

Abstract
RAS-driven malignancies remain a major therapeutic challenge. The two-stage 7,12-dimethylbenz(a)anthracene (DMBA)/12-o-tetradecanoylphorbol-13-acetate (TPA) model of mouse skin carcinogenesis has been used to study mechanisms of epithelial tumor development by oncogenic Hras. We used mice with a HrasG12V knock-in allele to elucidate the early events after Hras activation, and to evaluate the therapeutic effectiveness of farnesyltransferase (FTI) inhibition. Treatment of Caggs-Cre/FR-HrasG12V mice with TPA alone was sufficient to trigger papilloma development with shorter latency and a ~10-fold greater tumor burden than DMBA/TPA-treated WT controls. HrasG12V allele copy number was increased in all papillomas induced by TPA. DMBA/TPA treatment of HrasG12V knock-in mice induced an even greater incidence of papillomas, which either harbored HrasG12V amplification, or developed a HrasQ61L mutation in the second allele. Laser-capture microdissection of normal skin, hyperplastic skin and papillomas showed that amplification occurred only at the papilloma stage. HRAS mutant allelic imbalance was also observed in human cancer cell lines, consistent with a requirement for augmented oncogenic HRAS signaling for tumor development. The FTI SCH66336 blocks HRAS farnesylation and delocalizes it from the plasma membrane. NRAS and KRAS are not affected as they are alternatively prenylated. When tested in lines harboring HRAS, NRAS or KRAS mutations, SCH66336 delocalized, inhibited signaling and preferentially inhibited growth only of HRAS-mutant lines. Treatment with SCH66336 also induced near-complete regression of papillomas of TPA-treated HrasG12V knock-in mice. These data suggest that farnesyl transferase inhibitors should be reevaluated as targeted agents for human HRAS-driven cancers, such as those of bladder, thyroid and other epithelial lineages.
Keywords: RAS, Papilloma, Farnesyl Transferase Inhibitor
Introduction
Hras, KrasA, KrasB and Nras are plasma membrane GTPases that exist in an active, GTP-bound, or inactive, GDP-bound, state. Many human tumors have a predilection for mutations in one RAS gene family member. HRAS mutations are less common overall, but they have a particularly high prevalence in cancers of the upper aerodigestive tract, skin, thyroid and urinary bladder. KRAS mutations predominate in pancreas, lung, and colorectal malignancies, whereas NRAS mutations are found in melanomas and hematopoietic tumors (1, 2).
Ras isoforms differ in their respective C-terminal hypervariable regions responsible for lipid modification, subcellular localization, intracellular processing and trafficking (3–7). It is clear that they have distinct functions, as knockout mice of each Ras gene display different phenotypes (8–10). However, there is no definitive explanation for the predilection for individual RAS oncogenes in different tumor lineages.
The two-stage model of mouse skin carcinogenesis has been extensively used for the study of tumor initiation, promotion and progression. In 7,12-dimethylbenz(a)anthracene (DMBA) -treated mouse skin, repeated topical applications of the tumor promoter 12-o-tetradecanoylphorbol-13-acetate (TPA) triggers skin papilloma development and progression into carcinomas. Mutant Hras alleles, mainly Q61L, are found in a high proportion of benign papillomas initiated by DMBA (11, 12). Targeted deletion of the Hras gene decreases the papilloma burden following a DMBA/TPA carcinogenesis protocol (13, 14), further establishing the importance of Hras in papilloma formation. These are often accompanied by an increase in copy number of the mutant allele (15). However, it is not clear whether Hras allelic imbalance is an obligate step in skin papilloma development. An increase of the mutant-to-wild-type Hras allelic ratio has been implicated in progression from squamous to spindle cell carcinomas (16). Loss of the wild-type Hras allele is also seen in papillomas after chemical skin carcinogenesis, leading to the presumption that the wild-type protein may function as a tumor suppressor. Recent data suggest that loss of the wild-type Hras allele may promote tumor progression rather than initiation (14). Despite the well-established role of Hras mutations in papilloma initiation in the skin 2-stage carcinogenesis model, Schuhmacher et al reported that papillomas failed to develop after topical administration of TPA to mice with an HrasG12V knock-in allele (17). In this paper we revisit this question in an independently derived HrasG12V knock-in mouse model of Costello’s syndrome that spontaneously developed papillomas (18), and exploit this system to evaluate targeted therapeutics of Hras-driven tumors.
All Ras isoforms are farnesylated. Farnesyl transferase inhibitors (FTIs) block the addition of an isoprenoid group to the C-terminal portion of Ras to prevent formation of active Ras. FTIs block Hras farnesylation, membrane localization, and inhibit oncogenic Hras-driven cellular transformation in vitro (19, 20) and in vivo (21). However, in most clinical trials FTIs showed no significant antitumor activity in patients with advanced solid tumors such as lung, pancreatic and colon cancers, which mainly harbor KRAS mutations (22–24), or with acute myeloid leukemia, which primarily have mutations of NRAS (25). The refractoriness to FTIs of RAS-driven cancers has been attributed to compensatory geranylgeranylprenylation of KRAS and NRAS, which preserves their membrane targeting and function (26–28). However, the HRAS selectivity of FTIs versus K- or NRAS-driven tumors has not been extensively studied in cells or in a mouse model, and no trial with an FTI has been done exclusively in patients with HRAS mutant tumors.
