

Abstract
The Ehlers-Danlos syndromes (EDS) form a clinically and genetically heterogeneous group of inherited connective-tissue disorders characterized by joint hypermobility, tissue fragility and skin abnormalities. Six subtypes have been well characterized based on clinical features and molecular genetic abnormalities. The arthrochalasia type EDS (formerly type VIIa and VIIb) is characterized by severe generalized joint hypermobility with multiple dislocations including congenital bilateral dislocation of the hips, muscular hypotonia and distinct dysmorphic features. The diagnosis of the arthrochalasia type EDS is of importance in the neonatal period because of consequences of physical disability in later life. However, the differential diagnosis may be difficult because of overlap with other hypermobility syndromes. In addition, the significant hypotonia may direct the physician towards various neuromuscular diagnoses. As patients get older, the hypotonia decreases and facial features become less distinct. In this report we describe seven patients at different ages. Timing of diagnosis varied from prenatal life to adult age. The diagnosis of EDS type VII was confirmed by biochemical studies or mutation analysis showing characteristic mutations in COL1A1 and COL1A2. These mutations result in skipping of exon 6, which leads to defective collagen synthesis. For physicians treating patients with EDS type VII, achieving mobility for the patient is the greatest challenge and it may be impossible due to recurrent dislocations of nearly all joints in severe cases.
Keywords: Ehlers-Danlos Syndrome (EDS), the arthrochalasia type EDS, EDS type VII, hypermobility, (sub)luxations
Introduction
The Ehlers-Danlos syndromes (EDS) are a clinically and genetically heterogeneous group of inherited connective-tissue disorders characterized by joint hypermobility, tissue fragility and skin abnormalities. Other organ involvement, depending on the type of EDS, include blood vessel, skeletal, gastrointestinal, dental, brain, genitourinary and other system. The most recent nosology has simplified the classification of EDS into 6 subtypes based on their clinical features and molecular abnormalities, with a seventh group of miscellaneous forms of EDS. (1) The prevalence of all forms of EDS is estimated to be 1:5000. Of these, the classic and hypermobility types (EDS I-III) account for ~90%, and the vascular type (EDS IV) for ~5%. All other subtypes are extremely rare. (2)
The arthrochalasia type EDS (formerly EDS VIIA and B; OMIM 130060) is characterized by severe generalized joint hypermobility with multiple dislocations including bilateral congenital dislocation of the hips, muscular hypotonia, mild dysmorphic features and discrete skin abnormalities. (3, 4) In the past EDS type VII was subclassified into types VIIA and VIIB, based on partial or complete loss of exon 6 of the COL1A1 or COL1A2 respectively. This exon-skip leads to loss of the procollagen N-proteinase cleavage site, as well as the critical cross-linking lysyl residue leading to the loss of N- terminal peptidase cleavage site and abnormal processing of type-I procollagen to type-I collagen, the principal component of ligaments, tendons, dermis, bone and dentin. In contrast, homozygous mutations in the procollagen I N-terminal peptidase, lead to dermatosparaxis type EDS, formerly known as EDS type VII-C (OMIM 225410), that is characterized by redundancy and severe fragility of the skin. (5)
In addition to fragility of skin and joint laxity that are observed in other forms of EDS, the arthrochalasia type is also characterized by other anomalies that serve as a major diagnostic criterion, e.g. the congenital dislocation of the hips and distinct facial features.
EDS in general is a poorly recognized and under-diagnosed condition. Diagnosis of arthrochalasia type of EDS is further complicated by the neonatal phenotypic overlap with other skeletal dysplasias such as Larsen syndrome, pseudodiastrophic dysplasia, Desbuquois dysplasia, and other less-well delineated multiple joint dislocation or arthrogryposis syndromes and neuromuscular disorders. Although no curative treatments exist, a pre- or postnatal early diagnosis can be life saving and appropriate early intervention can alleviate physical and psychological suffering, for example omit invasive surgery that will be particularly unsuccessful in patients with arthrochalasia type of EDS.
We describe seven patients with the arthrochalasia type of EDS, and provide long term follow-up. We expand the phenotypic spectrum of this rare syndrome including its prenatal presentation.
Case reports
Patient 1
This patient is the second child of a non-consanguineous Kaukasian couple. The patient’s mother reports absence of scalp hair until the age of approximately 1 year. She required corrective surgery for micrognathia at age three years. Her heigt is 165cm, which is normal based on parental height. Her Beighton-score is 4/9. There are no other dysmorphic features in the mother. The first child of this couple died in utero at 22 weeks gestation.
Patient 1 (Figure 1 and 2) was born at 35 weeks gestation by vaginal breech delivery with hypotonia and dysmorphic features that included micrognathia and sparse hair. Examination of the limbs of patient 1 showed severe hypermobility of large and small joints with dislocations, even after gentle manipulation. She required nCPAP for the first two days because of hypoventilation due to the generalized hypotonia, and the differential diagnosis included a neurological disorder (particularly congenital myotonic dystrophy) or Larsen syndrome. Congenital myotonic dystrophy was ruled out by mutation analysis. Other neurological disorders seemed less likely over time, since the hypotonia improved and luxations are not known to occur to such extent in those disorders. Since clinical features were more compatible with arthrochalasia type EDS than Larsen syndrome, mutation-analysis of the Filamin B gene (causing Larsen syndrome) was not performed.
Figure 1.
