004 | .462-2.180 | .992 | 1.429 | .619-3.298 | .402 |
| Low CHD7 Expression | .122 | .035-.420 | .001 | .086 | .025-.292 | <.0001 |
Abbreviations: Recurrence-free survival (RFS), Overall Survival (OS), Perineural invasion (PNI), Lymphovascular invasion (LVI)
Multivariate analysis includes all clinically relevant covariates with p value < .2 on univariate analysis.
Bold font denotes statistical significance
Table 3. Multivariateα Cox Regression Analyses for patients receiving adjuvant therapy (N=59).
| Outcome | ||||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| RFS | OS | |||||
|
|
|
|||||
| HR | 95% CI | P | HR | 95% CI | P | |
| Tumor Size | 1.622 | 1.228-2.144 | .001 μ | 1.527 | 1.130-2.063 | .006 |
| Positive Margins | .849 | .408-1.768 | .662 | .724 | .347-1.511 | .389 |
| Higher Grade | 1.051 | .579-1.907 | .870 | 1.057 | .592-1.887 | .851 |
| Positive Nodes | 1.638 | .777-3.452 | .194 | 1.729 | .818-3.652 | .151 |
| PNI | .410 | .161-1.044 | .062 | 2.028 | .591-6.960 | .261 |
| LVI | 1.064 | .529-2.139 | .862 | 1.082 | .537-2.178 | .826 |
| Low CHD7 Expression | .271 | .107-.687 | .006 | .203 | .085-.486 | <.0001 |
Abbreviations: Recurrence-free survival (RFS), Overall Survival (OS), Perineural invasion (PNI), Lymphovascular invasion (LVI)
Multivariate analysis includes all clinically relevant covariates with p value < .2 on univariate analysis.
Bold font denotes statistical significance
Discussion
In this study, we demonstrate a rationale-driven approach for identifying novel biomarkers for outcome in patients with early-stage resected PAC treated with adjuvant gemcitabine. Using a synthetic lethal screen to identify genetic determinants of gemcitabine sensitivity in human pancreatic cancer cells, we identified 93 genes which, when silenced, demonstrate gemcitabine sensitization, including CHD7. CHD7 deficiency caused gemcitabine sensitization in PAC cells and delayed pancreatic tumor xenograft growth in mice treated with gemcitabine. We further found that CHD7 knockdown impaired ATR dependent phosphorylation of CHK1 and increased DNA damage induced by gemcitabine, revealing a novel function for CHD7 as a DDR protein, which maintains genome integrity in response to gemcitabine. We examined CHD7 as a potential biomarker based on its dysregulation and differential expression in a panel of PAC cell lines and tissues. Finally, we found that low CHD7 expression is associated with improved RFS and OS in patients with early-stage resected PAC treated with adjuvant gemcitabine. These findings support our rationale-driven approach in exploiting dysregulated DDR pathways in PAC to identify genetic determinants of gemcitabine sensitivity that can be translated to novel biomarkers or drug targets.
A third of the genes identified in our primary gemcitabine sensitivity screen are linked to the DDR, including ATR signaling pathway genes ATR, CHK1, RAD9, RAD1, HUS1, and CDK9 (37) and nucleotide metabolism genes RRM1 and RRM2. CHD7 was previously identified as a putative ATM/ATR substrate (17). Our finding that CHD7 silencing in human pancreatic cancer cells potentiates gemcitabine-induced DNA damage and impairs CHK1 Ser317 phosphorylation in response to gemcitabine treatment suggests that CHD7 also functions in the ATR signaling pathway and helps to explain at least in part why CHD7 knockdown causes gemcitabine sensitization in cells and in vivo. Still, the cell cycle effects of CHD7 expression require further understanding through future studies, which remain ongoing. For example, CHK1 inhibition has been shown to potentiate gemcitabine-induced cytotoxocity by inducing premature mitosis (38). A number of genes identified in our screen, including RRM1, RRM2, and CHK1 have previously been shown to determine gemcitabine sensitivity in human pancreatic cancer cells (39), and low RRM2 expression has been shown to be associated with improved outcome in patients with PAC (24) and specifically those treated with adjuvant gemcitabine (28), providing validation for our screen in identifying gemcitabine sensitivity genes that may function as potential biomarkers. Several of the gemcitabine sensitivity genes, including PLK1 and AURKB, are involved in mitotic progression that is in part targeted by nanoparticle albumin bound (nab)-paclitaxel (Abraxane, Celgene, Summit, NJ), which potentiates gemcitabine sensitivity and improves survival in patients with metastatic PAC treated with gemcitabine (40, 41). It is thus possible that the gemcitabine sensitivity genes reported in this study may also be novel druggable targets to be used in combination with gemcitabine. Indeed, PARP2, a target of PARP inhibitors that sensitizes pancreatic cancer cells to gemcitabine (42, 43), was also identified in our screen.