The FTI class of anti-cancer drugs has gone into disfavor in part because of the failure of clinical trials in patients that were not appropriately selected based on the oncogenic driver of the tumor. The data provided in this study present a strong preclinical rationale for revisiting the potential efficacy of FTIs in HRAS mutant tumors.
Results
HrasG12V is sufficient to initiate skin tumorigenesis
We treated the skin of Caggs-Cre/FR-HrasG12V mice, which have a knock-in HrasG12V allele that is globally expressed at endogenous levels (18), with the tumor promoter TPA only and examined them for tumor development. As shown in Fig 1, topical administration of TPA alone to HrasG12V mice is sufficient to trigger rapid papilloma development. The tumor latency is shorter and the incidence about 7-fold higher than in wild-type mice subjected to sequential DMBA/TPA treatment. However, combined DMBA/TPA treatment of HrasG12V mice resulted in about 1.8-fold more tumors than those treated with TPA alone, indicating that DMBA cooperated with the endogenously activated mutant Hras allele in tumor initiation. Histological examination at week 15 showed TPA-induced hyperplastic changes in the epidermis and typical papillomas associated with increased Ki67 staining in HrasG12V/ mice (Supplementary Fig 1). These observations indicate that oncogenic Hras is a tumor initiator and that it is sufficient to trigger the skin neoplastic process.
Fig. 1. Topical administration of TPA to HrasG12V mice triggers papilloma development.
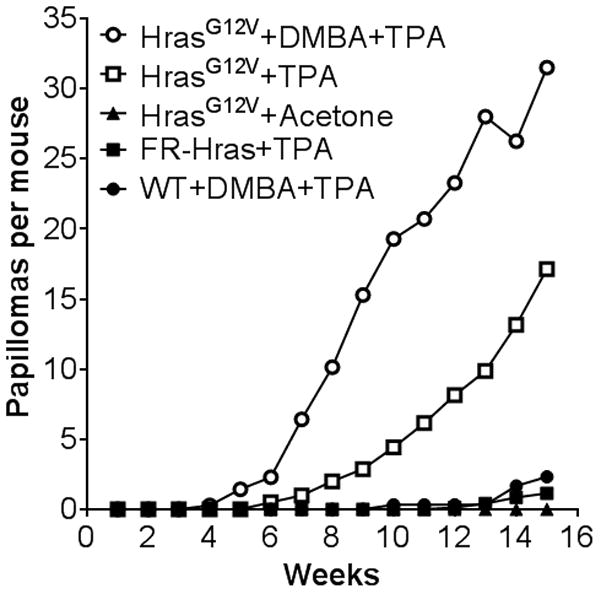
Average number of tumors per mouse during the time course of the study. There were five groups of mice: HrasG12V mice treated with a single dose of DMBA followed by TPA treatment twice per week (n=7), Hras G12V mice treated with TPA only (n=7), wild type mice treated DMBA followed by TPA (n=7), Hras G12V mice treated with the vehicle acetone (twice per week) (n=8) and FR-Hras mice (harboring the targeted Hras allele, unrecombined, because of the absence of Caggs-Cre) treated with TPA (n=7).
Hras allelic imbalance is an obligate step in papilloma development and occurs only at the papilloma stage
We previously reported that Hras gene copy number changes are almost always associated with the development of spontaneously-arising skin and forestomach papillomas, as well as angiosarcomas in HrasG12V mice (18). As shown in Fig 2A, 10/10 papillomas arising in HrasG12V mice treated with TPA alone had increased copies of the mutant Hras allele. By contrast, 7/10 randomly-selected papillomas induced by DMBA/TPA in HrasG12V mice had increased copies of the HrasG12V allele, whereas the remaining 3 had acquired an HrasQ61L mutation (Fig 2B,C), which was located in the second Hras allele (Supplementary Fig 2). This helps to explain the greater tumor burden induced by DMBA/TPA in HrasG12V mice compared to TPA alone (Fig 1). In DMBA/TPA-treated WT mice, 14/15 papillomas had only an HrasQ61L mutation, as expected (See Table 1). To determine the stage of tumor development at which Hras mutant allelic imbalance takes place we performed a PCR-based analysis of laser-capture microdissected specimens of normal skin, hyperplastic skin and papillomas from TPA-treated HrasG12V mice (Supplementary Fig 3). As shown in Fig 2D, there was no gain in Hras mutant allele copy number in hyperplastic samples, whereas the adjacent papillomas from the same animals had a higher number of HrasG12V alleles. Mutant Hras copy gain was also associated with increased expression of mutant Hras mRNA relative to wild-type (Supplementary Fig 4A and B). These data suggest that Hras-induced tumor formation requires augmented oncogenic Hras signaling, either through increased mutant Hras gene copy number or through acquisition of mutations in the second Hras allele. This also applies to the human cancer cell lines harboring HRAS mutations that we tested (Hth83, C643 and T24), all of which had two or more copies of mutant HRAS (Supplementary Fig 5).