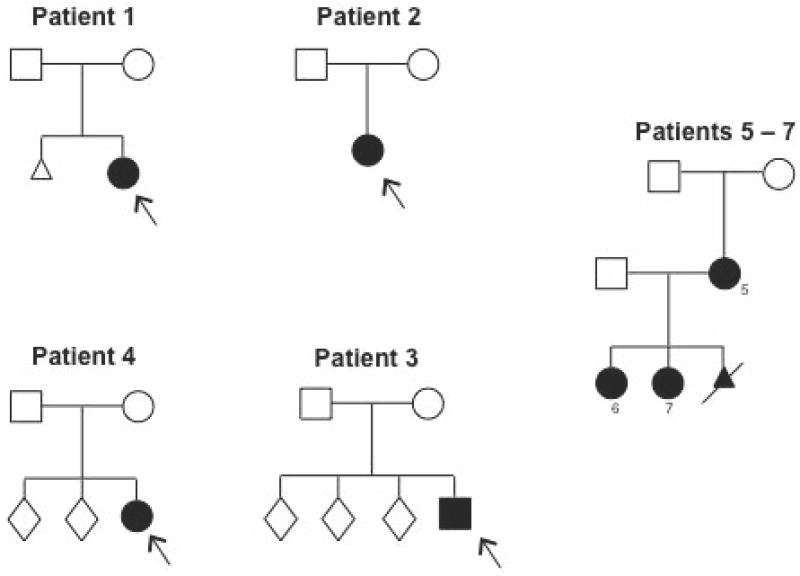
Pedigrees.
Pedigrees of all patients and their parents.
Figure 2.

Phenotypic features of patient 1.
A, muscular hypotonia on day 1 of life; B, hyperlaxity of ankle joints; C, facial features at age 3 months; D, micrognathia; E, hyperlaxity of knee; F, hypotonia and hyperlaxity at age 3 months.
At age three months, the dysmorphic features were still evident, and treatment for bilateral hip dislocation by a Pavlik-harness was unsuccesful. Hypermobility and dislocations of all joints persisted, and soft and velvety skin with criss-cross patterning of the palms and soles was noted. A diagnosis of the arthrochalasia type EDS was suspected and confirmed by mutation analysis.
At age 18 months, her motor development was significantly delayed, and she was unable to maintain good head balance, roll over, or sit, due to recurrent dislocations and hypotonia. Surgical treatment of the hip dislocations was deferred because of concern over abnormal wound healing and recurrent dislocations after surgery. Ankle-foot orthoses were prescribed to maintain a normal position of the joint. The use of these orthoses had to be discontinued because of skin abrasions and hematomas. Cognitive development has been normal.
In patient 1 a novel mutation in COL1A2 was discovered in lymphocyte DNA which was predicted to cause skipping of exon 6 and loss of the cleavage site for the telopeptidase. In the mother, no mutation in COL1A2 on lymphocyte DNA was identified.
Patient 2
This patient is the first child of healthy non-consanguineous Kaukasian parents. She was born at 40 weeks gestation. After birth extreme hyperlaxity of both small and large joints was noted, with bilateral hip dislocation, bilateral subluxation of the radius and a stable subluxation of vertebrae C1-C2 without spinal cord compression, in combination with neonatal hypotonia (Figure 1 and 3).(6) Because of these features combined with velvety skin and criss-cross patterning of the palms and soles, the diagnosis of arthrochalasia type EDS was considered. Diagnosis was confirmed by DNA analysis on day 17 after birth. Cardiac evaluation showed no abnormalities. Treatment of the hip dislocations by spica casts was unsuccessful. Presently, at the age of 18 months the patient is able to pull herself up on her feet but without correction is standing on the backs of her feet. Her right shoulder luxates repeatedly with minor manipulation. Mental development is normal. In patient 2, a mutation in COL1A2 was confirmed.
Figure 3.

Phenotypic features of patient 2.
A, muscular hypotonia on day 1 of life; B, facial features on day 1; C, criss-cross patterning of palms; D, facial features at age 1 year; E, hyperlaxity of feet.
Patient 3
This patient is the fourth child of healthy non-consanguineous Kaukasian parents, born at 39 weeks and 5 days of gestation. He was hospitalized 10 days after birth because of poor feeding and growth. At that time extreme hyperlaxity of both large and small joints, bilateral clubfeet (correctable after plaster casts and night splints), bilateral congenital dislocation of the hips and soft, velvety skin were noted (Figure 1 and 4). The arthrochalasia type EDS was suspected. Bilateral hip dislocations were treated with a Pavlik harness. Cardiac examination revealed no abnormalities. He developed an umbilical hernia in the first year of life. Presently, at the age of 4 years he is an active boy who is able to walk, run and climb stairs without luxations despite striking hyperlaxity of all joints. He is wearing orthopedic shoes to support his extremely flexible feet. He still has very loose, soft skin with piezogenic papules (herniation of fat through the dermis) along the lateral aspects of his heels. His mental development is normal. The patient’s father has hypermobile joints and soft skin with normal woundhealing and no joint pain. In patient 3 a previously reported mutation in COL1A2 was found. In the father, no mutation in COL1A2 was found in his lymphocytes or fibroblasts.
Figure 4.
