

Summary
JARID2 is an accessory component of Polycomb repressive complex-2 (PRC2) required for the differentiation of embryonic stem cells (ESCs). A role for JARID2 in the recruitment of PRC2 to target genes silenced during differentiation has been put forward, but the molecular details remain unclear. We identified a 30-amino acid region of JARID2 that mediates interactions with long noncoding RNAs (lncRNAs) and found that the presence of lncRNAs stimulated JARID2–EZH2 interactions in vitro and JARID2-mediated recruitment of PRC2 to chromatin in vivo. Native and crosslinked RNA immunoprecipitations of JARID2 revealed that Meg3 and other lncRNAs from the imprinted Dlk1-Dio3 locus, an important regulator of development, interacted with PRC2 via JARID2. Lack of MEG3 expression in human induced pluripotent cells altered the chromatin distribution of JARID2, PRC2, and H3K27me3. Our findings show that lncRNAs facilitate JARID2–PRC2 interactions on chromatin and suggest a mechanism by which lncRNAs contribute to PRC2 recruitment.
Introduction
Polycomb group (PcG) genes are key epigenetic regulators in multicellular organisms, as they maintain transcriptional repression of lineage-specific genes throughout development, thus contributing to the stability of cell identity (Schwartz et al., 2006). All mammalian PcG protein complexes identified so far perform their epigenetic function by acting on chromatin (Lanzuolo and Orlando, 2012); in particular, the Polycomb repressive complex 2 (PRC2) is responsible for di- and tri-methylation of lysine 27 in histone H3 (H3K27me2/3) (Margueron and Reinberg, 2011), a hallmark of facultative heterochromatin (Trojer and Reinberg, 2007).
One of the outstanding questions regarding mammalian PRC2 function is that of specificity of action: how are certain genes selected for repression, while others are unaffected? How can the same molecular machinery silence different genes in different cell lineages? Because none of the core components of PRC2 (EZH2, EED, SUZ12, RBBP4/7) possess a DNA binding domain (Margueron and Reinberg, 2011), it is believed that chromatin targeting must be specified elsewhere, either by interactions with DNA-binding factors (Boulay et al., 2012; Kim et al., 2009), pre-existing histone methylation (Margueron et al., 2009), chromatin-associated long noncoding RNAs (lncRNAs) (Rinn et al., 2007; Tsai et al., 2010), or a combination thereof (Margueron and Reinberg, 2011)
One essential factor for proper recruitment of PRC2 during the early phases of embryonic stem cell (ESC) differentiation is the Jumonji family, ARID domain-containing protein JARID2 (Landeira et al., 2010; Li et al., 2010; Pasini et al., 2010; Peng et al., 2009; Shen et al., 2009), which is often deleted in chronic myeloid malignancies (Puda et al., 2012). In the absence of JARID2, PRC2 is recruited late and incompletely to its target genes and its enzymatic function is diminished (Li et al., 2010; Son et al., in press), which results in failure to follow the differentiation program. Although JARID2 target sites are enriched for CGG- and GA-containing sequences (Peng et al., 2009), its DNA binding preferences lack the specificity to explain its distribution on chromatin (Li et al., 2010). Therefore, the nature of the recruitment pathway for JARID2 and the mode by which JARID2 regulates downstream steps of PRC2 assembly and function remain unclear.
Noncoding RNAs have been implicated in the regulation of epigenetic pathways, from early work on the lncRNA Xist in X chromosome inactivation (Brockdorff et al., 1992; Brown et al., 1992) and antisense transcripts in imprinted loci (John and Surani, 1996), to the more recent discovery of HOTAIR (Rinn et al., 2007) and its proposed role as a scaffold for chromatin-modifying “supercomplexes” (Tsai et al., 2010). Mammalian genomes contain thousands of lncRNAs (Guttman et al., 2009), most of which remain functionally uncharacterized. Because of their large size, potential for tertiary structure formation, and ability to form sequence-specific interactions with DNA, lncRNAs appear well suited to exchange information between chromatin-modifying complexes and the genomic sequence (Bonasio et al., 2010; Rinn and Chang, 2012). Polycomb repressive complex-1 (PRC1), PRC2, and the MLL complex interact with the lncRNAs ANRIL, HOTAIR and HOTTIP, respectively and these interactions facilitate their recruitment to chromatin (Rinn et al., 2007; Wang et al., 2011; Yap et al., 2010). However, the molecular details and downstream consequences of these RNA–protein interactions remain poorly understood. For example, the RNA-binding activity of PRC2 has been attributed to both EZH2 (Kaneko et al., 2010; Zhao et al., 2010) and SUZ12 (Kanhere et al., 2010), and, in unbiased analyses, large portions of the transcriptome were reported to bind to PRC2 (Kaneko et al., 2013; Khalil et al., 2009; Zhao et al., 2010), raising the question of how specificity is achieved in vivo.
Here, we show that the PRC2 accessory subunit JARID2 binds to lncRNAs in vivo and in vitro and that its interaction with MEG3, a lncRNA encoded by the imprinted DLK1–DIO3 locus, is necessary for proper recruitment and assembly of PRC2 at a subset of target genes in pluripotent stem cells.
Results
JARID2 binds to RNA in vitro
Despite a requirement for JARID2 during development (Takeuchi et al., 1995) and its key role in PRC2 recruitment and function during differentiation of mouse ESCs (Li et al., 2010; Pasini et al., 2010; Peng et al., 2009; Shen et al., 2009), the mechanisms by which JARID2 is targeted to chromatin and orchestrates PRC2 function remain poorly understood. Given that several PcG and PcG-associated proteins interact with lncRNAs, which in some cases regulate their recruitment to chromatin (Kanhere et al., 2010; Rinn et al., 2007; Yap et al., 2010), and based on our own preliminary observations in vitro (Kaneko et al., 2010), we hypothesized that lncRNAs might also regulate the function of JARID2.
We previously mapped an RNA-binding region (RBR) of EZH2, a core component of PRC2, and found that phosphorylation of a threonine within that region stimulated binding to lncRNAs (Kaneko et al., 2010). We performed similar in vitro RNA-binding assays on JARID2 using a bait spanning nucleotides 1–333 of HOTAIR, a lncRNA that regulates PRC2 function (Tsai et al., 2010), and detected an affinity for RNA within an internal fragment of JARID2, but not in the N-terminal or C-terminal regions (Figure 1A–B). Further mapping experiments revealed that the deletion of residues 332–358 resulted in a severe decrease of RNA binding in vitro (Figure 1C, Figure S1A–B). The sequence spanning these residues is conserved strongly in vertebrates (Figure S1C), but only very weakly with Drosophila (Figure S1D), suggesting that JARID2 may have acquired an additional layer of regulation in vertebrates.
Figure 1. Identification of the RNA-binding region of JARID2.