Fig. 2. Hras allelic imbalance in papillomas from HrasG12V mice.

A) PCR of genomic DNA of 10 papillomas or tails from TPA-treated HrasG12V mice with primers that distinguish mutant from WT Hras alleles. W: wild-type 622 bp allele; M: 666bp targeted allele. The mutant Hras allele copy number was increased in all 10 papillomas induced by TPA alone. B) PCR of DNA from 10 randomly selected papillomas from DMBA/TPA-treated HrasG12V mice, tails from HrasG12V mice and papillomas from DMBA/TPA treated WT mice. 7/10 papillomas had M>W allelic imbalance. The three lanes marked by an asterisk had ~ 1/1 ratio of M/W alleles. C) Representative Hras Sanger sequence traces of PCR products of tumor DNAs from panel B: Left: Tumor with M>W Hras allelic imbalance, showing the Hras G12V mutation, and a WT Hras Q61 codon. Right: Tumor (*) without Hras allelic imbalance (M=W), harboring both Hras G12V and Q61L mutations. G12V and Q61L are located in different alleles (Suppl Fig 2). D) Laser-capture microdissection (LCM) of normal skin epithelium, hyperplasia and papilloma. PCR of genomic DNA of LCM of tissues from TPA-treated HrasG12V mice. As indicated, Hras mutant allele amplification occurred only at the papilloma stage. N: normal skin epithelium; H: hyperplasia; P: papilloma.
Table 1. Hras.
Gene in Papillomas of HrasG12V/+ Mice Treated With DMBA/TPA
| HrasG12V mutation | HrasG12V copy number increase | HrasQ61L mutation | |
|---|---|---|---|
| HrasG12V+DMBA+TPA | 10/10 | 7/10 | 3/10 |
| HrasG12V+TPA | 10/10 | 10/10 | 0/10 |
| HrasWT+DMBA+TPA | 0/15 | nd | 14/15 |
The farnesyl transferase inhibitor SCH 66336 preferentially inhibits growth of HRAS-mutant human cancer cell lines
Covalent lipid modification of G proteins is required for their membrane localization. All RAS isoforms are farnesylated, whereas NRAS and KRAS, but not HRAS, are also weak substrates for geranylgeranylprenyltransferase-1 (GGTase-1) (26–28). The disappointing results of clinical trials with farnesyl transferase inhibitors (FTI) are thought to be due at least in part to the fact that they were tested in patients with KRAS or NRAS mutant cancers, in which the oncoprotein could still be prenylated and hence remain functional. We tested the effects of SCH 66336 on growth of 15 cell lines with different RAS mtations: 5 HRAS, 5°NRAS and 5 KRAS. As shown in Fig 3A, the 5 cancer cell lines with HRAS mutations were sensitive to the compound, with IC50 ranging from 34 - 197nM. By contrast, the cell lines with NRAS or KRAS were mostly resistant, with 6 of them having an IC50 > 600nM. These differences in FTI-responsiveness of HRAS vs K- or NRAS lines were also reflected in the growth curves (Fig 3B). Accordingly, SCH6636 also caused a profound inhibition of DNA synthesis in HRAS, but not in KRAS or NRAS mutant cells (Fig 3C and Fig 3D).
Fig. 3. Growth of human cancer cell lines with HRAS mutation is preferentially inhibited by SCH 66336.

A) IC50 of each cell line for SCH 66336. The indicated cell lines were incubated with different concentrations of SCH 66336 for 4 days and the cell number counted. All 5 HRAS mutant cell lines have lower IC50 for FTI compared to cells with NRAS or KRAS mutation. B) Growth of cell lines in the presence of SCH 66336. The indicated cell lines were incubated with SCH 66336 (25nM, 100nM) or vehicle for 2, 4 and 6 days. C) Left panel: FACS for FITC-labeled BrdU vs 7-AAD for representative HRAS (SKmel31) and NRAS (Hth7) mutant cell lines treated for 3 days with 100 nM SCH 66336. Right panel: SCH 66336 decreased DNA synthesis in the 4 HRAS mutant but not in NRAS or KRAS mutant cell lines.