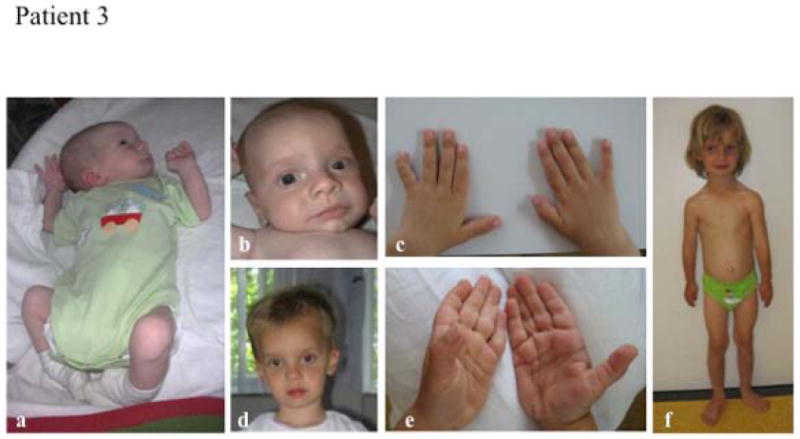
Phenotypic features of patient 3.
A, mild muscular hypotonia at age 3 months; B, facial features at age 3 months; C, hands at age 2 years; D, facial features at age 2 years; E, hands at age 2 years; F, full body view at age 5 years.
Patient 4
This patient is a 28-year-old woman, who is the third child of a healthy non-consanguineous Kaukasian couple. She was born at 36 weeks after premature rupture of membranes. At birth she presented with bilateral congenital dislocation of the hips, severe joint hypermobility and large fontanels. The neonatal period, infancy and childhood were characerized by delayed motor development secondary to hypotonia and recurrent large and small joint dislocations including ankles and patellae. Her cognitive development is normal. Her skin findings include very fragile skin with slow wound healing, easy bruising and a history of slowly healing fractures, ever since she was a child. Upon physical examination at age 28, she manifested joint hypermobility (Beighton score of 8/9), genua valga, flexible pes planus and hyperlordosis of the lower spine (Figure 1 and 5). She showed mild facial dysmorphism with epicanthic folds and microretrognathia. Her skin was very soft with some hyperextensibility and several cigarette-paper-like scars. Chest skin was translucent with a visible venous pattern. Echocardiogram revealed no abnormalities. Radiographic evaluation of the skeleton revealed presence of Wormian bones, thoracic scoliosis and generalized osteopenia. Bone densitometry however yielded normal values for DEXA Z-scores at the level of the femur and the lumbar spine. The diagnosis of arthrochalasia type EDS was confirmed on the biochemical and genetic level.
Figure 5.

Patient 4. A, extensive skin laesions, thin skin and deformities of the feet; B, hyperextension of the fifth finger, consistent with extreme laxity of the small joints; C, dislocations of small joints in second finger with swan neck deformity.
Patients 5-7
This family consists of an affected mother (patient 5) and two affected daughters (Figure 1 and 6). A third affected female fetus was lost during the third trimester. The mother was born at term via vaginal delivery, there were no pregnancy-associated complications. Her parents were non-consanguinous and were healthy (both of normal height).
Figure 6.
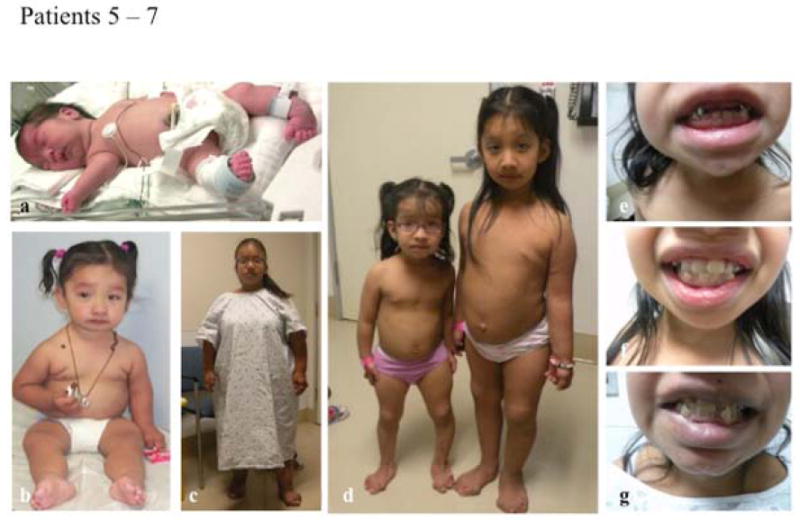
Phenotypic features of patients 5, 6 and 7.
A, muscular hypotonia and hyperlaxity on day 1 of life in patient 7; B, patient 6 at age 2 years; C, patient 5 at age 25 years; D, patients 6 and 7 at age 4 and 6 years respectively; E – G, abnormal dentition in patients 5, 6 and 7 respectively.
She had bilateral congenital hip dislocation, hypotonia and delayed achievement of motor milestones. She underwent bilateral hip surgeries at age 1. At the time of her first appointment with a geneticist she was 20 years old and pregnant with her first child, and a skeletal dysplasia was suspected because of short stature, joint hypermobility and small-joint contractures. On physical examination the skin was hyperextensibile, soft and thin and stretch marks were noticed on the lower back. There were multiple ecchymoses on the shins with accompanying dystrophic, cigarette-paper scars. The patient reported easy bruising and abnormally slowly healing wounds, beginning in childhood. Dental exam revealed transparent teeth with crowding, interpreted as dentinogenesis imperfecta-like changes (Figure 1 and 6). She has severe scoliosis, reports bilateral knee dislocations, and severe limitation in activity.