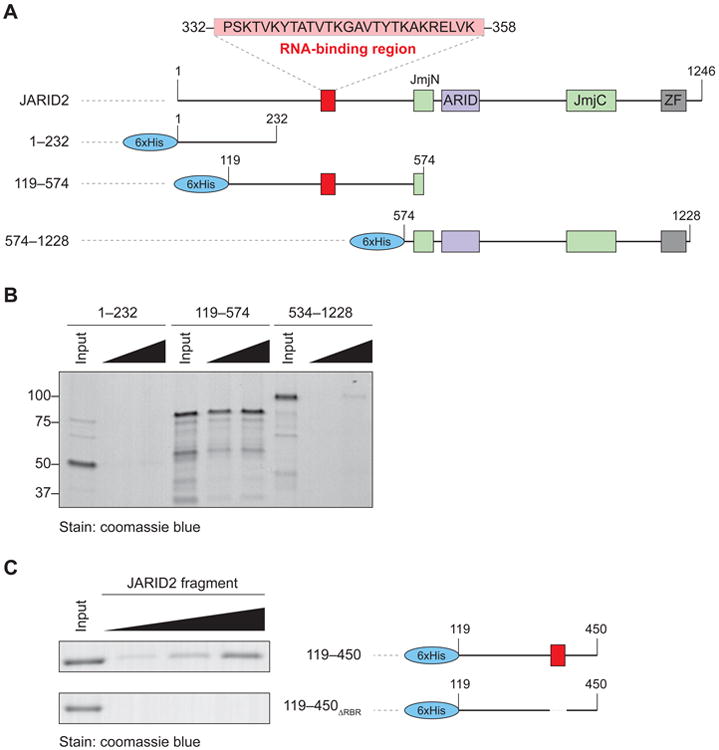
(A) Domain organization of human JARID2 and scheme of the 6xHis-fused truncations utilized in the mapping experiments.
(B) In vitro streptavidin pull-down after incubation of increasing concentrations of the indicated JARID2 recombinant fragments with biotinylated HOTAIR1–333. Input, 2 μg; titration, 2 and 4 μg.
(C) High resolution mapping of the residues of JARID2 necessary for RNA binding in vitro. The two indicated fragments (right) were incubated with HOTAIR1–333 and assayed as in (B). Input, 2 μg; titration, 1, 2, and 4 μg.
See also Figure S1.
JARID2 binds to EZH2, the catalytic component of PRC2 (Margueron and Reinberg, 2011) and stimulates its histone methyltransferase activity (Li et al., 2010; Son et al., in press). These functions require the same internal fragment that contains the RBR (Figure S1E), but can be uncoupled from the latter, given that deletion of residues 332–358 did not affect the ability of JARID2 to interact with PRC2 in vivo (Figure S1F), nor to stimulate its enzymatic activity (Figure S1G), whereas the 349–574 fragment did not bind to RNA (Figure S1A) but retained the ability to interact with nucleosome (Figure S1E) (Son et al., in press). Therefore, although partially overlapping regions of JARID2 are required for these various functions, the only activity that we could uniquely attribute to the 332–358 fragment is that of binding RNA in vitro. Henceforth, we will refer to these residues as the RBR of JARID2 and to the mutant protein lacking these residues as JARID2ΔRBR.
JARID2 and EZH2 bind to lncRNAs in vivo, including Meg3
To determine whether JARID2 makes direct contacts with RNA in vivo, we utilized the PAR-CLIP technique, which crosslinks RNA that has incorporated 4-thiouridine (4-SU) to proteins in vivo (Hafner et al., 2010). Consistent with our hypothesis, we detected a strong PAR-CLIP signal in immunoprecipitations (IPs) with our JARID2 antibody (Li et al., 2010) in extracts from E14 ESCs (Figure 2A). These IPs were conducted in presence of 2% lauryldimethylbetaine, a zwitterionic detergent that almost completely abolished the PRC2–JARID2 interaction while preserving antibody reactivity (Figure 2B). Furthermore, the radioactive signal we observed must have originated from RNA crosslinked to JARID2 because it was dependent on the incorporation of 4-SU, and was erased by treatment with increasing concentrations of RNase and by Jarid2 knockdown (Figure 2C).
Figure 2. JARID2 and EZH2 share interacting lncRNAs in vivo, including Meg3.
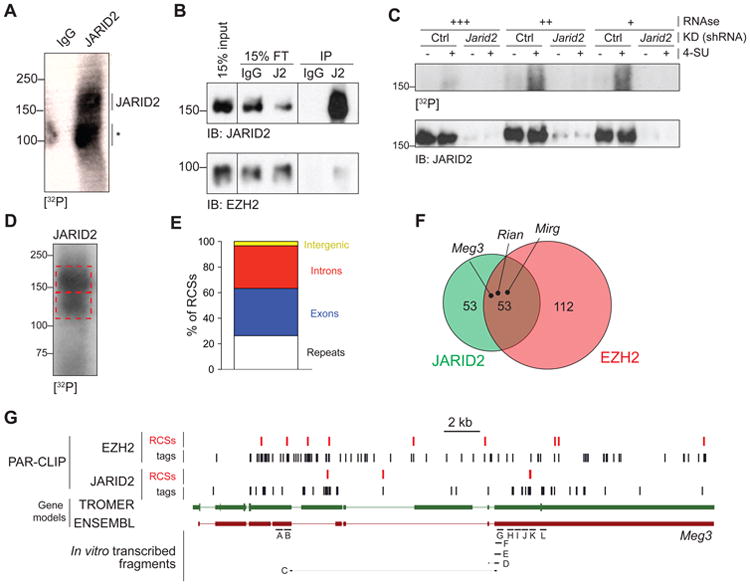
(A) PAR-CLIP with JARID2 antibodies or IgG in E14 ESC cells. The position of full length JARID2 is indicated. The asterisk marks a prfesumed degradation product.
(B) Immunoblot on the same material utilized for the autoradiography in (A). J2, anti-JARID2 antibody.
(C) PAR-CLIP (top) and immunoblot for JARID2 (bottom) performed in cells pulsed (+) or not pulsed (-) with 4-SU and stably transfected with an shRNA against Jarid2 or a control shRNA. Extracts were treated with increasing concentration of a cocktail of DNase-free RNase A and T1.
(D) PAR-CLIP-seq blot for JARID2.
(E) Distribution of JARID2 RCSs identified by PARalyzer in the genome. The stacked columns represent % of total RCSs. “Repeats” include all features listed in the RepMask database.
(F) Venn diagram of lncRNAs containing RCSs for JARID2, EZH2, or both. PAR-CLIP data for EZH2 were taken from GSE49433 (Kaneko et al., 2013).
(G) Genome browser view of JARID2 and EZH2 CLIP tags (black bars) or RCSs identified by PARalyzer (red bars) mapping to the Meg3 lncRNA. Gene models for Meg3 according to both TROMER and ENSEMBL are shown. Meg3 fragments tested for in vitro binding are indicated at the bottom.
See also Figure S2 and Table S1.