HRAS-specific delocalization and inhibition of MAPK signaling by SCH 66336
We next investigated whether the HRAS-selective effects on cell growth were also associated with corresponding effects on membrane targeting of the three RAS isoforms. As shown in Fig. 4A, SCH 66336 delocalized only HRAS from the membrane compartment to the cytosol of Hth83 cells, whereas KRAS and NRAS were unaffected. Treatment of Hth83 cells with SCH 66336 resulted in a time-dependent shift in migration of HRAS, which also accumulated over time (Fig 4B). This was already apparent 6h after addition of the compound, and peaked at about 48h. The timing of inhibition of pMEK closely paralleled HRAS defarnesylation, whereas cleavage of PARP was only apparent when pMEK was profoundly inhibited. We next performed dose-response studies of 4 HRAS and 4 KRAS or NRAS mutant lines incubated with SCH 66336 for 72h. SCH 66336 abrogated MAPK signaling, as determined by pMEK and pERK levels, in all 4 HRAS mutant lines in a dose-dependent manner, whereas AKT signaling was not decreased (Fig 4C). Interestingly, there was a paradoxical increase of pS473-AKT by the FTI in the HRAS-mutant Hth83 cells. This is likely an off-target effect, since knockdown of HRAS in these cells by RNA interference inhibited AKT phosphorylation at this site (Supplementary Figure 6B). SCH 66336 had no effect on MAPK signaling in cells with KRAS or NRAS mutation, despite inhibiting the farnesylation of wild-type HRAS (Fig 4D). Interestingly, although the NRAS mutant lines Hth7 and Act1 were relatively sensitive to the growth inhibitory effects of the compound (IC50 of ~ 200 nM; Fig 3), SCH 66336 did not inhibit pMEK in these lines even at the highest concentrations (1 μM). As Rheb GTPases are also farnesylated proteins, it is conceivable that the growth inhibitory effect of SCH 66336 on these NRAS mutant lines was exerted through their delocalization from endomembranes. This would be predicted to decrease mTOR activity, which was not observed, based on no detectable effect on phosphorylation of its substrate ribosomal S6. The mutant RAS dependency for signaling and growth is further demonstrated in Supplementary Fig 6 and Supplementary Fig 7. Thus, knock-down of mutant HRAS inhibited MAPK signaling and proliferation of Hth83 cells, whereas depletion of WT KRAS or NRAS had no appreciable effect on signaling and only a modest inhibitory effect on proliferation. Accordingly, in KRAS-mutant Cal62 cells and NRAS mutant Hth7 cells, only the respective mutant knockdown inhibited cell signaling and growth, whereas knockdown of the WT RAS isoforms was without effect on signaling and only modestly inhibited cell growth.
Fig. 4. SCH 66336 disrupts HRAS membrane localization and inhibits MAPK signaling in HRAS mutant lines.

A) Western blots of subcellular fractions of Hth83 cells treated with 250nM SCH 66336 for 48hr, probed with the indicated antibodies. SCH 66336 delocalized HRAS, but not KRAS or NRAS. C: Cytoplasmic; M: Membrane; N: Nucleus. B) SCH 66336 inhibits HRAS signaling with a delayed time course in Hth83 cells. Hth83 cells were incubated with 250nM SCH 66336 for the indicated time. The cell lysates were Western blotted with the indicated antibodies. U: unfarnesylated HRAS; F: farnesylated HRAS. C) SCH 66336 blocked MAPK signaling in a concentration-dependent manner. Western blots of 4 HRAS mutant cell lines treated with the indicated concentration of SCH 66336 for 72hr probed with indicated antibodies. D) SCH 66336 had no effect on MAPK signaling in cell lines with KRAS or NRAS mutations, despite inhibiting farnesylation of WT HRAS in these lines.
SCH 66336 induces regression of papillomas in Hras G12V mice
We next tested the effects of SCH 66336 on papillomas arising in 8 – 12 week-old HrasG12V mice treated with TPA for 12 to 16 weeks. Mice were continuously treated with topical TPA, and randomized to vehicle or SCH66336 80mg/kg BID by gavage, and tumor growth monitored every 2–3 days by caliper measurements. As shown in Fig 5A and 5B, treatment of tumor-bearing mice with SCH 66336 induced an almost complete regression of the papillomas within 10 days. By contrast, tumors from mice in the vehicle-treated group continued to increase in size. Body weight was unchanged between both treatment groups. Immunohistochemical analysis showed that SCH66336-treated tumors had decreased Ki67 staining and lower pERK positivity (Fig 5C), consistent with the in vitro findings. Hepatocytes are the only cell type exhibiting clearly increased downstream signaling following endogenous expression of HrasG12V in vivo (18). As shown in Fig 5D, SCH 66336 inhibited Hras farnesylation and decreased pERK in HrasG12V mouse livers, consistent with appropriate targeting of the drug in vivo.
Fig. 5. SCH 66336 induces regression of papillomas in HrasG12V mice.

