

Abstract
Novel mutations in myelin and myelin-associated genes have provided important information on oligodendrocytes and myelin and the effects of their disruption on the normal developmental process of myelination of the central nervous system (CNS). We report here a mutation in the folliculin-interacting protein 2 (FNIP2) gene in the Weimaraner dog that results in hypomyelination of the brain and a tract-specific myelin defect in the spinal cord. This myelination disruption results in a notable tremor syndrome from which affected dogs recover with time. In the peripheral tracts of the lateral and ventral columns of the spinal cord, there is a lack of mature oligodendrocytes. A genome-wide association study of DNA from three groups of dogs mapped the gene to canine chromosome 15. Sequencing of all the genes in the candidate region identified a frameshift mutation in the FNIP2 gene that segregated with the phenotype. While the functional role of FNIP2 is not known, our data would suggest that production of truncated protein results in a delay or failure of maturation of a subpopulation of oligodendrocytes.
Keywords: Weimaraner, hypomyelination, FNIP2, autosomal recessive
Introduction
The normal myelination of the central and peripheral nervous systems (CNS and PNS, respectively) requires intricate interactions between glial cells and axons during development. The molecular control of this process is complex and the genes involved must be expressed in the correct temporal sequence for proper glial cell differentiation, migration, and myelin production to occur. An important approach to unraveling the factors involved in glial cell maturation and association with axons is the study of naturally occurring animal mutants in which the process has gone awry (Griffiths, 1996; Lunn et al., 1995; Werner et al., 1998). Collectively, these animals have been called the myelin mutants, some of which are excellent models of human disease. Thus, mutants of the proteolipid protein gene (Plp1) (e.g. the jimpy mouse, myelin deficient rat, and shaking pup) are models of Pelizaeus-Merzbacher disease (PMD) (Duncan et al., 2011; Griffiths, 1996) while mutations in the mouse peripheral myelin protein 22 gene (Pmp22) in the trembler and tremblerj mouse provide models for Charcot-Marie-Tooth disease type 1E (Scherer and Wrabetz, 2008). Mutations in novel genes associated with myelination provide new opportunities for the study of myelination in animals, and offer the possibility that similar mutations may be the cause of previously uncharacterized human myelin disorders.
We present here a novel mutation in the Weimaraner dog that is associated with an early onset of tremor and a unique zone of disruption of myelination in the spinal cord (Kornegay et al., 1987). Analyses of pedigrees of affected dogs have previously suggested that the disease is an autosomal recessive trait. This disorder appears to be widespread in the breed in the USA and has also been described in Europe (Comont et al., 1988; Millan et al., 2010). An apparently identical disorder has been described in the Chow Chow breed of dog (Vandevelde and Braund, 1981; Vandevelde et al., 1978).
To identify the causative mutation of this disease, we undertook a genome-wide association (GWA) study using DNA from three unrelated pedigrees of Weimaraners as well as unrelated animals. The gene was mapped to canine chromosome 15 within a 3.75 Mb region that contains 17 genes. As there were no clear candidate genes, all genes in the interval were sequenced. A single point mutation predicted to cause a frameshift and result in a truncated protein was found within the gene encoding folliculin-interacting protein 2 (FNIP2). The function of this protein in myelination remains to be identified but its disruption may be responsible for a delay in migration or differentiation of a subpopulation of oligodendrocyte progenitor cells (OPCs).
Materials and Methods
Animal Tissue
This study was performed using protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Wisconsin at Madison. Blood samples were obtained from breeders with affected and closely related Weimaraners. Brains and spinal cords were collected from affected dogs that were donated for research with the owners’ consent. Pups were perfused at 14 and 16 days of age (two at each time point) after euthanasia, with 4% paraformaldehyde, and the brain and spinal cord removed. The brains and spinal cords were trimmed and sections cut for myelin staining or for in situ hybridization using a 35S-labeled riboprobe to Plp1 and methods previously described (Duncan and Hoffman, 1997). Brain sections were also labeled for myelin basic protein using a previous protocol (O’Connor et al., 2000). The spinal cords were trimmed and processed for embedding in plastic resin and 1 micron sections were stained with toluidine blue. Affected Chow pups were perfused at 2, 3, 5, and 8 weeks of age (one at each time point). Affected dogs from the mating of recovered (homozygous) Weimaraner and Chow were similarly perfused and spinal cords collected. We also had access to tissue from 1-year and 2-year-old recovered Chows.