To identify the RNAs bound to JARID2 in vivo, we excised 32P-labeled bands from the PAR-CLIP membranes (Figure 2D), eluted the crosslinked RNA and sequenced it. In addition to the major, full-length band, we excised a faster migrating band that also reacted with JARID2 antibodies in WT but not in Jarid2-/- cells (data not shown). We obtained 90,000–200,000 unique CLIP tags in each replicate and analyzed their distribution using the PARalyzer software, which takes advantage of the T>C transitions caused by 4-SU crosslinking to discriminate signal from noise (Corcoran et al., 2011). PARalyzer identified 9,050 putative RNA–protein contact sites (RCSs) for JARID2, of which ∼26% overlapped by more than 50% with `repeats (Figure 2E) and were discarded. Among the 2,057 lncRNAs annotated in the mouse genome (ENSEMBL release 67), we identified 106 that contained at least one non-repetitive RCS for JARID2 (Table S1). This bioinformatic pipeline applied to our previously generated EZH2 PAR-CLIP data (Kaneko et al., 2013) revealed that, in the same ESCs, EZH2 interacted with 165 lncRNAs, of which 53 were in common with JARID2 (Figure 2F, Table S1), including Meg3/Gtl2, Rian, and Mirg, three lncRNAs encoded within the imprinted Dlk1–Dio3 locus.
We focused our attention on the lncRNA Meg3 (also known as Gtl2), because of previous reports linking it to pluripotency (Stadtfeld et al., 2010), imprinting (da Rocha et al., 2008), and PRC2 (Zhao et al., 2010). Consistent with our PARalyzer analysis, several tags and RCSs from both JARID2 and EZH2 PAR-CLIP data mapped to Meg3 (Figure 2G). Although some of the JARID2 CLIP tags mapped to a 5′ region annotated as an intron by ENSEMBL (red track), the existence of an exon in this region is supported by the TROMER database (Benson et al., 2004) (green track), and our own RNA-seq (data not shown). Another cluster of JARID2 CLIP tags mapped to the 3′ exon and accumulated in a region where PARalyzer identified an RCS (Figure 2G). Few CLIP tags mapped to highly expressed genes such as Nanog (Figure S2A) or Gapdh (Figure S2B), suggesting that the presence of Meg3 tags reflected direct interactions in vivo.
The RBR of JARID2 contributes to Meg3 binding in vitro and in vivo
Having identified Meg3 as a JARID2-interacting lncRNA by CLIP, we next tested this interaction in vitro. We performed pull-down assays with in vitro-transcribed fragments of Meg3 (Figure 2G, bottom) obtained from a previously generated clone (Zhao et al., 2010). Although all tested fragments bound to a recombinant JARID2 fragment spanning the RBR (Figure 3A), the interaction was stronger for fragments originating from the 3′ end of the clone (Figure 3B), including one (fragment “K”) that spanned the only RCS identified in this region (Figure 2G). We analyzed structural predictions for these fragments but found no obvious similarities among those that bound with higher affinity, except a trend for less stable structures (Figure S3). Importantly, a JARID2 fragment lacking the RBR displayed a pronounced reduction in binding affinity, at least toward the Meg3 fragment tested (fragment “F”, Figure 3A–B).
Figure 3. The JARID2 RBR mediates interactions of the Meg3 lncRNA with PRC2.
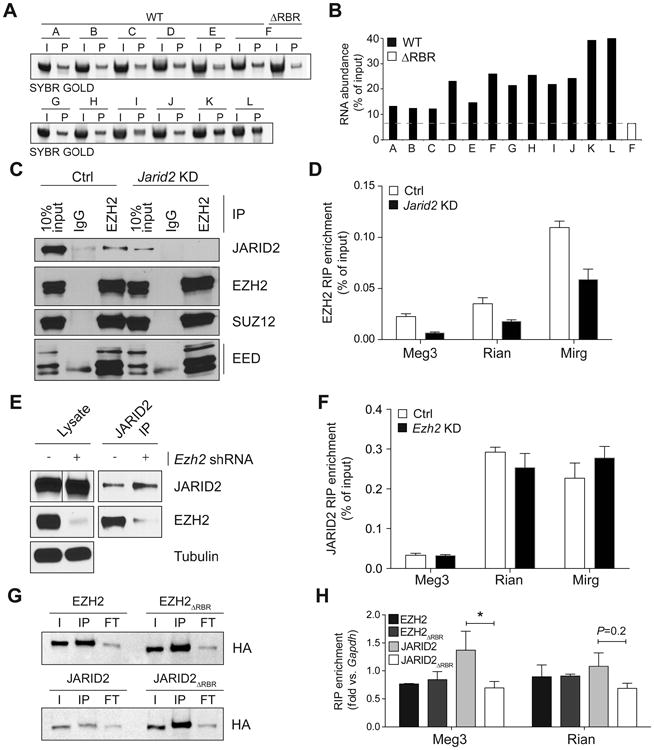
(A) In vitro pull-down assay with different fragments of Meg3 using 4 μg of GST-JARID2 119-450 WT or ΔRBR.
(B) Quantification of bands shown in (A).
(C) RIPs for EZH2 were performed in E14 ESCs stably transfected with an shRNA against Jarid2 (KD) or empty vector (ctrl). Co-precipitated proteins were revealed by western blot. 10% input and IgG lanes are shown as control.
(D) RT-qPCR on EZH2 RIPs from control E14 ESCs (white bars) or Jarid2 knockdown E14 ESCs (black bars). Data is shown as % of RIP input. Bars represent the mean of 4 replicates + s.d.
(E and F) As in (C) and (D) but Ezh2 was knocked down and the RIP were performed with JARID2 antibodies.
(G) Western blots for HA RIPs from nuclear extracts of KH2 transiently transfected with N3-tagged EZH2, EZH2ΔRBR (top), JARID2, and JARID2ΔRBR (bottom). I, input; IP, HA immunoprecipitation; FT, flow-through.
(H) RT-qPCR normalized to Gapdh levels on HA RIPs described in (E). Bars indicate the mean of 3 biological replicates + s.e.m. *, P < 0.05 by Mann-Whitney U test.
See also Figure S3 and S4.
To validate these JARID2–RNA interactions with a technique more quantitative than PAR-CLIP, we resorted to native RNA immunoprecipitations (RIPs) followed by qPCR. Consistent with our PAR-CLIP results and previous reports (Zhao et al., 2010), Meg3 was enriched in PRC2 RIPs performed with EZH2 antibodies, and so were two other lncRNAs encoded within the same imprinted locus, Rian and Mirg (Figure 3C–D). However, when we depleted JARID2 by shRNA-mediated knockdown we observed a considerable decrease in the amounts of these lncRNAs co-precipitating with PRC2 (Figure 3C–D), and similar results were obtained with SUZ12 antibodies (Figure S4A–B). Importantly, knockdown of Ezh2 did not affect the ability of JARID2 to bind to Meg3, Rian, or Mirg (Figure 3E–F), suggesting that the JARID2–Meg3 interaction makes the largest contribution to the affinity of Meg3 for PRC2.
We then asked whether Meg3 bound to JARID2 via the RBR. To this end, we transiently expressed in mouse ESCs HA-tagged EZH2, EZH2 lacking the previously identified RBR (EZH2ΔRBR) (Kaneko et al., 2010), JARID2, and JARID2ΔRBR, and performed HA RIPs followed by RT-qPCRs. Consistent with the results presented above, the interaction of Meg3 with JARID2 was significantly decreased when its RBR was removed, whereas EZH2 and EZH2ΔRBR co-precipitated Meg3 with equal efficiencies (Figure 3G–H). We detected a similar trend for Rian, although in that case the difference did not reach statistical significance (Figure 3H). Similarly, human JARID2 but not human JARID2ΔRBR bound to Meg3 in mouse ESCs (Figure S4C–D).