A) Representative photos of mice after treatment with vehicle or SCH 66336 for the indicated times. HrasG12Vmice were first treated with TPA for 12 to 16 weeks to induce papilloma development. Mice then were treated with 80mg/kg SCH 66336 b.i.d. (n=5) or vehicle (n=6) by gavage for the indicated times. B) Tumor volumes at the indicated times during treatment with vehicle (n=10) or SCH 66336 (n=11) from the mice described above. Tumor size was measured on days 0, 3, 5, 7 and 10. Bars represent mean ± SEM. C) SCH 66336 decreased cell proliferation and pERK in papillomas. Representative H&E and IHC staining for Ki67 and pERK in sections of mouse papillomas treated with vehicle or SCH 66336. D) Western blot of liver tissues from HrasG12Vmice treated with vehicle (n=4) or SCH 66336 (n=4), probed with the indicated antibodies. SCH 66336 decreased MAPK signaling and inhibited Hras farnesylation in livers of HrasG12V mice.
Discussion
Our studies show that endogenous expression of mutant Hras is likely sufficient to initiate skin tumorigenesis, as exposure of HrasG12V mice to TPA alone resulted in papilloma development with shorter latency and greater tumor burden compared to DMBA/TPA treated Hras-wt mice. These results differ from those of a prior study in an independently-derived HrasG12V knock-in mouse model, in which topical administration of TPA failed to induce papillomas (17). Although this could be due to a mouse strain effect, in our view it is more likely that the co-expressed IRES-β-geo cassette included in the 3′UTR of the HrasG12V targeting vector in the Schuhmacher study may have interfered with endogenous expression of the oncoprotein, so that the required threshold for tumor initiation was not achieved. This is also supported by the fact that the HrasG12V-IRES-β-geo mouse model had an attenuated developmental phenotype compared to the one described here.
Tumor initiation can be achieved by activating mutations or copy gains of oncogenes. We find that Hras-induced transformation is universally associated with copy gains of the genomic locus containing the mutant allele, suggesting that Hras allelic imbalance is obligate for tumor development. The evidence for this is quite compelling. Thus, 10/10 papillomas arising in HrasG12V mice treated with TPA alone had increased copies of the mutant Hras allele. Moreover, all 10 randomly-selected papillomas induced by DMBA/TPA in HrasG12V mice had either increased copies of HrasG12V or an acquired Q61L mutation in the second Hras allele. All spontaneously arising skin papillomas, forestomach papillomas and angiosarcomas arising in these mice also had HrasG12V amplification (18). Hras mutant copy number increases have also been observed in DMBA/TPA-induced papillomas from wild-type mice (15). We also found HRAS copy gains in 2/3 human cancer cell lines we tested, with the other having a homozygous HRAS mutation, possibly due to uniparental disomy at that locus. HRAS activating mutations are also associated with gene copy gains in human Spitz nevi tumors (29) and human thyroid neoplasms (30). As might be predicted, Hras mutant allelic imbalance was not present in normal skin or in hyperplastic specimens, whereas it was consistently found in papilloma tissues. The association of oncogene mutation and amplification is not uncommon. For instance, an increase in EGFR (31) or KRAS (32) mutant allele copy number is seen in many non-small cell lung cancers. However, mutant allelic imbalance is not required for transformation, as many NSCLCs harboring these mutations have a single copy of the oncogene.
The role of wild-type Ras alleles in cell transformation is complex (33). On the one hand, wild type Kras and Nras can act as tumor suppressors in cells with a corresponding mutant Ras allele (34), an effect that has been shown to be dose-dependent (35, 36). Our results suggest that loss of the wild-type Hras allele is not required for transformation of the skin epithelium by mutant Hras, as only a minor subset of papillomas had loss of WT Hras in HrasG12V knock in mice (18). Recent data from the Balmain lab is consistent with these findings, as Hras heterozygosity appears to be primarily involved in malignant transformation of skin tumors, rather than on tumor initiation (14). We found that WT-HRAS alleles are present in most human cancer cell lines harboring HRAS mutations. Instead, our results suggest that intensification of mutant Hras signaling through copy-number imbalances may play a key, and possibly rate-limiting, role in papilloma initiation, as supported by the fact that this imbalance is only seen at the papilloma stage. This was also noted in Rat 1 fibroblasts harboring a single HrasG12V mutant allele introduced by homologous recombination (37). Finney and Bishop found that endogenous HrasG12V expression was not sufficient to induce Rat 1 fibroblast transformation in vitro, whereas cells commonly had excess copies of the mutant Hras allele when transformed clones developed after serial passaging.
We found that knockdown of Hras or Kras in cell lines expressing the corresponding mutant oncoprotein markedly decreased basal MAPK signaling, whereas individual knockdown of the wild type Ras isoforms did not. These data do not challenge the conclusions of recent studies that point to an important supportive role of wild-type Ras in signaling activated by oncogenic Ras (38–40), since we did not attempt to deplete all wild-type Ras isoforms. Silencing of oncogenic Ras in three different RAS-mutant cell lines inhibited cell growth more profoundly than depletion of each of the wild-type Ras isoforms, although the latter also dampened cell growth to a modest extent, consistent with a cooperative role of wild-type Ras proteins in tumorigenesis (38–40).