Genotype Data
We selected 48 Weimaraners for genotyping: 35 from three unrelated pedigrees (Fig. 1) (9 affected animals, 18 known carriers, and 8 phenotypically normal “unaffected” animals who may or may not be carriers) and 13 unrelated “singleton” animals (five affected animals, one known carrier, and seven phenotypically normal animals). DNA was isolated from blood using the Wizard Genomic DNA Purification Kit (Promega Corp., Madison, WI). DNA concentrations were checked using Quant-iT PicoGreen (Invitrogen, Carlsbad, CA) and standardized to 80 ng/μL in TE buffer. Genotyping was performed with 5 μL (400 ng) DNA using the Illumina CanineHD BeadChip (Illumina Inc., San Diego, CA) at the Southern California Genotyping Core facility (University of California, Los Angeles, CA) following the manufacturer’s instructions. SNP genotypes were called following the manufacturer’s recommended protocol using the Illumina GenomeStudio (version 1.0) software. Genotypes from these 48 animals were then combined with genotype data from an additional 36 animals who did not present with the tremor phenotype that had been previously genotyped on the Illumina CanineHD BeadChip. Genotypes were obtained for each of the 84 animals (40 males and 44 females) in the combined data set at greater than 99.34% of the 172,115 SNP markers.
FIGURE 1.

The three Weimaraner pedigrees studied. The symbols on the right identify the gender, phenotype and those dogs from which the DNA was obtained.
As the tremor phenotype showed a clear autosomal recessive pattern of inheritance in the three pedigrees (Fig. 1), our analysis considered only the 166,583 SNP markers located on the 38 autosomal chromosomes. The autosomal SNP genotypes were filtered to remove lower-quality SNPs: we excluded 75 SNPs that had at least one Mendelian inconsistency in the data set, 69,429 SNPs that had a minor allele frequency less than 5%, and 1,089 SNPs that had missing genotypes in ≥10% of the animals. The final genotype data set used in the genome-wide association (GWA) analysis was comprised of 95,990 high-quality autosomal SNPs for which we had genotypes in 84 animals (40 males and 44 females) of whom 14 were affecteds, 19 were known carriers, and 51 were unaffecteds.
Genome-Wide Association and Haplotype Homozygosity Analyses
We performed genome-wide association mapping to identify the region harboring the causative genetic mutation using EMMAX (Kang et al., 2010), which corrects for sample stratification (e.g. cryptic relatedness) and the nonindependence of related individuals using a pairwise identity-by-state (IBS) kinship matrix. A region of association spanning from 57.3 to 62.3 Mb on chromosome 15 was identified. Within this region, homozygous segments in each animal were defined as runs of homozygous genotypes (ROH) identified using the homozyg option in PLINK (Purcell et al., 2007) (v.1.07) with a sliding window size of 50 SNPs and default criteria; ≤1 heterozygous genotype and ≤5 missing genotypes required for a window to be called homozygous, and the proportion of overlapping windows that must be called homozygous to define any given SNP as in an ROH of ≥0.05. Using this approach, ROH were identified in 45 of the 84 animals, including all 14 affecteds. Homozygous haplotype analysis defined a critical interval from 57.3 to 61.0 in which all affected animals shared a single homozygous haplotype. To rule out the presence of a copy number variant (CNV) as the possible cause of the observed haplotype homozygosity, potential CNVs were identified in each animal using PennCNV (Wang et al., 2007) (v.2010May01) from the normalized intensity data (log R ratio) and allele frequency data (B allele frequency) of each SNP calculated using the Illumina GenomeStudio (version 1.0) software.