In our clinical data, low CHD7 expression was associated with increased OS and RFS in all patients receiving adjuvant therapy, although this was likely driven by the inclusion of patients receiving gemcitabine. The association of low CHD7 expression with increased survival was magnified in patients receiving gemcitabine as a component of their adjuvant therapy despite smaller patient numbers, indicating that low CHD7 expression may indeed be associated with gemcitabine sensitivity in these patients. In contrast, CHD7 expression in patients not receiving gemcitabine was not statistically significant. This analysis is underpowered with limitation of small sample size and selection bias, but our findings provide valuable hypothesis-generating data suggesting that CHD7 may have predictive value in these patients.
Given the evidence that patients with low CHD7 expression demonstrate improved outcomes, it is possible that adjuvant therapy regimens could be tailored to individualize patient treatment based on CHD7 expression. This should be examined in future prospective trials and in larger secondary analyses of completed prospective studies. While adjuvant chemotherapy for patients with PAC is advantageous, the ideal drug regimen remains unclear. The benefit of adjuvant gemcitabine compared with adjuvant 5-fluorouracil (5-FU) in patients with early-stage resected PAC patients has not been demonstrated in any large trials. Both the ESPAC-3 trial, which randomized patients with resected PAC to adjuvant gemcitabine versus 5-FU, and the RTOG 97-04 trial, which randomized patients with resected PAC to adjuvant pre and post chemoradiotherapy gemcitabine versus 5-FU, reported no significant difference in disease-free survival (DFS) or OS between the two arms (3, 44). Our finding that low CHD7 expression is associated with improved outcome in early-stage PAC patients treated with adjuvant gemcitabine suggest that, once validated, CHD7 expression could potentially be used as a predictive biomarker to individualize adjuvant therapy for these patients. Additionally, the optimal radiation dose and fractionation for patients with resected pancreatic adenocarcinoma remains unknown, and molecular biomarkers to guide adjuvant therapy decisions are essential (45). The potential utility of CHD7 expression as a prognostic and potentially predictive biomarker still remains a hypothesis-generating observation and requires validation in a prospective clinical trial, in which regimen dosing and duration are more homogenous.
Interest in genetic sequencing data, such as with The Cancer Genome Atlas (TCGA) and other similar projects, continues to increase (46, 47), leading to rapidly increasing knowledge of genes expressed and mutated in specific cancer types including PAC. As this knowledge becomes available, it is crucial that an approach be developed to help identify those genes that may serve as clinically relevant prognostic or predictive biomarkers or potential drug targets for novel therapeutics. The successful identification and validation of CHD7 as a novel gemcitabine sensitivity gene that is associated with outcome in early stage PAC patients treated with adjuvant gemcitabine is evidence that our approach may be successful in identifying other clinically relevant biomarkers or drug targets.