These data supported the conclusion that the Meg3 interacts with PRC2 mainly through the RBR of JARID2, which led us to speculate that this lncRNA may participate in the function of JARID2 on chromatin.
MEG3 regulates PRC2 occupancy in trans
In light of the connection between MEG3 and pluripotency (Stadtfeld et al., 2010), we turned our attention to a set of 8 human induced pluripotent stem cell (hiPSC) lines that differ greatly in their levels of MEG3 expression (Nishino et al., 2011), thus offering a natural experimental system in which to study the effects of MEG3 on PRC2 function. We classified these lines into 5 MEG3+ and 3 MEG3- (Figure S5A–B) and confirmed that EZH2, SUZ12, and JARID2 were expressed at similar levels in MEG3+ and MEG3- lines (Figure S5C–E). Importantly, pluripotent markers OCT3/4 and NANOG were also expressed at comparable levels in all lines, and in all cases at much higher levels than in differentiated cells, such as foreskin fibroblasts (Figure S5F–G).
Next, we performed chromatin IP (ChIP) followed by deep sequencing (ChIP-seq) for JARID2, EZH2, and H3K27me3, the product of PRC2 catalysis. We identified enriched regions (ERs) for all three features and compared normalized read densities in MEG3+ vs. MEG3- cells. The genome-wide analysis revealed 29, 89, and 268 differentially bound regions (DBRs) with an FDR < 0.1 in MEG3+ vs. MEG3- cells for JARID2, EZH2, and H3K27me3, respectively (Figure 4A–C). At most of these MEG3-dependent DBRs, PRC2 exhibited stronger binding in the hiPSC lines that expressed MEG3, suggesting that the lncRNA had a stimulatory function (Figure 4A–C, Figure S6A–C). Genes near the MEG3-dependent DBRs were enriched for functional terms related to the regulation of transcription during embryonic development and differentiation (Table S2), even taking into account the enrichment of developmental and transcription-related terms in the background population comprising all JARID2, EZH2, and H3K27me3 ERs (Table S3).
Figure 4. Decreased occupancy of PRC2 at some chromatin targets in MEG3- cells.

(A-C) MA plots for JARID2 (A), EZH2 (B), and H3K27me3 (C) occupancy, as determined by the normalized and input-corrected read densities in MEG3+ (above dotted line) vs. MEG3- (below dotted line) hiPSC lines. Each dot represents an ER in common between at least two hIPSC lines. DBRs with an FDR < 0.1 are displayed in red.
(D) Venn diagram for DBRs with FDR < 0.1.
(E) RT-qPCR analysis of PRC2 targets in MEG3+ and MEG3- hiPSCs. Bars represent the mean RNA abundance (as % of GAPDH) in the 5 MEG3+ and 3 MEG3- lines tested. *; P < 0.05, as calculated by Mann-Whitney U test.
(F) RT-qPCR for Meg3 24 h after transfection of KH2 ESCs with control (white bar) or Meg3 siRNAs.
(G and H) ChIP-qPCR for JARID2 (G) or EZH2 (H) with primers mapping to PRC2 peaks near the indicated genes in KH2 ESCs treated with control (white bars) or Meg3 (black bars) siRNAs. Bars represent the mean of 3 replicates + s.e.m. *, P < 0.05 by Mann-Whitney U test.
See also Figure S5, S6, S7, Table S2 and S3.
Of the 29 MEG3-dependent DBRs for JARID2, 21 overlapped with EZH2 DBRs (Figure 4D), and of these 16 also overlapped with H3K27me3 DBRs, a fraction much larger than expected by chance alone (P-value < 10-20, hypergeometric distribution). Among the regions with lower JARID2 occupancy in the absence of MEG3 were the loci encoding the transcription factors ZIC5, NR4A2, ZIC2, and PITX, as well as the neuronal gene PCDHGC5 (Figure S6D and data not shown), all of which function in differentiating or differentiated cells and must therefore be silenced in pluripotent stem cells. In all loci tested, the MEG3-dependent loss of PRC2 targeting resulted in transcriptional derepression (Figure 4E).
It has been suggested that, in mouse ESCs, Meg3 exerts a cis-repressive effect on the adjacent Dlk1 gene by recruiting PRC2 (Zhao et al., 2010). To determine whether a similar mechanism was conserved in hiPSCs, we analyzed the DLK1 promoter in MEG3+ vs. MEG3- hiPSCs. Unexpectedly, we observed no differences in JARID2 or PRC2 occupancy (data not shown) and no evidence of a reciprocal correlation in the levels of MEG3 and DLK1 RNA in these cells (Figure S5H) or with the protein-coding RNA at the other extremity of the imprinted locus, DIO3 (Figure S5J).
Because the DLK1-DIO3 locus encodes multiple ncRNAs (MEG3, RIAN, and MIRG), all repressed in MEG3- hiPSCs, we examined the effect of altering MEG3 levels alone on PRC2 localization. We overexpressed MEG3 (or GFP RNA as a control) in MEG3- hiPSCs and analyzed the distribution of JARID2 and EZH2 by ChIP-seq. A caveat of this experiment is that MEG3 was expressed at levels ∼10 times higher than in the average MEG3+ line (Figure S7A); nonetheless, we observed increased densities of JARID2 at some MEG3-dependent DBRs but not at control targets (Figure S7B). However, we did not detect recovery of JARID2 occupancy at all JARID2 DBRs (Figure S7C), suggesting that the regulation afforded by endogenous MEG3 could not be fully recapitulated by providing large amounts of the lncRNA in trans. Interestingly, PRC2 was preferentially depleted from the EZH2 DBRs upon overexpression of MEG3 (Figure S7D). Although this depletion of EZH2 was unexpected, the fact that the EZH2 DBRs were preferentially depleted by the manipulation of MEG3 levels is consistent with the idea that occupancy at these sites is selectively regulated by this lncRNA.
To further verify that the observed changes in PRC2 occupancy were directly related to the changes in MEG3 levels, we performed transient knockdown of the orthologous lncRNA in mouse ESCs. Despite only partial knockdown efficiency (∼50%; Figure 4F) 6 out of 9 DBRs tested lost JARID2 and EZH2 after Meg3 depletion (Figure 4G–H), suggesting that the regulation of PRC2 by Meg3 is conserved between human and mouse. PRC2 occupancy at Plek2, Mbnl3, and Cdx4, was not affected by Meg3 knockdown (data not shown), despite the fact that orthologous loci were among the MEG3-dependent DBRs in hiPSCs. However, it is not surprising that subtle differences in gene regulation would exist across this species barrier, especially given that mouse and human stem cells are not equivalent (Ginis et al., 2004).
Together, our ChIP experiments in hiPSCs and mouse ESCs support the conclusion that MEG3 acts in trans on PRC2 and JARID2 by facilitating their recruitment to a subset of target genes.