Mutation Analysis of Candidate Genes
We used the Table View option in NCBI Map Viewer (Build 2.2) to identify all possible genes that mapped to the candidate interval on chromosome 15. Primers were designed manually to amplify every exon of each gene with the parameters of requiring them to be at least 20 nucleotides in length and have a GC content of 50%. Amplified exons were subjected to electrophoresis on agarose gels, and if multiple products were observed the single band of the expected size was purified using the Quick Gel extraction and PCR purification combo kit (Life Technologies, Inc., Carlsbad, CA); if only a single band was observed, amplified exons were directly purified using Econospin columns (Epoch Biolabs, Houston, TX). Purified PCR products were sequenced by dideoxy-sequencing and the resulting sequences aligned against the CanFam2 reference sequence using Genetyx-Mac (v. 12.0.7) to identify variants. To determine carrier frequency of the mutation identified in exon 8 of the FNIP2 gene, the primers, FNIP2_Forward: 5′-CATCCCCTGGCATTACAGTT-3′ and FNIP2_Reverse: 5′-GGAAATTCCTTTGTGCGTCT-3′ were used to generate PCR products of 208 bases that flank the I294L mutation. PCR was carried out in a 20 μL reaction volume containing 40ng DNA, 0.2 mM dNTPs, 0.8 μM forward and reverse primers in 10× PCR buffer, and 1 unit AmpliTaq Gold® polymerase (Applied Biosystems, Foster City, CA). PCR conditions were as follows: 5 mins at 95°C followed by 35 cycles of 30 s at 95°C, 30 s at 60°C, and 1 min at 72°, with a final extension of 10 min at 72°C. PCR products were purified as described above, and sequenced using the Big Dye® terminator mix on an ABI 3500 Genetic Analyzer. Sequence alignment against the reference FNIP2 gene sequence was performed using the Vector NTI software (Life Technologies, Grand Island, NY).
Expression of FNIP2 in OPCs
Rat CG4 oligodendrocyte progenitor cells were cultured as previously described (Bourikas et al., 2010) except for supplementation with either 5% bovine growth serum (Hyclone) or 10% fetal bovine serum and passaged twice weekly. Three to five million confluent cells were independently transfected with 40 nmols of either Ambion (Life Technologies, Grand Island, NY) control siRNA (siControl #2, catalog #AM4613) or Sox10 siRNA (siSox10 #1, catalog #s131239) using the Amaxa Nucleofection system as reported previously (Gokey et al., 2012; Jones et al., 2011). For gene expression analysis, RNA was purified from CG4 cells 48h after transfection using Ambion Tri Reagent (Life Technologies, Grand Island, NY), and cDNA synthesized and analyzed by quantitative RT-PCR using the StepOne Plus system (Applied Biosystems, Foster City, CA). Relative amounts of each gene were determined by the comparative Ct method and normalized with 18S rRNA (Livak and Schmittgen 2001). RT-qPCR primer sequences are as follows: 18S rRNA, 5′-CGCCGCTAGAGGTGAAATTCT-3′ and 5′-CCAACCTCCGACTTTCGTTCT-3′; Sox10, 5′-CGAATTGGGCAAGGTCAAGA-3′ and 5′-CACCGGGAACTTGTCATCGT-3′; Fnip2, 5′-GCGATGAGAAGCTGAAGCAGT-3′ and 5′-TGCACCGTGTGGACCAGAT-3′.
Results
Clinical Findings
A generalized tremor was first observed when the animals were between 12 and 14 days of age. The severity of the tremor varied between litter members. However, all affected pups were able to ambulate, usually with a “hopping” gait in the hind limbs. The tremor was not present when the pups were at rest or asleep and diminished with time by 3 to 4 months of age, though some dogs retained a persistent fine tremor of the hind limbs. The disease was similar in Chow pups but was more severe and persisted longer, often up to 4 to 5 months of age.