All Ras isoforms (Hras, Kras and Nras) are farnesylated. Although ~30% of human cancers harbor RAS mutations, the therapeutic efficacy of FTIs in the treatment of cancer patients has been disappointing (22–25). We found that human cancer cell lines harboring HRAS mutations are more sensitive to the antiproliferative effects of the FTI SCH 66336 than those carrying KRAS or NRAS mutations. Using FTI SCH 66336 across a panel of 5 different cell lines from different tissues with HRAS mutation, the FTI reduced growth at IC50s ranging from 34 – 197nM. By contrast, the cell lines with NRAS or KRAS were mostly resistant, with six of them having an IC50 > 600nM (Fig. 3). Treatment of HrasG12V mice harboring papillomas with SCH66336 resulted in profound tumor regression associated with inhibition of Hras downstream signaling. Although this effect is likely exerted through inhibition of Hras signaling in the transformed epithelial cells, we cannot exclude an effect of the FTI on stromal cells or on the tumor vasculature, as the mutant allele is ubiquitously expressed.
The failure of clinical trials with FTIs can now be explained based on a better understanding of their mechanism of action. Unlike HRAS, which is exclusively farnesylated, KRAS and NRAS can be prenylated by geranylgeranyl transferases upon blockade of farnesylation, and remain functional (27, 28). Consistent with this, we found that SCH 66336 delocalized HRAS, but not KRAS or NRAS, from the membrane to the cytoplasm, and abrogated its downstream signaling via MAPK. Clinical trials with FTIs did not consider this mutational specificity in their criteria for selecting patients for enrolment. This was in part due to preclinical studies that showed that some cell lines with KRAS or NRAS mutations, or even cells that were wild-type for all RAS oncogenes, were variably growth inhibited by FTIs. Bernhard et al showed that an FTI inhibitor, when used at concentrations that inhibited farnesylation but not geranylgeranylation, increased the radiosensitivity of two HRAS mutant lines but not two KRAS mutant cell lines (41). By contrast, Sepp-Lorenzino et al noted that the effects of a peptidomimetic FTI were unrelated to the RAS mutation status of the cells. In hindsight, this is not surprising as there were no HRAS mutant cell lines used in their experiments (42). The FTI SCH 66336 was found to selectively delocalize HRAS and induce G1/S arrest in cell lines expressing oncogenic HRAS, while showing inhibitory effects on the G2-M transition and on anchorage-independent growth in cells with NRAS mutation or that were wild type for RAS (43). These and other preclinical reports, and the fact that KRAS and NRAS are mutated far more frequently in human cancers than HRAS, may have led to these agents being tested in trials with patients selected irrespective of tumor genotype. Hence, the clinical activity of these compounds has not yet been evaluated in patients harboring the cancers that are most likely to respond.
A recent clinical trial with the FTI lonafarnib shows that benefits can be derived when the patient population is selected based on a better understanding of the therapeutic target. Children with the Hutchinson-Gilford progeria syndrome harbor a germline mutation in LMNA which disables a proteolytic cleavage site in lamin A required to remove its farnesylated C-terminus during its post-translational processing. Treatment with the FTI lonafarnib showed improvement of vascular stiffness, bone structure and audiological status in a significant fraction of these children (44). According to the COSMIC database, the frequency of HRAS mutations is about 15 % in salivary tumors, 10% in tumors of the urinary tract, 8% in tumors of the cervix and upper aerodigestive tract and about 4% in prostate and thyroid cancers. In certain subtypes of thyroid cancer the prevalence of HRAS mutation is particularly high, such as in sporadic medullary thyroid cancers (45, 46). Based on all these considerations, we propose that FTIs should be reappraised in properly designed clinical trials against their validated therapeutic targets.
Materials and Methods
Experimental Animals, Skin Carcinogenesis and Drug administration
FR-Hras G12V mice, which harbor a latent HrasG12V knock-in allele under the regulatory control of its endogenous gene promoter, were crossed with Caggs-Cre mice to obtain widespread endogenous expression levels of oncogenic Hras (18). Six to eight weeks old mice were shaved before a single 25 μg dose of DMBA (Sigma-Aldrich) was applied to the dorsal skin. A week later, TPA (12.5 μg, Sigma-Aldrich) was applied twice per week for 15 weeks. The farnesyl transferase inhibitor SCH 66336 (Merck) was dissolved in 20% (wt/vol) 2-hydroxylpropyl-β-cyclodextrin and administered to mice b.i.d by oral gavage at a dose of 80mg/kg. Skin tumors were monitored and recorded and measured twice per week for the duration of the studies.
Tissue preparation, histopathology and immunohistochemistry
Mice were killed by CO2 asphyxiation. Normal and tumor tissue lysates were prepared for extraction of RNA, DNA or protein as described (18). Histology was performed on H&E-stained formalin-fixed paraffin embedded sections. Antibodies used for IHC are listed in the SI section. Animal care and all procedures were approved by the MSKCC Institutional Animal Care and Use Committee.
Hras Allelic Imbalance Analysis
The genomic DNA from tissues was used as a template for PCR amplification with primers that distinguish mutant (666 bp product due to insertion of loxp) from WT Hras alleles (622 bp). The primers and PCR conditions were previously described (18).