It is worth noting that recent advances in chemotherapy have increased the use of FOLFIRINOX therapy in the metastatic setting, impacting the potential utility of this study. Still, NCCN guidelines in the metastatic setting equivalently recommend FOLFIRINOX or two gemcitabine-based regimens (gemcitabine with the addition of erlotinib or nab-paclitaxel), both with category one evidence (48). Additionally, gemcitabine or fluoropyrimidine therapies are still recommended in the adjuvant setting, which is where this study's clinical focus remains. Future studies should evaluate the predictive role of CHD7 in a larger, randomized prospective trial to validate potential gemcitabine sensitivity genes in a similar fashion to other identified predictive biomarkers (49). The current study suggests that CHD7 may be a useful biomarker for determining which patients will derive greater benefit from gemcitabine therapy, providing clinicians a way to better select patients for specific adjuvant therapy regimens in the future.
Supplementary Material
Acknowledgments
We thank members of the Yu lab and Chris Crane for helpful comments during article preparation. This work was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (ULl TR000454 to LEC and SBF) and (TLlTR000456 to LEC); Pancreatic Cancer Action Network/American Association for Cancer Research (16982 to DSY); Department of Defense/Peer Reviewed Cancer Research Program (CA110535 to DSY); and Georgia Cancer Coalition (11072 to DSY). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding Source: Supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Numbers ULl TR000454 to LEC and SBF and TLlTR000456 to LEC, PanCAN/AACR 16982 to DSY, DOD/PRCRP CA110535 to DSY, and Georgia Cancer Coalition 11072 to DSY. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62(1):10–29. doi: 10.3322/caac.20138. Epub 2012/01/13. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V, Haas M, Boeck S. Systemic treatment of advanced pancreatic cancer. Cancer treatment reviews. 2012;38(7):843–53. doi: 10.1016/j.ctrv.2011.12.004. Epub 2012/01/10. [DOI] [PubMed] [Google Scholar]
- 3.Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, Goldstein D, et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2010;304(10):1073–81. doi: 10.1001/jama.2010.1275. Epub 2010/09/09. [DOI] [PubMed] [Google Scholar]
- 4.Karhu R, Mahlamaki E, Kallioniemi A. Pancreatic adenocarcinoma -- genetic portrait from chromosomes to microarrays. Genes, chromosomes & cancer. 2006;45(8):721–30. doi: 10.1002/gcc.20337. Epub 2006/05/12. [DOI] [PubMed] [Google Scholar]
- 5.Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2007;297(3):267–77. doi: 10.1001/jama.297.3.267. Epub 2007/01/18. [DOI] [PubMed] [Google Scholar]
- 6.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 7.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 8.Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, et al. Cosmic 2005. British journal of cancer. 2006;94(2):318–22. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nature genetics. 2004;36(9):955–7. doi: 10.1038/ng1407. Epub 2004/08/10. [DOI] [PubMed] [Google Scholar]
- 10.Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. American journal of human genetics. 2008;83(4):511–9. doi: 10.1016/j.ajhg.2008.09.005. Epub 2008/10/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. British journal of cancer. 2004;91(2):355–8. doi: 10.1038/sj.bjc.6601894. Epub 2004/06/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6(269):11. doi: 10.1126/scisignal.2004088. Epub 2013/04/04. 1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. Epub 2012/05/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463(7283):958–62. doi: 10.1038/nature08733. Epub 2010/02/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Human molecular genetics. 2010;19(18):3491–501. doi: 10.1093/hmg/ddq265. Epub 2010/07/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nature genetics. 2011;43(6):607–11. doi: 10.1038/ng.825. Epub 2011/05/03. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 18.Batsukh T, Schulz Y, Wolf S, Rabe TI, Oellerich T, Urlaub H, et al. Identification and characterization of FAM124B as a novel component of a CHD7 and CHD8 containing complex. PloS one. 2012;7(12):e52640. doi: 10.1371/journal.pone.0052640. Epub 2013/01/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagaraju GP, Zhu S, Wen J, Farris AB, Adsay VN, Diaz R, et al. Novel synthetic curcumin analogues EF31 and UBS109 are potent DNA hypomethylating agents in pancreatic cancer. Cancer letters. 2013;341(2):195–203. doi: 10.1016/j.canlet.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Harsha HC, Kandasamy K, Ranganathan P, Rani S, Ramabadran S, Gollapudi S, et al. A compendium of potential biomarkers of pancreatic cancer. PLoS medicine. 2009;6(4):e1000046. doi: 10.1371/journal.pmed.1000046. Epub 2009/04/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maupin KA, Sinha A, Eugster E, Miller J, Ross J, Paulino V, et al. Glycogene expression alterations associated with pancreatic cancer epithelial-mesenchymal transition in complementary model systems. PloS one. 2010;5(9):e13002. doi: 10.1371/journal.pone.0013002. Epub 2010/10/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer cell. 2009;16(3):259–66. doi: 10.1016/j.ccr.2009.07.016. Epub 2009/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maithel SK, Coban I, Kneuertz PJ, Kooby DA, El-Rayes BF, Kauh JS, et al. Differential expression of ERCC1 in pancreas adenocarcinoma: high tumor expression is associated with earlier recurrence and shortened survival after resection. Annals of surgical oncology. 2011;18(9):2699–705. doi: 10.1245/s10434-011-1610-x. Epub 2011/03/02. [DOI] [PubMed] [Google Scholar]
- 24.Fisher SB, Patel SH, Bagci P, Kooby DA, El-Rayes BF, Staley CA, 3rd, et al. An analysis of human equilibrative nucleoside transporter-1, ribonucleoside reductase subunit M1, ribonucleoside reductase subunit M2, and excision repair cross-complementing gene-1 expression in patients with resected pancreas adenocarcinoma: Implications for adjuvant treatment. Cancer. 2013;119(2):445–53. doi: 10.1002/cncr.27619. Epub 2012/05/10. [DOI] [PubMed] [Google Scholar]
- 25.Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW, et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer. 2013;119(17):3148–55. doi: 10.1002/cncr.28144. Epub 2013/05/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall WA, Petrova AV, Colbert LE, Hardy CW, Fisher SB, Saka B, et al. Low CHD5 expression activates the DNA damage response and predicts poor outcome in patients undergoing adjuvant therapy for resected pancreatic cancer. Oncogene. 2013 doi: 10.1038/onc.2013.488. Epub 2013/11/28. [DOI] [PubMed] [Google Scholar]
- 27.Basturk O, Singh R, Kaygusuz E, Balci S, Dursun N, Culhaci N, et al. GLUT-1 expression in pancreatic neoplasia: implications in pathogenesis, diagnosis, and prognosis. Pancreas. 2011;40(2):187–92. doi: 10.1097/MPA.0b013e318201c935. Epub 2011/01/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, Nakata K, et al. Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia. 2010;12(10):807–17. doi: 10.1593/neo.10458. Epub 2010/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cotta-Ramusino C, McDonald ER, 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332(6035):1313–7. doi: 10.1126/science.1203430. Epub 2011/06/11. 10.1126/science.1203430332/6035/1313 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Molecular cell. 2009;35(2):228–39. doi: 10.1016/j.molcel.2009.06.021. Epub 2009/08/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agata N, Ahmad R, Kawano T, Raina D, Kharbanda S, Kufe D. MUC1 oncoprotein blocks death receptor-mediated apoptosis by inhibiting recruitment of caspase-8. Cancer research. 2008;68(15):6136–44. doi: 10.1158/0008-5472.CAN-08-0464. Epub 2008/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akada M, Crnogorac-Jurcevic T, Lattimore S, Mahon P, Lopes R, Sunamura M, et al. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(8):3094–101. doi: 10.1158/1078-0432.CCR-04-1785. Epub 2005/04/20. [DOI] [PubMed] [Google Scholar]