In the litter produced by the mating of the recovered, homozygous Weimaraner (male) and Chow (female), five pups were born. Two pups developed a mild tremor around 3 weeks of age, while two appeared normal. The fifth pup died shortly after birth.
Developmental Myelin Changes in the CNS
As previously described (Kornegay et al., 1987), the hallmark abnormality in affected Weimaraners is the peripheral zone of dysmyelination in the lateral and ventral columns of the spinal cord, most notably at the cervical and thoracic levels (Fig. 2). Myelination of these areas increased between 14 and 26 days, especially in the ventral column, but the dorsal spinocerebellar tract of the dorsolateral column was most severely affected and remained notably dysmyelinated. The sub-pial area had a normal-appearing glial cell density and cells with the light microscopic appearance of oligodendrocytes and astrocytes were present. However, PLP in situ hybridization showed that there were fewer mature oligodendrocytes than in the adjacent deeper white matter of the cord or in the same sub-pial areas of controls (Fig. 2). The brains of affected pups showed less myelin throughout the white matter than controls (Fig. 2), though no tract or focal disruptions in myelination were seen. The folia as well as the medulla of the cerebellum of affected dogs had fewer mature oligodendrocytes when compared with controls as seen by in situ hybridization with the PLP probe (Fig. 2).
FIGURE 2.

Myelin defects in the brain and spinal cord. A–D Thoracic spinal cord from a normal 25-day-old dog (A and C) and an age-matched affected dog (B and D). In the affected dog (B), there is an obvious zone of disrupted myelination beneath the pia in the lateral and ventral columns while the dorsal columns appear normal. On higher power of the lateral column (D) there are only scattered myelinated axons compared with the control (C) which is normally myelinated. Bars = 0.5 mm (A and B), 20 μm (C and D). E–H In situ hybridization from the spinal cord (E and F) and cerebellum (G and H) of a 4-week-old normal dog (E and G) and age-matched affected dog (F and H), labeled with a PLP antisense riboprobe. There are fewer labeled cells (mature oligodendrocytes) in the periphery of the spinal cord (F) and cerebellar folia (H) in affected dogs than in controls. Bars = 10 μm. I and J Coronal sections of whole brain from a 5-week-old normal Weimaraner (I) and an affected dog (J). In the affected dog, there is a decrease in staining density of myelin throughout the brain (as seen in the corpus callosum), compared with controls suggesting a generalized paucity of myelin. Bars = 0.5 mm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Study of the myelination defect in the spinal cord of affected Chows showed that the myelination defect was more severe and persisted longer than in the Weimaraner (Fig. 3). Study of Chows was additionally informative as more time points were available than for the Weimaraners. There was a slow but progressive increase in myelin from 2 weeks to 2 years, though at 11 months, the dorsolateral tracts contained mainly hypomyelinated axons and at 2 years there still appeared to be a major difference in myelin sheath thickness between the affected dog and controls. Likewise, axons of the deep white mater of the cord were more thickly myelinated than those at the periphery. Four out of five pups resulting from the mating of the recovered (homozygous) Chow with the Weimaraner were studied. All four had a notable but variable defect in myelination of the superficial dorsolateral tracts of the cervical and thoracic cord (data not shown).
FIGURE 3.