The patient’s first child (patient 6) was born at 39 weeks gestation after an uneventful pregnancy. At the age of 7 months, she was evaluated because of congenital bilateral hip dislocation, hypotonia, joint laxity, and redundant skin. Additional dysmorphic features were noted: hypertelorism, epicanthal folds, frontal bossing, flat midface, and micrognathia (Figure 1 and 6). She had open sutures with large fontanelles, bilateral ectopia lentis, criss-cross patterning of palms and soles, contractures of the fingers (caused by scar formation within connective tissue due to prolonged and/or recurrent dislocation of small joints), bilateral club feet, umbilical hernia and short stature (3rd percentile, with weight and head circumference at 75th percentile). Radiological examination showed diminished mineralization of long bones, and no Wormian bones. At age 14 months there were no new findings, and her umbilical hernia was resolving. Height, weight and head circumference were at the 50th percentile. At the age of 14 months biochemical analysis suggested the diagnosis of the arthrochalasia type EDS, because of retention of the aminoterminal propeptide of the pro alpha 1(I) chains of type I collagen (see also section on biochemical studies further on in this paper).
She underwent a bilateral open hip reduction surgery at age 18 months but the right hip immediately redislocated. Follow-up at age 26 months shows similar findings, with no independent walking and normal speech. At age 8 years, imaging of the pelvis showed that the right femoral head was dislocated while the left femoral head positioned in the acetabulum. The right knee demonstrated moderate tricompartmental osteoarthritic changes. The left knee was normal. The patient reports recurrent dislocation of her left knee, right knee pain, is very limited in her activity and tires easily. She also has dental abnormalities similar to her mother.
The patient’s second child (patient 7) was delivered with normal Apgar scores at 40 weeks gestation by cesarean section due to fetal intolerance to labor and failure to progress. Because of the family history of EDS, the pregnancy was closely monitored and a prenatal diagnosis confirmed recurrence of EDS. Ultrasound at 19 weeks gestation showed no significant abnormalities, but at 22 weeks, features suggesting a skeletal dysplasia appeared: ulnar deviation of digits with shortening of first and second digits, talipes on the right, and measurements of long bones which were 1 standard deviation below mean with micrognathia evident on profile view. At 26 weeks gestation polyhydramnios was noted with no new skeletal findings, and at 32 weeks multiple joint dislocations were seen, with hyperextended legs and bilateral equinovarus. At birth, physical examination revealed bilateral hip dislocations, redundant neck skin and soft, hyperextensible skin with similar dysmorphic features as her sister, including micrognathia. Due to severe equinus contractures and tight bilateral Achilles’ tendons she underwent corrective surgery as well as Achilles’ tonotomies at 6 month of age. Follow-up at age 8 months showed normal development for her age. There was frontal bossing with wide-open sutures and fontanelles, mild flattening of the midface, mild hypertelorism and epicanthal folds. The sclerae were tinged blue-gray. At age 6, imaging studies revealed a right dislocated hip and severely subluxated left hip. The patient has scoliosis (20 degrees) in the thoracic and the lumbar regions. She tends to walk with an antalgic gait and complains of subluxations of her right knee and chronic knee and hip pain. The parents third child showed similar prenatal ultrasound findings as their second daughter: several dislocations of large and small joints with bilateral club feet. Again, arthrochalasia type EDS was suspected and the parents decided to terminate the pregnancy.
Biochemical studies and mutation analysis
Patients 1 – 3
DNA was isolated from peripheral blood using standard techniques. Sequence analysis of exon 6 of the COL1A1 and COLA2 genes was performed using the Big Dye VS 3.0 kit (Applied Biosystems). PCR primer sequences 5’to 3’ COL1A1: GTTGCAGCACACCAGGAAGTGCATGATGTCAG (forward; intron 5) and AAACAAGACCCAGGCCTGGGAGTTCTTC (reverse; intron 8); COL1A2 GTTGCAGTCGGCCAAGTTTTTGACGTACAGCT (forward; intron 5) and AAACTGGCGTGGTAAAATGTGACATAAAA (reverse; intron 6).
In patient 1 a novel mutation in COL1A2 was discovered (VD34771: c.279+2T>G) which is predicted to cause skipping of exon 6 and loss of the cleavage site for the telopeptidase (Figure 7). In patient 2, a mutation in COL1A2 caused loss of the splice acceptor site and usage of a cryptic site 15 bases into the next exon, resulting in loss of 5 amino acids (VD30882: c.226-2A>G). (5) In patient 3 a similar and previously reported mutation of COL1A2 was found (VD17256: c.279+1G>T). Because of mild features of EDS in the father of patient 3, mutation analysis in blood, fibroblasts and saliva was performed. Somatic mosaicism for this IVS6+1G>T mutation was not demonstrated.
Figure 7.