RNA facilitates JARID2–PRC2 interactions
Given that loss of MEG3 caused defects in PRC2 recruitment and that several lncRNAs crosslinked to both JARID2 and EZH2 in vivo, we hypothesized that RNA–mediated scaffolding could stabilize JARID2–PRC2 interactions. To test this hypothesis in vitro, we first examined the lncRNA HOTAIR, which functions as a scaffold between the LSD1/CoREST/REST complex and PRC2 (Tsai et al., 2010). Low amounts of HOTAIR stimulated the interaction between recombinant EZH2 and JARID2 fragments in vitro (Figure 5A–B, compare lanes 1–3), whereas higher concentrations of the RNA resulted in a return to baseline interaction levels, suggesting the possibility of squelching (Figure 5A–B, compare lanes 3–5). This stimulation was considerably reduced for JARID2 fragments that lacked the RBR (Figure 5A–B, lanes 6–10) but retained the ability to interact with EZH2 (Figure 5A, compare lanes 1 and 6), or when we replaced the HOTAIR lncRNA fragment with a shorter version, a control pre-miRNA that forms 2 stem-loops, or double-stranded DNA containing the HOTAIR lncRNA sequence (Figure 5C–F). Stimulation of binding was also observed when we incubated recombinant EZH2 and JARID2 with those Meg3 fragments that displayed maximum affinity in the in vitro pull-down assay (Figure 5G). Therefore, stimulation of JARID2–EZH2 interactions may be a general mechanism of action for lncRNAs that bind to these proteins.
Figure 5. HOTAIR stimulates EZH2-JARID2 interactions via the JARID2 RBR.
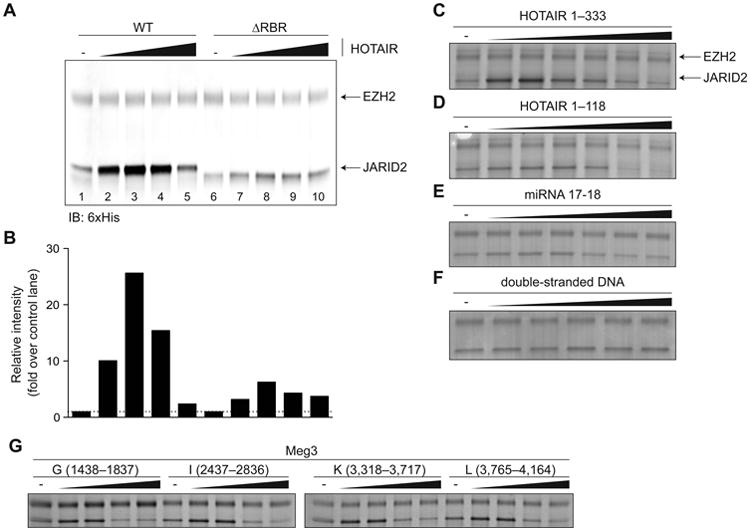
(A) FLAG-6xHis-tagged recombinant EZH2 (20 pmol) was incubated with 6xHis-tagged JARID2119–450 WT or ΔRBR (40 pmol) in presence of increasing amounts of HOTAIR1–333 (0–12 pmol). Pull-down was performed with anti-FLAG beads and proteins revealed by 6xHis immunoblot.
(B) Densitometric quantification of the signal for JARID2 shown in (A). WT and ΔRBR lanes were normalized each to lane 1 and lane 6 (no HOTAIR control), respectively.
(C-F) In vitro interaction stimulation assays with recombinant 6xHis-tagged JARID2119–574 and FLAG-6xHis-tagged full-length EZH2 in presence of increasing concentrations of HOTAIR1–333 (C), a smaller 5′ truncation of HOTAIR (D), a microRNA (E), or double stranded DNA (F). Pulldown was performed with anti-FLAG beads and proteins stained with Coomassie blue.
(G) Same as (C-F) using Meg3 fragments.
The JARID2 RBR stimulates PRC2 assembly on chromatin
Having discovered that lncRNAs stimulate JARID2–PRC2 interactions in vivo (Figure 4) and in vitro (Figure 5) and knowing that the RBR was required for the latter (Figure 5A–B), we asked whether it was also required for the former. To this end, we overexpressed JARID2 or JARID2ΔRBR in human foreskin fibroblasts, which express high levels of HOTAIR (Rinn et al., 2007), and measured its accumulation at known genomic targets by ChIP-qPCR. WT and mutant JARID2 were expressed at comparable levels in lentivirally-transduced fibroblasts (Figure 6A, top) and did not affect EZH2 levels (Figure 6A, middle). Upon JARID2 overexpression, we observed increased accumulation of the protein at known chromatin targets, such as HOXD8 and GATA2, compared to controls (Figure 6B). JARID2ΔRBR accumulated at these targets with the same efficiency as the WT protein but, unlike the WT, it was incapable to recruit additional EZH2, which remained at the same levels as observed in the mock-transfected cells (Figure 6C). Given that these targets were already silent in the mock-transfected controls, we did not attempt to detect further repression at the transcriptional level.
Figure 6. JARID2 recruits EZH2 to chromatin in an RBR-dependent manner.
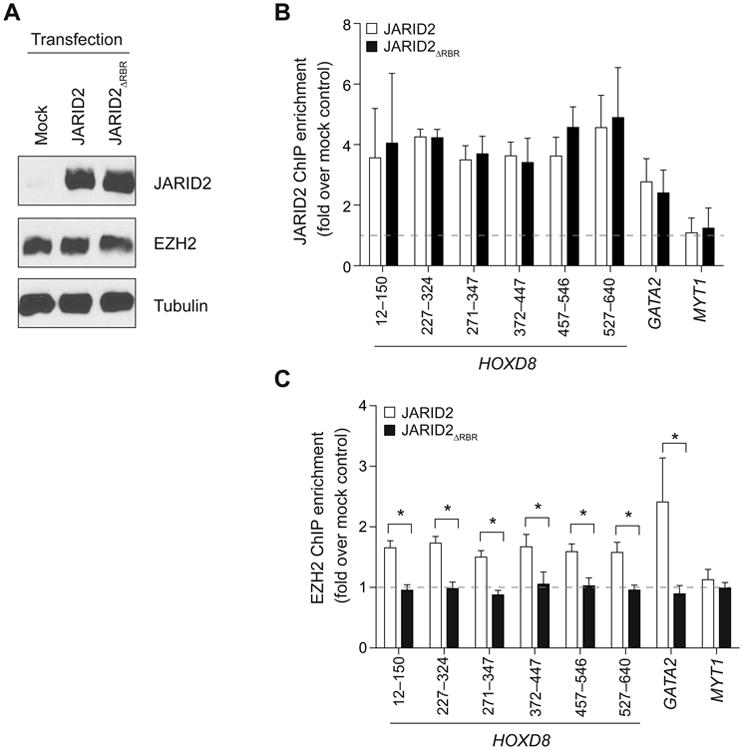
(A) Western blots for foreskin fibroblasts transduced with lentiviruses expressing JARID2, JARID2ΔRBR, or mock-transduced.
(B and C) ChIP-qPCR with antibodies against JARID2 (B) or EZH2 (C) at known PRC2 chromatin targets in foreskin fibroblasts transduced as in (A). Several primer sets are shown for HOXD8. The primers for MYT1 were designed at a distal location, devoid of PRC2, and serve as a negative control. The ChIP enrichment is normalized against that obtained with the same antibodies in mock-transfected control cells (dotted line). Bars represent mean of 4 technical replicates + s.d (B) or 3 biological replicates + s.e.m (C). *, P < 0.05 by Mann-Whitney U test.