Laser-Capture Microdissection
Mouse skin was promptly excised after killing and immediately frozen with liquid N2. Frozen sections (8 μm were used to microdissect skin epithelial cells of normal, hyperplastic and papilloma tissues by laser capture using an Arcturus microdissection system. DNA was extracted using DNeasy Blood & Tissue Kit (QIAGEN).
Cell culture
The following cancer cell lines were used in this study: HRAS mutant: Hth83 and C643 (thyroid), Hs578T (breast), SK-Mel-31 (melanoma), T24 (bladder); KRAS mutant: Cal62 (thyroid), H441, A549 and H2030 (lung), Hct116 (colorectal); NRAS mutant: ACT1 and Hth7 (thyroid), Sk-Mel-30, Sk-Mel-130 and SK-Mel-173 (melanoma). Culture conditions are described in the SI materials and methods. For proliferation assays, cells were plated in triplicate into 6-well plates at 5×104cells/well, and treated the next day with or without SCH 66336 for the indicated times, with media changes every day. Cells were collected by trypsinization and counted in a Vi-Cell cell viability analyzer (Beckman Coulter). IC50 values were calculated using Prism software. For Western blotting, cell extracts and immunoblotting were performed as described (18). For antibody details, see Supplementary Information.
Bromodeoxyuridine (BrdU) labeling
Cells were seeded in 6-well plates with 1.5×105 cells per well 1 day before treatment with SCH 66336 for 72 h. Cells were then incubated with 40 μM BrdU for 4 h. Cell fixation and labeling with anti-BrdU fluorescein isothiocyanate (FITC) conjugated antibody and 7-aminoactinomycin D (7-AAD) were performed using the FITC BrdU Flow Kit (BD Pharmingen) according to the manufacturer’s instructions. The percentage of BrdU positive cells was determined by fluorescence –activated cell sorting analysis (FACS) using a Cell Lab Quanta flow cytometer (Beckman Coulter).
Cell Fractionation
Subcellular fractionation was carried out through sequential incubation with cytosolic, membrane and nuclear fraction extraction buffers, using the Subcellular Protein Fractionation Kit of Thermo Scientific. Fractions were subjected to Western blotting with the indicated antibodies.
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1-CA72597 and T32-DK07313, the Margot Rosenberg Pulitzer Foundation, the Byrne fund and the Lefkofsky Family Foundation. We are indebted to the Molecular Cytology and Comparative Pathology Core facilities of Memorial Sloan Kettering Cancer Center. We are also grateful to Drs. Paul Kirschmeier and W. Robert Bishop (formerly of Schering-Plough) for providing us with SCH 66336.
References
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 3.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–31. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011;2:216–31. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leon J, Guerrero I, Pellicer A. Differential expression of the ras gene family in mice. Mol Cell Biol. 1987;7:1535–40. doi: 10.1128/mcb.7.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2011;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475–87. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esteban LM, Vicario-Abejon C, Fernandez-Salguero P, Fernandez-Medarde A, Swaminathan N, Yienger K, et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 2001;21:1444–52. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plowman SJ, Williamson DJ, O’Sullivan MJ, Doig J, Ritchie AM, Harrison DJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23:9245–50. doi: 10.1128/MCB.23.24.9245-9250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–81. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 12.Balmain A, Ramsden M, Bowden GT, Smith J. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature. 1984;307:658–60. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- 13.Ise K, Nakamura K, Nakao K, Shimizu S, Harada H, Ichise T, et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene. 2000;19:2951–6. doi: 10.1038/sj.onc.1203600. [DOI] [PubMed] [Google Scholar]
- 14.To MD, Rosario RD, Westcott PM, Banta KL, Balmain A. Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene. 2012:10. doi: 10.1038/onc.2012.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bremner R, Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990;61:407–17. doi: 10.1016/0092-8674(90)90523-h. [DOI] [PubMed] [Google Scholar]
- 16.Buchmann A, Ruggeri B, Klein-Szanto AJ, Balmain A. Progression of squamous carcinoma cells to spindle carcinomas of mouse skin is associated with an imbalance of H-ras alleles on chromosome 7. Cancer Res. 1991;51:4097–101. [PubMed] [Google Scholar]
- 17.Schuhmacher AJ, Guerra C, Sauzeau V, Canamero M, Bustelo XR, Barbacid M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J Clin Invest. 2008;118:2169–79. doi: 10.1172/JCI34385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Mitsutake N, LaPerle K, Akeno N, Zanzonico P, Longo VA, et al. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc Natl Acad Sci U S A. 2009;106:7979–84. doi: 10.1073/pnas.0900343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bishop WR, Bond R, Petrin J, Wang L, Patton R, Doll R, et al. Novel tricyclic inhibitors of farnesyl protein transferase. Biochemical characterization and inhibition of Ras modification in transfected Cos cells. J Biol Chem. 1995;270:30611–8. doi: 10.1074/jbc.270.51.30611. [DOI] [PubMed] [Google Scholar]