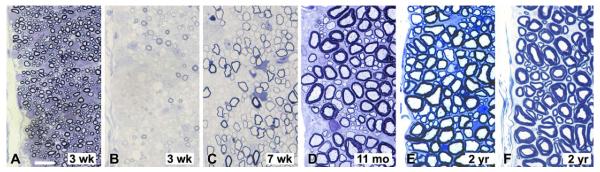
Partial recovery of myelination with time. The spinocerebellar tract in the dorsolateral column from control dogs at 3 weeks (A) and 2 years (F) and affected Chows at 3 (B) and 7 weeks (C), 11 months (D) and 2 years (E). At 3 weeks, very little myelin is present in the mutant (B) while the control dog is normally myelinated at this time. While more axons become myelinated with time (C–E), at 2 years of age axons are clearly more thinly myelinated in the mutant (D and E) than in the control (F). Scale bar for A-F = 20 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Long-Term Changes in Myelin in the Chow
Two mature Chows were studied, one aged 11 months, the other 2 years. Both had severe tremor as pups but recovered over a 3- to 4-month period, though they retained a persistent positional tremor of the hind legs. In the case of the 11-month-old dog, the superficial tracts of the ventral and lateral columns of the spinal cord were clearly hypomyelinated compared with the deeper white matter and the dorsal columns (Figs. 3 and 4). A striking difference from the younger dogs however was the notable presence of scattered vacuolated myelin sheaths throughout these hypomyelinated areas only (Fig. 4C), most of which contained intact axons. Occasional degenerate axons were seen. Many nonmyelinated axons were seen, predominately in the ventral column (Fig. 4). In the dorsolateral column, most axons were myelinated but surrounded by much thinner myelin sheaths than in controls. In the trapezoid body in the brain stem, many axons close to the pia were not myelinated suggesting even slower myelin repair than in the spinal cord (Fig. 5). The myelin defect was less obvious in the 2-year-old Chow though many thinly myelinated fibers were present, especially in the ventral and dorsolateral columns of the cervical and thoracic cord (Figs. 3 and 6A). Vacuolated myelinated fibers were noted but were less frequent than at the 11 month time point. However, there was clear evidence of myelin breakdown with preservation of axons within these vacuoles (Fig. 6), confirming a demyelinating component to the disease. Indeed, scattered medium and large diameter demyelinated axons were seen in peripheral white matter of the ventral and lateral columns as well as axons with thin myelin sheaths thought to represent remyelination (Fig. 6).
FIGURE 4.

Myelin vacuolation develops with time and axons remain non-myelinated. (A) The ventral column of a control mature dog shows even myelination throughout the white matter compared with an 11-month-old Chow that has a peripheral zone of abnormality of the superficial tracts (B). On higher power (C) many myelin vacuoles are seen close to the pia. Three differing patterns of myelination are present in the ventral column (1, 2, 3 arrows) shown on higher power below. The deep white matter is normally myelinated (1) while an adjacent area (2) contains mainly hypomyelinated fibers, and closer to the ventral surface of the cord (3), many axons remain nonmyelinated. Scale bars, A and B = 0.5 mm, C = 20 μm, 1–3 = 20 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FIGURE 5.
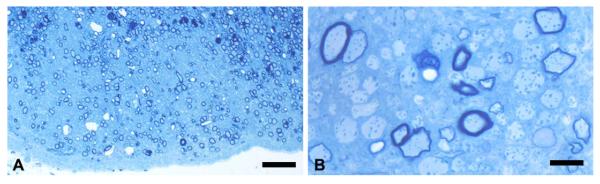
Myelin deficits persist in the brain of the mature dog. The trapezoid body close to the pia in the 11 month old Chow. This structure—which is normally completely myelinated—shows a paucity of myelin (A) and on higher power, many non-myelinated axons can be seen (B). Scale bars, A = 20 μm, B = 10 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FIGURE 6.