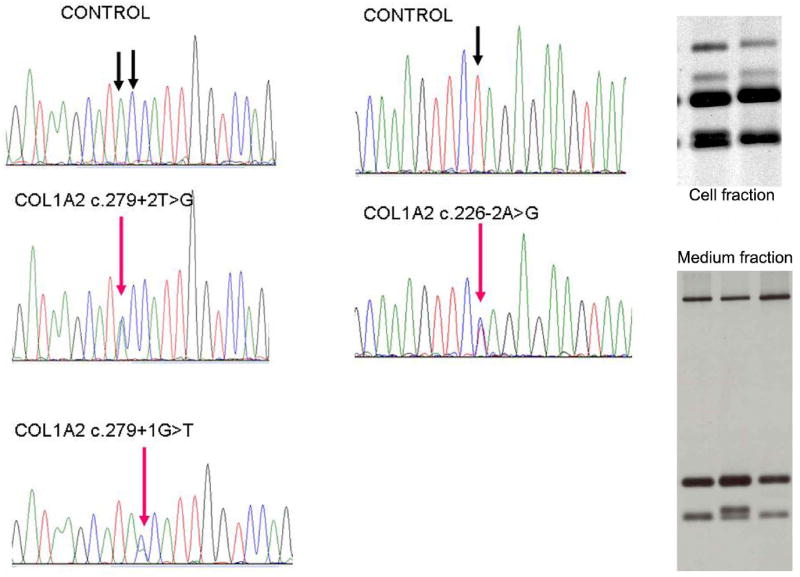
Sequence traces of mutations in the COL1A2 gene from patients 1 - 3. Only reverse sequence are shown, because due to a long T stretch near the 5’ end of exon 6. This exon cannot be sequenced in forward direction. Patient 1: c.279+2T>G. Patient 2: c.226+1A>G. Patient 3: c.279+1G>T.
SDS-PAGE analysis of 14C-labeled collagen molecules in dermal fibroblasts from patient 4. Top right figure: collagen medium fraction. Left and right lane: control, middle lane: patient. Top band corresponds to the α1(I) collagen chain, the lowest band corresponds to the α2(I) collagen chain. In the patient there is an additional band, which corresponds to the uncompletely processed pNα2(I) chains, consistent with defective collagen synthesis. Bottom right figure: cell fraction. Similar bands visible. Left lane: control, right lane: patient.
Patient 4
SDS-PAGE analysis of the pepsin-digested collagen retained in the cells and secreted into the medium showed the presence of an additional band migrating between the α1(I) and the α2(I) chains of type I collagen and resembling the mobility of pNα2(I) chains (Figure 7). SDS-PAGE of medium procollagens showed evidence for accumulation of pNα1(I) and pNα2(I) bands. The conversion of a pulse of [3H]-procollagen to collagen was followed over 5 days. In comparison with processing of procollagen from control cell cultures, amino propeptide processing was substantially delayed in this proband.
Molecular studies were performed using the following technique: RNA was reverse-transcribed, and the N-terminal regions of the COL1A1 and the COL1A2 gene were PCR-amplified in the probands. The amplimers were separated in agarose, followed by direct sequencing. In cells from the proband direct sequencing of the RT-PCR product revealed heterozygous skipping of the entire sequence of exon 6 of COL1A2. At the genomic level, this was the result of a point mutation in the splice donor site (IVS 6 +1 G>C).
To complete the studies performed on material from patient D, transmission electron microscopy was performed. Fibril density was lower in the dermis than in control, and occasional cauliflower deformations of the fibrils were seen. Collagen fibrils were irregular in contour and had distinctly smaller diameters than in control samples (43.82 +/- 6.09 nm).
Patients 5 – 7
Fibroblasts were obtained from the eldest child (patient 6) at the age of 14 months and used for biochemical analysis of type I collagen. The assay revealed retention of the aminoterminal propeptide of the pro alpha 1(I) chains of type I collagen, suggesting abnormal conversion of products of one COL1A1 allele, suggesting the diagnosis of arthrochalasia type EDS. Similar finding were identified on fibroblasts from the mother and second child.
Discussion
We describe seven patients with the arthrochalasia type EDS (formerly Ehlers-Danlos type VIIa and b). This form of EDS is very rare with only 27 cases reported in the literature to date.(3-5, 7-17) It is characterized by generalized joint hypermobility with recurrent dislocations of other joints including congenital bilateral hip dislocation, muscular hypotonia and mild dysmorphic features. Motor development is significantly delayed in the neonatal and postnatal period due to muscular hypotonia and recurrent joint luxations, but cognitive development is usually normal.
As mentioned before, arthrochalasia type EDS was formerly known as type VII, which was subclassified into types VIIA and VIIB, based on the gene that is mutated. However, due to the limited number of cases reported, it is not clear whether EDS type VIIA & B are separate clinical entities. A previous report suggests that COL1A1 mutations lead to a more severe phenotype than COL1A2 mutations (4) based on procollagen molecular structure that contains two pro alpha 1(I) chains and only one pro alpha 2(I) chain.(5) However, according to our series of patients, the phenotype seems to be similar in patients with COL1A1 (patients 5-7) and COL1A2 mutations (patients 1-4), at least in the neonatal and postnatal periods. Part of this similarity can be attributed to the fact that patients 5-7 all come from the same family, but also in literature the phenotypical difference between COL1A1 and COL1A2 mutations seems less clear. Within the complete group of arthrochalasia type EDS patients there is significant phenotypical variability, clearly demonstrated by the differences between the severely affected patient 1 and mildly affected patient 3, who both have mutations in COL1A2. Intriguingly, the clinical phenotype of the arthrochalasia EDS is considerably different from that of dermatosparaxis type (VIIC), caused by mutations in the procollagen I N-proteinase, ADAMTS2, that prevent proper processing of the type I procollagen and in which extreme skin fragility and easy bruising are the predominant clinical features. Since ADAMTS2 is also involved in processing of type III procollagen, this might explain some of the phenotypical differences.(18) Yet, there are clinical features that are shared with types VIIA and VIIB, such as the abnormal dentition and skin fragility. (19) The mechanism underlying these variations is still obscure.