- 20.Kohl NE, Mosser SD, deSolms SJ, Giuliani EA, Pompliano DL, Graham SL, et al. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science. 1993;260:1934–7. doi: 10.1126/science.8316833. [DOI] [PubMed] [Google Scholar]
- 21.Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med. 1995;1:792–7. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 22.Van CE, van d V, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–8. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 23.Rao S, Cunningham D, de GA, Scheithauer W, Smakal M, Humblet Y, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–7. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 24.Johnson BE, Heymach JV. Farnesyl transferase inhibitors for patients with lung cancer. Clin Cancer Res. 2004;10:4254s–7s. doi: 10.1158/1078-0432.CCR-040016. [DOI] [PubMed] [Google Scholar]
- 25.Harousseau JL, Martinelli G, Jedrzejczak WW, Brandwein JM, Bordessoule D, Masszi T, et al. A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood. 2009;114:1166–73. doi: 10.1182/blood-2009-01-198093. [DOI] [PubMed] [Google Scholar]
- 26.Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci U S A. 1989;86:8323–7. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang FL, Kirschmeier P, Carr D, James L, Bond RW, Wang L, et al. Characterization of Ha-ras, N-ras, Ki-Ras4A, and Ki-Ras4B as in vitro substrates for farnesyl protein transferase and geranylgeranyl protein transferase type I. J Biol Chem. 1997;272:10232–9. doi: 10.1074/jbc.272.15.10232. [DOI] [PubMed] [Google Scholar]
- 28.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–64. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 29.Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol. 2000;157:967–72. doi: 10.1016/S0002-9440(10)64609-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Namba H, Gutman RA, Matsuo K, Alvarez A, Fagin JA. H-ras protooncogene mutations in human thyroid neoplasms. J Clin Endocrinol Metab. 1990;71:223–9. doi: 10.1210/jcem-71-1-223. [DOI] [PubMed] [Google Scholar]
- 31.Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:6829–37. doi: 10.1200/JCO.2005.01.0793. [DOI] [PubMed] [Google Scholar]
- 32.Modrek B, Ge L, Pandita A, Lin E, Mohan S, Yue P, et al. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7:1244–52. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- 33.Hayes TK, Der CJ. Mutant and wild-type Ras: co-conspirators in cancer. Cancer Discov. 2013;3:24–6. doi: 10.1158/2159-8290.CD-12-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Wang Y, Vikis HG, Johnson L, Liu G, Li J, et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 35.Diaz R, Lue J, Mathews J, Yoon A, Ahn D, Garcia-Espana A, et al. Inhibition of Ras oncogenic activity by Ras protooncogenes. Int J Cancer. 2005;113:241–8. doi: 10.1002/ijc.20563. [DOI] [PubMed] [Google Scholar]
- 36.Diaz R, Ahn D, Lopez-Barcons L, Malumbres M, Perez DCI, Lue J, et al. The N-ras proto-oncogene can suppress the malignant phenotype in the presence or absence of its oncogene. Cancer Res. 2002;62:4514–8. [PubMed] [Google Scholar]
- 37.Finney RE, Bishop JM. Predisposition to neoplastic transformation caused by gene replacement of H-ras1. Science. 1993;260:1524–7. doi: 10.1126/science.8502998. [DOI] [PubMed] [Google Scholar]
- 38.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–9. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013;3:112–23. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- 40.Jeng HH, Taylor LJ, Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun. 2012;3:1168. doi: 10.1038/ncomms2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernhard EJ, McKenna WG, Hamilton AD, Sebti SM, Qian Y, Wu JM, et al. Inhibiting Ras prenylation increases the radiosensitivity of human tumor cell lines with activating mutations of ras oncogenes. Cancer Res. 1998;58:1754–61. [PubMed] [Google Scholar]
- 42.Sepp-Lorenzino L, Ma Z, Rands E, Kohl NE, Gibbs JB, Oliff A, et al. A peptidomimetic inhibitor of farnesyl: protein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer Res. 1995;55:5302–9. [PubMed] [Google Scholar]
- 43.Ashar HR, James L, Gray K, Carr D, McGuirk M, Maxwell E, et al. The farnesyl transferase inhibitor SCH 66336 induces a G(2) --> M or G(1) pause in sensitive human tumor cell lines. Exp Cell Res. 2001;262:17–27. doi: 10.1006/excr.2000.5076. [DOI] [PubMed] [Google Scholar]
- 44.Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109:16666–71. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boichard A, Croux L, Al GA, Broutin S, Dupuy C, Leboulleux S, et al. Somatic RAS mutations occur in a large proportion of sporadic RET-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J Clin Endocrinol Metab. 2012;97:E2031–E2035. doi: 10.1210/jc.2012-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moura MM, Cavaco BM, Pinto AE, Leite V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab. 2011;96:E863–E868. doi: 10.1210/jc.2010-1921. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.