These results suggest that RNA–protein interactions via the JARID2 RBR contribute to the recruitment and assembly of PRC2 on chromatin. Interestingly, depletion of HOTAIR in foreskin fibroblasts also impairs PRC2 recruitment at these loci (Rinn et al., 2007; Tsai et al., 2010), suggesting that it may act through interactions with the JARID2 RBR.
The fact that JARID2ΔRBR is incapable of recruiting EZH2 to target sites is compatible with a model by which lncRNAs modulate JARID2–PRC2 interactions and orchestrate the distribution and activity of PRC2 on chromatin.
Discussion
The results presented above allow us to add JARID2 to the growing list of chromatin-associated proteins that interact with lncRNAs and offer further support to the hypothesis that lncRNAs are a component of the Polycomb axis in mammals.
We mapped the JARID2 RBR to an N-terminal fragment that contains no annotated features or domains, despite the fact that, in addition to RNA binding, it is also responsible for interactions with SUZ12, EZH2, nucleosomes, and PRC2 stimulation (Kim et al., 2003; Li et al., 2010; Pasini et al., 2010; Son et al., in press). Although we assigned these biochemical activities to distinct protein fragments (Figures S1), they do map to adjacent regions, and it is tempting to speculate that this vicinity might reflect a functional cross-talk, by which, for example, RNA binding could stimulate nucleosomal binding and contribute to PRC2 regulation.
Among the lncRNAs identified by PAR-CLIP, we focused our functional analysis on Meg3 because of its connections with ESC pluripotency, imprinting, and PRC2 function (da Rocha et al., 2008; Stadtfeld et al., 2010; Zhao et al., 2010). We demonstrated JARID2–Meg3 interactions using RIP-qPCR, PAR-CLIP and in vitro pull-down assays. Importantly, we also found RCSs for EZH2 within Meg3 in our previously published EZH2 PAR-CLIP dataset (Kaneko et al., 2013) (Figure 2F–G). This is consistent with earlier results obtained by formaldehyde crosslinking (Zhao et al., 2010) and supports a model by which Meg3 contacts both JARID2 and EZH2 and stimulates their interaction. Without JARID2 the interaction between Meg3 and PRC2 is much weaker (Figure 3D), suggesting that of the two contact points, the one on JARID2 makes the larger contribution to the affinity for Meg3. We note that by using Ezh2-/- cells as a control Zhao et al. could not have detected this requirement for JARID2, because in absence of EZH2 the IP performed with an anti-EZH2 antibody would not have recovered JARID2.
We envision a model in which some lncRNAs function as scaffold to stimulate assembly of PRC2 at JARID2 target sites (Figure 7A). In addition, the existence of DBRs that lose JARID2 occupancy in absence of MEG3 (Figures 4A) suggests that, at certain sites, lncRNAs might also be required for the initial recruitment of JARID2 (Figure 7B). In both scenarios, the net result of lncRNA action is to increase PRC2 occupancy and H3K27me3 deposition (Figure 7A–B). We have demonstrated this model using MEG3 and genomic targets in hiPSCs and mouse ESCs (Figure 4); however, the recovery of other lncRNAs crosslinked to both JARID2 and EZH2 by PAR-CLIP-seq (Figure 2F, Table S1) allows us to speculate that this mode of action might not be limited to MEG3. In fact, the JARID2ΔRBR mutant does not recruit PRC2 to the HOX locus in foreskin fibroblasts, despite the fact that this locus is not MEG3-dependent. We propose that other lncRNAs function as scaffold for JARID2–PRC2 interactions in this setting and we note that foreskin fibroblasts express high levels of HOTAIR (Rinn et al., 2007), which is also able to stimulate JARID2–EZH2 interactions in vitro (Figure 5).
Figure 7. Proposed model for the interplay of lncRNAs, JARID2, and PRC2.

(A) At some target genes, the presence of JARID2 by itself (I) is not sufficient for maximum PRC2 recruitment, which requires scaffolding by lncRNAs (II). The presence of both JARID2 and lncRNAs stimulates further recruitment and assembly of PRC2 on chromatin, resulting in increased H3K27me3 (III). The structure of lncRNAs bound to JARID2 (and PRC2) remains to be elucidated and the one shown here is only for the purpose of illustration.
(B) In some cases, lncRNAs might contribute to the initial recruitment of JARID2 to chromatin (I). Because JARID2 also binds PRC2 via protein–protein interactions, this results in increased PRC2 recruitment and H3K27 methylation (II).
Not all lncRNAs bind equally to JARID2 and EZH2. Our reanalysis of EZH2 PAR-CLIP tags (Kaneko et al., 2013) identified a number of lncRNAs not shared with JARID2 (Figure 2F, Table S1) that, likely, regulate PRC2 function in JARID2-independent ways. In addition to lncRNAs, EZH2 binds to a variety of coding transcripts in vivo and in vitro with high affinity but seemingly low specificity (Davidovich et al., 2013; Kaneko et al., 2013). Those interactions appear to constitute a distinct regulatory mechanism from the one described here, as they occur mostly at promoters of transcribed genes, which have low PRC2 occupancy (Davidovich et al., 2013; Kaneko et al., 2013) and are largely devoid of JARID2 (data not shown). However, we cannot exclude that nascent RNAs could compete with lncRNAs for binding to EZH2, which might help explain their apparent inhibitory function.
Our gain-of-function experiments in hiPSCs confirmed that at least some JARID2 DBRs identified in MEG3+ vs. MEG3- hiPSCs can be rescued by supplying MEG3 lncRNA in trans to otherwise MEG3-negative hiPSCs (Figure S7B). The ectopic expression of MEG3 lncRNAs caused EZH2 depletion rather than increased occupancy at the previously identified MEG3-dependent DBRs (Figure S7D), which was in contrast to our expectations. However, considering that lentiviral transduction resulted in 10-fold higher MEG3 levels compared to endogenous MEG3 (Figure S7A), we speculate that such an excess of RNA molecules acted in a dominant negative fashion through squelching, as seen in vitro (Figure 5). It is also possible that the random integration of the lentivirally-encoded MEG3 at ectopical sites within the genome may explain its unphysiological behavior in these experiments, given that the genomic location of lncRNA genes appears to play a pivotal role in their function (Ulitsky et al., 2011). Nonetheless, the fact that MEG3-dependent DBRs were selectively affected confirmed that occupancy at these targets is indeed regulated by MEG3, which was further supported by the results of transient Meg3 knockdown in mouse ESCs (Figure 4G–H).
Genomic imprinting of MEG3 is unstable in human ESCs, and several hiPSC lines, regardless of their parental cell type, maintain repression at this locus even after continuous passaging (Nishino et al., 2011). This is likely mediated by aberrant DNA hypermethylation (Nishino et al., 2011)(data not shown). Therefore, it is possible that current reprogramming protocols fail to set the appropriate epigenetic state at the DLK1–DIO3 locus, which is an important consideration given that improper regulation of this imprinted regions leads to developmental abnormalities in mouse (Stadtfeld et al., 2010; Takahashi et al., 2009) and human (Kagami et al., 2008). The mechanistic details of the epigenetic alteration at this imprinted locus during reprogramming remains elusive, but our data suggest that MEG3 interactions with PRC2 might play an important role.