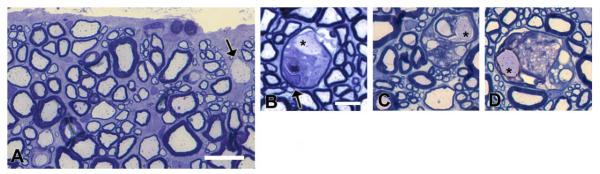
Late-onset myelin degeneration with axonal preservation. In the 2-year-old Chow, demyelinated axons (arrow) are scattered throughout areas of previous hypomyelination with numerous thinly myelinated axons suggestive of remyelination (A). Myelin vacuoles are present and in many of these, degenerating myelin is seen (B–D) but the axon appears intact (*). In one instance a likely macrophage (B, arrow) is seen within the myelin sheath. Scale bars, A = 10 μm, B–D = 5 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Genome-wide association analysis
In order to map the locus underlying the myelination defect, a genome-wide association analysis was performed using 95,990 autosomal SNPs with genotypes in 84 Weimaraner (14 affected, 19 known carriers, and 51 unaffected). A significant association was detected for a region on the q-arm of chromosome 15 (Fig. 7A) over a 5 Mb region from 57,271,315 to 62,270,464 bp (Fig. 7B). The lowest P value was obtained with SNP BICF2S23612490 (P = 1.79 × 10−14) at position 59,514,062 bp. Inspection of the log R ratio and B Allele Frequency values across this 5 Mb region in each animal ruled out the presence of a CNV as the possible cause of the disease in these animals (data not shown). Haplotype homozygosity analysis identified a 3.75 Mb homozygous region (57,271,315–61,016,318 bp) in which all affected dogs were homozygous for a single haplotype (Fig. 7B, grey box).
FIGURE 7.

Genome-wide association maps disease gene to chromosome 15. (A) Manhattan plot of −log10-transformed P values obtained upon GWA analysis of genotypes at 95,990 autosomal SNPs in the 84 Weimaraner samples. A single peak is evident on chromosome 15q with the most strongly associated SNP at 59,514,062 bp (BICF2S23612490, P = 1.79 × 10−14). (B) Locuszoom plot (Pruim et al., 2010) of critical interval on chromosome 15q and candidate genes within the interval. Top, −log10-transformed P values at individual SNPs colored by the strength of their correlation (r2) with the most significant SNP in the region (BICF2S23612490, colored purple). Also shown are the proportion of affected (solid blue line) and unaffected (dashed blue line) animals that were homozygous for the common haplotype in affected animals. Bottom, genes in the region, with the 3.75 Mb region in which all affected animals are homozygous for a single haplotype shown by the grey box and the gene containing the causal mutation is identified in red. Pairwise r2 were estimated using unrelated unaffected animals only. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Identification of a Mutation Underlying Hypomyelination and Tremor in the Weimaraner
The critical interval defined by association mapping of the hypomyelination phenotype extended from 57.27 Mb to 62.27 Mb on chromosome 15 contained 17 genes (Fig. 7B). We initially sequenced GRIA2 as it was the only gene known to exhibit clear and functional CNS expression, however, no mutations were identified. We therefore designed 338 primers to amplify all 199 putative exons of the remaining 16 genes for sequencing. Fifty-four variants were identified; all except one were either in an intron, in the untranslated regions, or resulted in no change in the amino acid sequence. Only one variant was found to have deleterious consequences on the protein and consisted of a deletion of a single A nucleotide within exon 9 of the gene encoding folliculin-interacting protein 2 (FNIP2). This mutation at nucleotide 880 of the coding sequence (c.880delA, XM_532705) causes a frameshift, and the amino acid sequence is altered beginning at codon 294, with a premature translation stop signal introduced at codon 296 (Fig. 8). A highly truncated protein of 295 amino acids compared with the wild-type protein of 1,106 amino acids is predicted to result from this p.Ile294fsX296 mutation.
FIGURE 8.