The most important clinical features for diagnosis of the arthrochalasia type EDS are those features known for all other EDS types: hypermobility and recurrent joint dislocations (Table 1). Characteristic for the arthrochalasia type is bilateral congenital hip dislocation that is difficult to treat. The cutaneous features are milder than in most other EDS types. There is a certain degree of skin fragility and some bruising, but only the minority of the patients have abnormal scarring and only slightly delayed wound healing. As with the other EDS types, there is significant skin redundancy, causing one of the key features of arthrochalasia type EDS: a criss-cross pattern that is seen on the palms and soles.
Table 1.
Summary of clinical features of all patients compared to the literature.
| Patients | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Total | LIT | NA |
|---|---|---|---|---|---|---|---|---|---|---|
| Feature | ||||||||||
| Hyperlaxity | + | + | + | + | + | + | + | 7 / 7 | 27 / 27 | - |
| (Sub)luxations | + | + | + | + | + | + | + | 7 / 7 | 24 / 24 | 3 |
| Hip dislocation | + | + | + | + | + | + | + | 6 / 7 | 25 / 25 | 2 |
| Scoliosis | + | - | - | + | - | - | + | 3 / 7 | 15 / 20 | 7 |
| Velvety skin | + | + | + | + | + | + | + | 7 / 7 | 12 / 12 | 15 |
| Haematomas | + | + | - | + | - | - | + | 4 / 7 | 8 / 16 | 11 |
| Abnormal scarring | - | - | - | + | - | - | + | 2 / 7 | 5 / 13 | 14 |
| Criss-cross patterning | + | + | + | + | + | + | + | 6 / 7 | 2 / 2 | 25 |
| Blue sclerae | - | - | - | - | ± | + | + | 3 / 7 | 2 / 5 | 22 |
| Fractures | - | - | - | + | - | - | - | 0 / 7 | 6 / 15 | 12 |
| Osteopenia | NA | NA | NA | + | + | NA | NA | 2 / 2 | - | 27 |
| Wormian bones | NA | NA | NA | + | - | NA | NA | 1 / 2 | 2 / 2 | 25 |
| Abnormal dentition | ± | NA | - | - | + | + | + | 4 / 6 | 1 / 3 | 24 |
| Frontal bossing | ± | + | + | - | + | + | + | 6 / 7 | 2 / 5 | 22 |
| Micrognathia | + | + | + | + | + | + | + | 7 / 7 | 4 / 5 | 22 |
| Large fontanel | + | NA | NA | - | + | + | NA | 3 / 3 | 4 / 5 | 22 |
| Short stature | - | - | - | + | + | + | + | 4 / 7 | 2 / 6 | 21 |
| Other | + | - | - | + | + | + | + | - | + | |
| Gene | COL1A2 | COL1A2 | COL1A2 | COL1A2 | COL1A1 | COL1A1 | COL1A1 |
Abbreviations: NA: data not tested in our patients or not available in original publication. LIT: literature, number of patients with certain phenotypic features and total number of patients for which there is mention of this particular feature
too young to be able to assess dentition.
The differences between the phenotype of arthrochalasia type EDS in comparison with other EDS types is particularly clear when looking at other features than skin and joints. Arthrochalasia type EDS patients have dysmorphic features that are also among the distinguishing features of the syndrome. Some dysmorphic features are present in all our patients and the majority of patients in the literature (Table 1). These features include mild hypertelorism, bilateral epicanthic folds, large fontanels (mainly the anterior fontanel), and especially pronounced micrognathia. A second difference between arthrochalasia type EDS and other types is the neonatal hypotonia which is often severe. It is not clear whether the hypotonia is solely caused by the laxity of the connective tissue or whether there is a muscular component. Remarkably, the hypotonia seems to diminish with increasing age, regardless of the level of joint hypermobility at that time. The mental development of all patients is normal.
The presence of the dysmorphic features with hypotonia results in consideration of other syndromes besides EDS, as reported for patient 1. Many children with arthrochalasia type EDS are suspected of having another connective tissue disorder (particularly Larsen syndrome), a neurological disorder or some type of skeletal dysplasia. Patient 7 (mother of patients 5 and 6) was not diagnosed until her eldest daughter was evaluated for similar problems.
Considering the overlap with other syndromes, it is interesting to note that other mutations in the COL1A1 and COL1A2 genes are responsible for osteogenesis imperfecta, a severe disorder of bone structure and development (OMIM 166200, 166210 & 166220). Clinically, there is some overlap between OI and EDS type VII.(20) Several of our patients (patients 1 and 5-7) have blue sclerae and some have abnormal dentition, features known to be present in OI. In reverse, hypermobility is a feature seen in most patients with different types of OI, even though it is difficult to evaluate during physical examination because of the risk of bone fractures. Fractures are not a distinct feature of EDS type VII, but there are some reports of decreased bone mineral density (BMD) in patients with several types of Ehlers-Danlos syndrome, including the arthrochalasia type.(2, 21) In the literature there is no mention of fractures in patients with arthrochalasia type EDS. Some of our patients displayed decrease bone mineral density, but no fractures. Given these findings it is important to be alert to the possibility of osteopenia / osteoporosis in patients with arthrochalasia type EDS and to maintain a low treshold for testing and, if necessary, repeat this screening every 1 to 2 years.