Although our findings suggest a molecular mechanism by which MEG3 contributes to JARID2 and PRC2 function, further investigations are required to address: (1) whether and how this mechanism extends to the other lncRNAs identified by PAR-CLIP; and (2) whether and how lncRNAs regulate de novo recruitment of their interacting proteins to distant loci. Our in vitro binding analyses of Meg3 lncRNA suggest that JARID2 does not bind to RNA in a sequence-specific manner, consistent with the fact that the primary sequence of lncRNAs is not well conserved (Ulitsky et al., 2011)
In conclusion, we have demonstrated that JARID2, an essential regulatory component of PRC2 in pluripotent stem cells, contains an RNA-binding region that mediates, at least in part, its interaction with the imprinted lncRNA MEG3. This—and possibly other—RNA interaction contributes to proper recruitment and assembly of PRC2 on target genes and likely plays an important role in orchestrating the epigenetic regulation of gene expression that accompanies the transition from stem cell pluripotency to differentiation.
Experimental Procedures
For information about antibodies, oligonucleotides, and plasmids see Table S4, S5, S6 and Supplemental Experimental Procedures.
Cells
Human iPSCs were cultured as described previously (Nishino et al., 2011). Cells were harvested at passage 34 (UtE-iPS-4), 41 (UtE-iPS-11), 45 (UtE-iPS-7), 41 (AM-iPS-6), 50 (AM-iPS-8), 39 (UtE-iPS-6), 41 (MRC5-iPS-25) and 90 (Edom-iPS-2). Human foreskin fibroblasts (ATCC, #PCS-201-010, lot# 58490326) were cultured in fibroblast basal medium (ATCC) plus fibroblast growth kit-low serum (ATCC). KH2 ESCs expressing the reverse tetracycline-controlled transactivator (rtTA) (Hochedlinger et al., 2005) were maintained in standard mESC culture conditions. KH2 lines expressing Jarid2 and Jarid2ΔRBR were generated by transfection of the relevant pINTA-N3 construct and selecting with 50 μg/ml Zeocin (Invitrogen). Transgene expression was induced with doxycycline for 24 hours. E14Tg2A.4 mouse ES cell lines (E14 mESC), HEK293 and HEK293T cells were cultured as described (Kaneko et al., 2010). Jarid2 knockdown mESCs were described previously (Li et al., 2010).
In vitro binding assays
Protein purification, synthesis of HOTAIR lncRNA (1-333), and biotinylated RNA pull-down assays were described previously (Kaneko et al., 2010). All RNA fragments were generated by in vitro transcription; see Supplemental Information for details.
LncRNA-mediated stimulation of EZH2-JARID interactions was assayed by incubating FLAG-and 6xHis-tagged EZH2 (20 pmol) with increasing amounts (0–12 pmol) of lncRNAs in 100 μl of binding buffer (50 mM Tris-HCl, pH 7.9; 100 mM KCl; 0.1% NP-40) for 30 min at 25°C. 6xHis-tagged truncated JARID2 (40 pmol) was added to the reaction and incubated for 30 min at 25°C. Complexes were purified using FLAG-M2 affinity gel (Sigma), after washing with binding buffer.
For JARID2–Meg3 in vitro binding assays, 4 μg of GST-JARID2119–450 were incubated with a series of Meg3 fragments (1 μg, 400 nts each, see Table S7). Bound RNAs were purified with glutathione beads, resolved with 7M urea gels, stained with SYBR-gold (Invitrogen), and quantified with an ImageQuant LAS4000 (GE Healthcare Life Sciences).
Knockdowns
For conditional knockdown of Ezh2 in E14 mESCs, we generated stable clones with an integration of pTRIPZ lentiviral inducible shRNAmir targeting human and mouse Ezh2 (Open Biosystems #RHS4696-99635303). Selection was done by puromycin (1 μg/ml) and clones were screened by red fluorescent protein (RFP) expression after 3–4 days of doxycycline (1 μg/ml) induction.
For transient knockdown of Meg3, we tested four siRNAs from Qiagen (SI05169486, SI05169710, SI01060129, SI01060136) and used the siRNA resulting in the most efficient knockdown (SI05169486). See also Supplemental Information.
Chromatin immunoprecipitation
ChIP from hiPSCs, foreskin fibroblast, and mESCs was performed as described (Kaneko et al., 2007) with minor modification and library were constructed as described (Gao et al., 2012). Briefly, cells were cross-linked with 1% formaldehyde for 10 min and sonicated in ChIP buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 1% Triron X-100, 0.5% NP-40, 5 mM EDTA pH 8.0, 1 mM PMSF and protease inhibitors) with a Diagenode Bioruptor. Incubations with antibodies were carried out in an ultrasonic water bath for 30 min at 4°C. Samples were decrosslinked at 65°C for ∼16 hours for library construction or 95°C 10 min for ChIP-qPCR. See Supplemental information for more details.
RNA immunoprecipitation
For Figure 3G–H, nuclear extracts were obtained using an established protocol (Dignam et al., 1983) with minor modifications to minimize RNAse activity, lysates were diluted in RIP buffer (20 mM Tris pH 7.94°C, 200 mM KCl, 0.05% IGEPAL CA-630, 10 mM EDTA), cleared by centrifugation at 20,000g for 10 min, and incubated with depleting amounts of antibody for 3 h at 4°C. Immunocomplexes were recovered with protein G-coupled dynabeads (Invitrogen) for 1 h at 4°C. Beads were washed in RIP-W buffer (20 mM Tris pH 7.94°C, 200 mM KCl, 0.05% IGEPAL CA-630, 1 mM MgCl2) twice and incubated with 2 u of TURBO DNase (Ambion) in 20 μl RIP-W buffer for 10 min at room temperature, to avoid DNA bridging artifacts. After two additional washes RNA was eluted and purified with TRIzol (Invitrogen).
For Figure 2C–F RIPs were performed on whole cell lysates, as described before (Kaneko et al., 2010)
See also Supplemental Information.
ChIP-seq analysis
Sequenced reads from ChIP-seq experiments were mapped with Bowtie using parameters -v2 -m4 --best (Langmead et al., 2009). Normalized genome-wide read densities were computed and visualized on the UCSC genome browser. Bound regions (ERs) were identified using MACS 2.09 (Zhang et al., 2008) and default parameters. ERs were associated to gene targets using ChIPpeakAnno and ENSEMBL annotation 67.
PAR-CLIP
ESCs were pulsed with 100 μM 4-SU (Sigma) for 16-24h and crosslinked with 400 mJ/cm2 UVA (365 nm) using a Stratalinker UV crosslinker (Stratagene, CA). Cells were lysed for 10 min at 37°C in CLIP buffer (20 mM HEPES pH 7.4, 5 mM EDTA, 150 mM NaCl, 2% lauryldimethylbetaine) with protease inhibitors, 20 U/ml Turbo DNase (Life technologies), and 200 U/ml murine RNase inhibitor (New England Biolabs). IPs were carried out in CLIP buffer for 1 hour at 4°C. When necessary, extracts were treated with RNAse A + T1 cocktail (Ambion) for 5′ at 37°C. Immunocomplexes were recovered with protein G-coupled dynabeads for 45 min at 4°C. DNA was removed with Turbo DNase (2U in 20 μl). Crosslinked RNA was labeled by incubations with 5U Antarctic phosphatase and 5U T4 PNK (both from New England Biolabs) in presence of 10 μCi [γ-32P] ATP (PerkinElmer, MA). Labeled material was resolved on 8% bis-tris gels, transferred to nitrocellulose and exposed to autoradiography films for 1–24 hours.