- 33.Azorsa DO, Gonzales IM, Basu GD, Choudhary A, Arora S, Bisanz KM, et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. Journal of translational medicine. 2009;7:43. doi: 10.1186/1479-5876-7-43. Epub 2009/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong N, Wang M, Li H, Cui Y, Guo Q. Gemcitabine in combination with vinorelbine in elderly patients with anthracycline- and taxane-pretreated metastatic breast cancer. Cancer chemotherapy and pharmacology. 2012;69(5):1315–22. doi: 10.1007/s00280-012-1830-1. Epub 2012/02/07. [DOI] [PubMed] [Google Scholar]
- 35.O'Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, et al. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Molecular cell. 2010;40(4):645–57. doi: 10.1016/j.molcel.2010.10.022. Epub 2010/11/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446(7137):815–9. doi: 10.1038/nature05697. Epub 2007/04/13. [DOI] [PubMed] [Google Scholar]
- 37.Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y, et al. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO reports. 2010;11(11):876–82. doi: 10.1038/embor.2010.153. Epub 2010/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan MA, Parsels LA, Parsels JD, Lawrence TS, Maybaum J. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle. 2006;5(17):1983–8. doi: 10.4161/cc.5.17.3184. Epub 2006/08/26. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, Chen Z, Malysa A, Li X, Oliveira P, Zhang Y, et al. A kinome screen identifies checkpoint kinase 1 (CHK1) as a sensitizer for RRM1-dependent gemcitabine efficacy. PloS one. 2013;8(3):e58091. doi: 10.1371/journal.pone.0058091. Epub 2013/03/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Von Hoff D, Ervin TJ, Arena FP, Chiorean EG, Infante JR, Moore MJ, et al. Randomized phase III study of weekly nab-paclitaxel plus gemcitabine versus gemcitabine alone in patients with metastatic adenocarcinoma of the pancreas (MPACT) 2013 ASCO Gastrointestinal Cancers Symposium, abstract. 2012 [Google Scholar]
- 41.Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer discovery. 2012;2(3):260–9. doi: 10.1158/2159-8290.CD-11-0242. Epub 2012/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacob DA, Bahra M, Langrehr JM, Boas-Knoop S, Stefaniak R, Davis J, et al. Combination therapy of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide and gemcitabine shows strong antitumor activity in pancreatic cancer cells. Journal of gastroenterology and hepatology. 2007;22(5):738–48. doi: 10.1111/j.1440-1746.2006.04496.x. Epub 2007/04/21. [DOI] [PubMed] [Google Scholar]
- 43.Porcelli L, Quatrale AE, Mantuano P, Leo MG, Silvestris N, Rolland JF, et al. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Molecular oncology. 2012 doi: 10.1016/j.molonc.2012.10.002. Epub 2012/11/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Regine WF, Winter KA, Abrams RA, Safran H, Hoffman JP, Konski A, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2008;299(9):1019–26. doi: 10.1001/jama.299.9.1019. Epub 2008/03/06. [DOI] [PubMed] [Google Scholar]
- 45.Hall WA, Colbert LE, Liu Y, Gillespie T, Lipscomb J, Hardy C, et al. The influence of adjuvant radiotherapy dose on overall survival in patients with resected pancreatic adenocarcinoma. Cancer. 2013;119(12):2350–7. doi: 10.1002/cncr.28047. Epub 2013/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi: 10.1038/nature11547. Epub 2012/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iacobuzio-Donahue CA, Velculescu VE, Wolfgang CL, Hruban RH. Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(16):4257–65. doi: 10.1158/1078-0432.CCR-12-0315. Epub 2012/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fisher SB, Patel SH, Bagci P, Kooby DA, El-Rayes BF, Staley CA, 3rd, et al. An analysis of human equilibrative nucleoside transporter-1, ribonucleoside reductase subunit M1, ribonucleoside reductase subunit M2, and excision repair cross-complementing gene-1 expression in patients with resected pancreas adenocarcinoma: Implications for adjuvant treatment. Cancer. 2012 doi: 10.1002/cncr.27619. Epub 2012/05/10. [DOI] [PubMed] [Google Scholar]
- 49.Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136(1):187–95. doi: 10.1053/j.gastro.2008.09.067. Epub 2008/11/11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.