A single base pair deletion within exon 9 of the FNIP2 gene is associated with the disease. (A) Schematic of the canine FNIP2 gene with vertical bars representing exons of the gene and the width representative of the size of the exon, the lines between the exons representing the intronic sequence that is spliced to create the mature mRNA, and the position of the mutation within the genomic sequence indicated below the gene. (B) Chromatograms of an unaffected normal, a carrier, and an affected dog are shown with the vertical grey bar identifying the “A” nucleotide that is deleted on one haplotype in the carrier, and on both haplotypes in the affected dog. The amino acid residue affected by the mutation is shown in red in the amino acid sequence above the DNA sequence. The asterisk denotes the premature stop codon created by the frameshift mutation. (C) Schematic of the FNIP2 protein showing the PRKAA1 interaction domain. The red bar identifies the truncated protein created by the frameshift mutation. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DNA from 49 dogs, including the 48 that had been used for the initial genome scan and one additional “control” dog, were examined for the presence of the p.Ile294fsX296 mutation by DNA sequencing. All 14 affected dogs were homozygous for the mutation, 8 obligate and 15 predicted carriers were heterozygous for the mutation, and four unaffected animals from the two pedigrees as well as seven of eight unrelated “control” dogs did not bear the mutation. One unrelated “control” dog was found to be heterozygous for the mutation. In addition, 105 unrelated Weimaraners were tested to estimate the frequency of the FNIP2 I294L mutation within the breed. Nine out of 105 dogs had a single copy of the I294L mutation, providing an estimated carrier frequency of 4.285%. An additional 102 dogs from different breeds were also tested and no copies of the FNIP2 I294L mutation were identified within this sample set.
To assess the expression pattern of FNIP2, we examined the expression of its murine orthlogue in a previously published microarray data set of CNS gene expression based on sorting of cell types into neurons, astrocytes, and oligodendrocytes (Cahoy et al., 2008). This analysis showed that FNIP2 was expressed in oligodendrocytes and neurons at roughly equivalent, albeit relatively modest, levels (data not shown); expression in astrocytes was very minimal.
To probe this observation further, we analyzed the expression pattern in the CG4 oligodendrocyte cell line (Louis et al., 1992). Moreover, since Sox10 is a major determinant of oligodendrocyte differentiation, we used siRNA directed against Sox10 to investigate whether Fnip2 expression is regulated by this critical transcription factor. Quantitative RT-PCR analysis revealed expression of Fnip2 in CG4 cells, and depletion of Sox10 resulted in a 2-fold reduction of Fnip2 expression (Fig. 9). These data suggest that Fnip2 expression is regulated by Sox10 in a manner similar to that of many other myelin-associated genes in oligodendrocytes (Li et al., 2007; Stolt et al., 2002).
FIGURE 9.

Quantitative RT-PCR analysis to detect expression of levels of Fnip2 in CG4 oligodendrocyte cells transfected with either control or Sox10 siRNA. Data show average transcript levels of Fnip2 from three independent transfections (P < 0.05). Error bars indicate standard deviations.
Discussion
We have identified a mutation in the gene encoding folliculin-interacting protein 2 (FNIP2) in the Weimaraner breed of dog (with a likely synergistic allele present in the Chow breed) that results in defective myelination of the central nervous system. While there is a wealth of knowledge about genes that play a role in myelination, including those that encode proteins that are structural components of myelin or that encode factors that regulate the expression of myelin genes, there is still much that needs to be elucidated about the complex events that determine this process. Thus, identification of novel genes involved in oligodendrocyte development and myelination is instructive.
The single base deletion within FNIP2 is predicted to result in a highly truncated protein and thus, result in a complete absence of functional FNIP2. The fact that the tremor can abate in some affected dogs is puzzling given the severity of the mutation. Since a temporal examination of the myelination status in affected dogs is not practical, further examination of this phenomenon may require such an analysis in smaller animal models such as a mouse where FNIP2 expression is ablated.