As in our patients, the most important problems for children with arthrochalasia type EDS are orthopaedic, in particular the bilateral congenital hip dislocation, since this severely affects mobility. As seen in our patients stable reduction by a closed method, such as a Pavlik harness, orthosis or hip-spica cast, is rarely successful, as has been shown by others. (4) Operative procedures involving only the joint capsule are rarely successful as well. Only iliac osteotomy (e.g. Salter or Pemberton type) with or without femoral osteotomy has been shown to improve stability of the hips in other hyperlaxity syndromes.(22, 23)
The recurrent or persistent dislocation of other joints of both lower and upper extremities can cause a severe delay in motor development. In our patients generalized hypermobility seems to be worse in infancy, when there is also significant muscular hypotonia. This tends to improve with age, making the application of orthoses to stabilize the lower extremities more successful later in childhood. However, application of orthoses can occasionally lead to skin problems such as abrasions and hematomas, as seen in patient 1 in whom the use of orthoses had to be discontinued because of these skin lesions. For dislocations of upper extremity joints no ortheses or operative procedures are advised, since these are rarely successful. We believe only those patients with a substantial chance of achieving mobility should be treated. An osteotomy is, in our opinion, the only possibly successful method for these children. If skin problems are mild, the use of orthoses to stabilize knee, ankle and of foot joints can improve motor development.
Both autosomal dominant and autosomal recessive modes of inheritance have been described in arthrochalasia type EDS. Patients 5, 6 and 7, from one family, have a similar severity of symptoms. The pedigree is compatible with an autosomal dominant mode of inheritance. In comparison, the mother of patient 1 displays some characteristics of EDS type VII such as micrognathia, but mutation analysis on blood lymphocytes was normal. Somatic mosaïcism might be responsible for these phenotypic features. Germ line mosaicism has been reported for COL1A1 and COL1A2 mutations causing osteogenesis imperfecta. Studies base on clinical and biochemical data have shown that in at least 6% of the cases of lethal osteogenesis imperfecta type II, one of the parents of the affected child has germ-line mosaicism.(24) Somatic mosaicism has also been described for OI.(25) Together this suggests a significant recurrence risk for apparently sporadic OI. Possibly, some reported autosomal recessive cases of arthrochalasia type EDS can similarly be attributed to somatic mosaicism with percentages of affected cells as high as 65%, leading to a moderate recurrence risk.(26) This might have significant consequences for genetic counseling of the parents of patient 1.
In conclusion, we show that the arthrochalasia type of Ehlers-Danlos syndrome is a rare, but recognizable connective tissue disease. Certain features are significant clues to the diagnosis: the criss-cross patterning of the palms/soles, micrognathia with other mild dysmorphic features and severe neonatal hip problems with hypotonia. Mutations in COL1A1 and COL1A2 seem to cause similar phenotypes with clinical features becoming less severe with increasing age. Awareness of this syndrome may decrease the delay in diagnosing this syndrome, which eventually will benefit motor development of these patients and might prevent unnecessary surgical procedures.
Acknowledgments
The authors greatly appreciate the cooperation of the patients and their family to make this publication possible. Drs. Reinstein and Graham appreciate support from the Steven Spielberg Pediatric Research Center, the NIH/NICHD Program Project Grant (HD36657), the Medical Genetics NIH/NIGMS Training Program Grant (5-T32-GM08243), and the Cedars-Sinai General Clinical Research Center Grant (M01-RR00425) for samples collected under CSMC IRB Protocols 0463 and 4232. Fransiska Malfait is a post-doctoral research fellow from the fund for scientific research - Flanders. Prof. Anne De Paepe is supported by a Methusalem grant BOF08/01M01108 from the Flemish Government and the Ghent University. Prof. van Steensel acknowledges support from the Dutch Cancer Society KWF (grant UM2009-4352) and the Association for Internation Cancer Research AICR (grant 11-0687).