For PAR-CLIP-seq, 100 pmol of a 3′-blocked DNA adapter were ligated to the RNA after dephosphorylation and before 5′ labeling by incubating the beads with T4 RNA ligase 1 (New England Biolabs) for 1 hour at 25°C. After autoradiography, bands were excised and the RNA eluted with proteinase K for 30′ at 37°C and proteinase K in 3.5M urea for 30′ at 55°C. Custom 5′ adapters were ligated, and the products were size-selected on polyacrylamide or agarose gels, amplified and sequenced on an Illumina HiSeq 2000.
For the analysis, adapter sequences were removed and reads < 17 nt discarded. The remaining reads were mapped to the mm9 genome using BOWTIE (Langmead et al., 2009) allowing two mismatches and removing duplicates. RCSs were identified with PARalyzer (Corcoran et al., 2011) requiring at least two T->C conversions per RCS. For Figure 2F, RCSs were assigned to lncRNAs in ENSEMBL 67 when they overlapped anywhere within the gene body, to account for imprecisions in the annotation of lncRNAs.
RNA structural predictions
Structural predictions and minimum free energy calculations shown in Figure S3 were performed with the Vienna RNA Websuite using default settings (Gruber et al., 2008).
Supplementary Material
Highlights.
JARID2 binds lncRNAs
JARID2 and PRC2 interactions are stimulated by lncRNAs
The Meg3 imprinted lncRNA binds to PRC2 via JARID2
Loss of MEG3 in hiPSCs correlates with loss of JARID2 and PRC2 throughout the genome
Acknowledgments
We thank the Genome Technology Center at NYU for help with sequencing, Varun Narendra for bioinformatics analyses, and Matthias Stadtfeld for comments on the manuscript. This work was supported by grants from the National Institute of Health (GM-64844 and R37-37120) and the Howard Hughes Medical Institute (to D.R.). R.B. was supported by a Helen Hay Whitney Foundation postdoctoral fellowship and by the Helen L. and Martin S. Kimmel Center for Stem Cell Biology postdoctoral fellow award. R.S.M. was supported by a Ph.D. and an international research stay fellowship from CONACyT (213029).
Footnotes
Accession Numbers: The databank accession number for ChIP and CLIP sequences reported in this paper is GSE48518. EZH2 PAR-CLIP-seq data used for comparative analyses were taken from GSE49433 (Kaneko et al., 2013).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32:D23–26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Dubuissez M, Van Rechem C, Forget A, Helin K, Ayrault O, Leprince D. Hypermethylated in cancer 1 (HIC1) recruits polycomb repressive complex 2 (PRC2) to a subset of its target genes through interaction with human polycomb-like (hPCL) proteins. J Biol Chem. 2012;287:10509–10524. doi: 10.1074/jbc.M111.320234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, Swift S, Rastan S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992;71:515–526. doi: 10.1016/0092-8674(92)90519-i. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Hendrich BD, Rupert JL, Lafreniere RG, Xing Y, Lawrence J, Willard HF. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–316. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol. 2013;20:1250–1257. doi: 10.1038/nsmb.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Molecular cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginis I, Luo Y, Miura T, Thies S, Brandenberger R, Gerecht-Nir S, Amit M, Hoke A, Carpenter MK, Itskovitz-Eldor J, et al. Differences between human and mouse embryonic stem cells. Dev Biol. 2004;269:360–380. doi: 10.1016/j.ydbio.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin M, Feldser D, Huarte M, Zuk O, Carey B, Cassady J, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009 doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- John RM, Surani MA. Imprinted genes and regulation of gene expression by epigenetic inheritance. Curr Opin Cell Biol. 1996;8:348–353. doi: 10.1016/s0955-0674(96)80008-1. [DOI] [PubMed] [Google Scholar]
- Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, Yamamori S, Kishimoto H, Nakayama M, Tanaka Y, et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–242. doi: 10.1038/ng.2007.56. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Rozenblatt-Rosen O, Meyerson M, Manley JL. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes & development. 2007;21:1779–1789. doi: 10.1101/gad.1565207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol. 2013;20:1258–1264. doi: 10.1038/nsmb.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Viiri K, Araujo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira CF, Brookes E, Walker K, Bell GW, et al. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol Cell. 2010;38:675–688. doi: 10.1016/j.molcel.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil A, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein B, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kang K, Kim J. AEBP2 as a potential targeting protein for Polycomb Repression Complex PRC2. Nucleic Acids Res. 2009;37:2940–2950. doi: 10.1093/nar/gkp149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TG, Kraus JC, Chen J, Lee Y. JUMONJI, a critical factor for cardiac development, functions as a transcriptional repressor. J Biol Chem. 2003;278:42247–42255. doi: 10.1074/jbc.M307386200. [DOI] [PubMed] [Google Scholar]
- Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M, et al. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat Cell Biol. 2010;12:618–624. doi: 10.1038/ncb2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzuolo C, Orlando V. Memories from the polycomb group proteins. Annu Rev Genet. 2012;46:561–589. doi: 10.1146/annurev-genet-110711-155603. [DOI] [PubMed] [Google Scholar]
- Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010;24:368–380. doi: 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS genetics. 2011;7:e1002085. doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. 2010 doi: 10.1038/nature08788. [DOI] [PubMed] [Google Scholar]
- Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell. 2009;139:1290–1302. doi: 10.1016/j.cell.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puda A, Milosevic JD, Berg T, Klampfl T, Harutyunyan AS, Gisslinger B, Rumi E, Pietra D, Malcovati L, Elena C, et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. American journal of hematology. 2012;87:245–250. doi: 10.1002/ajh.22257. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annual review of biochemistry. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Y, Shadwell J, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nature reviews Genetics. 2006;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- Shen X, Kim W, Fujiwara Y, Simon MD, Liu Y, Mysliwiec MR, Yuan GC, Lee Y, Orkin SH. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell. 2009;139:1303–1314. doi: 10.1016/j.cell.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Shen SS, Margueron R, Reinberg D. Nucleosome binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. doi: 10.1101/gad.225888.113. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Okamoto A, Kobayashi R, Shirai M, Obata Y, Ogawa H, Sotomaru Y, Kono T. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Human molecular genetics. 2009;18:1879–1888. doi: 10.1093/hmg/ddp108. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Yamazaki Y, Katoh-Fukui Y, Tsuchiya R, Kondo S, Motoyama J, Higashinakagawa T. Gene trap capture of a novel mouse gene, jumonji, required for neural tube formation. Genes & development. 1995;9:1211–1222. doi: 10.1101/gad.9.10.1211. [DOI] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell. 2007;28:1–13. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.