References
- 1.Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Yen JL, Lin SP, Chen MR, et al. Clinical features of Ehlers-Danlos syndrome. J Formos Med Assoc. 2006;105:475–480. doi: 10.1016/S0929-6646(09)60187-X. [DOI] [PubMed] [Google Scholar]
- 3.Hass J, Hass R. Arthrochalasis multiplex congenita; congenital flaccidity of the joints. J Bone Joint Surg Am. 1958;40-A:663–674. [PubMed] [Google Scholar]
- 4.Giunta C, Superti-Furga A, Spranger S, et al. Ehlers-Danlos syndrome type VII: clinical features and molecular defects. J Bone Joint Surg Am. 1999;81:225–238. doi: 10.2106/00004623-199902000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Byers PH, Duvic M, Atkinson M, et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet. 1997;72:94–105. doi: 10.1002/(sici)1096-8628(19971003)72:1<94::aid-ajmg20>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 6.Schakel WaW FJ. Een neonaat met flexibele gewrichten. Nederlands Tijdschrift voor Geneeskunde. 2010;154:A1141. [PubMed] [Google Scholar]
- 7.Nicholls AC, Sher JL, Wright MJ, et al. Clinical phenotypes and molecular characterisation of three patients with Ehlers-Danlos syndrome type VII. J Med Genet. 2000;37:E33. doi: 10.1136/jmg.37.11.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitaker IS, Rozen WM, Cairns SA, et al. Molecular genetic and clinical review of Ehlers-Danlos Type VIIA: implications for management by the plastic surgeon in a multidisciplinary setting. J Plast Reconstr Aesthet Surg. 2009;62:589–594. doi: 10.1016/j.bjps.2008.11.119. [DOI] [PubMed] [Google Scholar]
- 9.Chiodo AA, Hockey A, Cole WG. A base substitution at the splice acceptor site of intron 5 of the COL1A2 gene activates a cryptic splice site within exon 6 and generates abnormal type I procollagen in a patient with Ehlers-Danlos syndrome type VII. J Biol Chem. 1992;267:6361–6369. [PubMed] [Google Scholar]
- 10.Weil D, D’Alessio M, Ramirez F, et al. Temperature-dependent expression of a collagen splicing defect in the fibroblasts of a patient with Ehlers-Danlos syndrome type VII. J Biol Chem. 1989;264:16804–16809. [PubMed] [Google Scholar]
- 11.Vasan NS, Kuivaniemi H, Vogel BE, et al. A mutation in the pro alpha 2(I) gene (COL1A2) for type I procollagen in Ehlers-Danlos syndrome type VII: evidence suggesting that skipping of exon 6 in RNA splicing may be a common cause of the phenotype. Am J Hum Genet. 1991;48:305–317. [PMC free article] [PubMed] [Google Scholar]
- 12.Weil D, D’Alessio M, Ramirez F, et al. Structural and functional characterization of a splicing mutation in the pro-alpha 2(I) collagen gene of an Ehlers-Danlos type VII patient. J Biol Chem. 1990;265:16007–16011. [PubMed] [Google Scholar]
- 13.Watson RB, Wallis GA, Holmes DF, et al. Ehlers Danlos syndrome type VIIB. Incomplete cleavage of abnormal type I procollagen by N-proteinase in vitro results in the formation of copolymers of collagen and partially cleaved pNcollagen that are near circular in cross-section. J Biol Chem. 1992;267:9093–9100. [PubMed] [Google Scholar]
- 14.Weil D, Bernard M, Combates N, et al. Identification of a mutation that causes exon skipping during collagen pre-mRNA splicing in an Ehlers-Danlos syndrome variant. J Biol Chem. 1988;263:8561–8564. [PubMed] [Google Scholar]
- 15.Pope FM, Nicholls AC, Palan A, et al. Clinical features of an affected father and daughter with Ehlers-Danlos syndrome type VIIB. Br J Dermatol. 1992;126:77–82. doi: 10.1111/j.1365-2133.1992.tb08409.x. [DOI] [PubMed] [Google Scholar]
- 16.D’Alessio M, Ramirez F, Blumberg BD, et al. Characterization of a COL1A1 splicing defect in a case of Ehlers-Danlos syndrome type VII: further evidence of molecular homogeneity. Am J Hum Genet. 1991;49:400–406. [PMC free article] [PubMed] [Google Scholar]
- 17.Cole WG, Chan D, Chambers GW, et al. Deletion of 24 amino acids from the pro-alpha 1(I) chain of type I procollagen in a patient with the Ehlers-Danlos syndrome type VII. J Biol Chem. 1986;261:5496–5503. [PubMed] [Google Scholar]
- 18.Le Goff C, Somerville RP, Kesteloot F, et al. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development. 2006;133:1587–1596. doi: 10.1242/dev.02308. [DOI] [PubMed] [Google Scholar]
- 19.Malfait F, De Coster P, Hausser I, et al. The natural history, including orofacial features of three patients with Ehlers-Danlos syndrome, dermatosparaxis type (EDS type VIIC) Am J Med Genet A. 2004;131:18–28. doi: 10.1002/ajmg.a.30299. [DOI] [PubMed] [Google Scholar]
- 20.Cabral WA, Makareeva E, Colige A, et al. Mutations near amino end of alpha1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing. J Biol Chem. 2005;280:19259–19269. doi: 10.1074/jbc.M414698200. [DOI] [PubMed] [Google Scholar]
- 21.Dolan AL, Arden NK, Grahame R, et al. Assessment of bone in Ehlers Danlos syndrome by ultrasound and densitometry. Ann Rheum Dis. 1998;57:630–633. doi: 10.1136/ard.57.10.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennet GC, Rang M, Roye DP, et al. Dislocation of the hip in trisomy 21. J Bone Joint Surg Br. 1982;64:289–294. doi: 10.1302/0301-620X.64B3.6212586. [DOI] [PubMed] [Google Scholar]
- 23.Steel HH, Kohl EJ. Multiple congenital dislocations associated with other skeletal anomalies (Larsen’s syndrome) in three siblings. J Bone Joint Surg Am. 1972;54:75–82. [PubMed] [Google Scholar]
- 24.Byers PH, Tsipouras P, Bonadio JF, et al. Perinatal lethal osteogenesis imperfecta (OI type II): a biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. Am J Hum Genet. 1988;42:237–248. [PMC free article] [PubMed] [Google Scholar]
- 25.Cabral WA, Marini JC. High proportion of mutant osteoblasts is compatible with normal skeletal function in mosaic carriers of osteogenesis imperfecta. Am J Hum Genet. 2004;74:752–760. doi: 10.1086/383252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zlotogora J. Germ line mosaicism. Hum Genet. 1998;102:381–386. doi: 10.1007/s004390050708. [DOI] [PubMed] [Google Scholar]