To date, the only definitive dysmyelination/hypomyelination disorder in humans is Pelizaeus-Merzbacher disease (PMD)/spastic paraplegia Type II that is caused by duplications or point mutations of the proteolipid protein (Plp1) gene (Griffiths, 1996). Plp1 is located on the X chromosome, hence only males are affected and the PLP protein is severely diminished in the CNS. Immunolabeling for both PLP and MBP revealed normal staining of myelinated axons (data not shown), hence both the genetics (non-X-linked as seen in PMD) and their normal expression ruled these out of consideration in the Weimaraner. Cases (both males and females) with PMD-like (PMDL) disease have been described. Eight percent of these cases have mutations, inherited in an autosomal recessive manner, in the gap junction protein, gamma-2 gene (GJC2) (Hobson and Garbern, 2012). It may be worthwhile examining those cases of PMDL that do not have a molecular diagnosis for mutations in the FNIP2 gene. Other white matter disorders in children, with the potential exception of Alexander’s disease (Messing et al., 2012), fall within the category of demyelinating leukodystrophies (Aicardi, 1993; Costello et al., 2009; Perlman and Mar, 2012) and hence are dissimilar to the disease of the Weimaraner and Chow dog breeds.
A previous transcriptome analysis of purified neurons, astrocytes, and oligodendrocytes shows that Fnip2 is expressed in both neurons and oligodendrocytes (Cahoy et al., 2008). Moreover, we found that Fnip2 is expressed in the CG4 oligodendrocyte cell line (Stolt and Wegner, 2010 Stolt et al., 2002), and its expression is diminished after siRNA-mediated reduction of Sox10, a major regulator of oligodendrocyte differentiation (Stolt et al., 2002). These data are consistent with a role of Fnip2 in oligodendrocyte function, although the region-specific nature of the myelin defect suggests that it is not absolutely required for myelination. Interestingly, the microarray data (Cahoy et al., 2008) indicate a similar expression level of the related Fnip1 gene in oligodendrocytes and neurons, which may partially compensate for the loss of Fnip2 activity. FNIP1 and FNIP2 interact with folliculin and this interaction is important in signaling events involving 5′AMP-activated protein kinase (AMPK), a negative regulator of the mammalian regulator of rapamycin (mTOR) (Baba et al., 2006). Activation of mTOR has been found to be essential for OPC differentiation (Tyler et al., 2009; Wood et al., 2013) and also for normal Schwann cell myelination of the PNS (Sherman et al., 2012). Although much earlier in development, it has recently been shown that folliculin and FNIP1/2 act through the negative regulation of the bHLH transcription factor, Tfe3, to maintain pluripotency in embryonic stem cells (Betschinger et al., 2013). In contrast, Tcf3, which is known to be involved in OPC differentiation (Kim et al., 2011), has the opposite effect on the transcriptional regulation of Estrogen-related receptor beta (Esrrb), a known factor involved in the maintenance of pluripotency in embryonic stem cells (Zhang et al., 2008). Thus, FNIP2 signaling may be implicated in a number of pathways early in neural development and at later times of OPC differentiation.
The unique nature of the myelin defect in the spinal cord may help explain the function of FNIP2 in myelination. Three potential defects in oligodendrocyte development could account individually, or in combination, for the spatial distribution of the defect. First, there could be premature death of the OPCs that migrate to the sub-pial zones of the ventral and lateral columns. Second, there could be a failure or delay in migration of OPCs toward this zone. Third, OPCs migrate normally to sub-pial zones but then do not differentiate. The first hypothesis seems unlikely as very few pyknotic cells were seen in the subpial neuropil at any time point examined (data not shown). While TUNEL labeling has not been performed, there were no more pyknotic nuclei than in control dogs, nor nuclei with chromatin margination indicating apoptosis. It seems unlikely that a failure in migration of OPCs is the underlying defect as the non/poorly myelinated zones contain many glial cell nuclei with variable appearance (i.e. not all are astrocytes; Fig. 2). However, we do not have a good marker of OPCs in the dog that would confirm this; therefore a deficiency in migration remains a possibility. A previous study demonstrated a decrease in Olig1-positive cells in the periphery of the spinal cord in Weimaraners (Millan et al., 2010). We show here a decrease in PLP-positive cells in the areas of hypomyelination (Fig. 2) suggesting that a delay or failure of oligodendrocyte differentiation occurs. This was also noted in the cerebellar folia.
Studies on the origin and spatial dissemination of OPCs in the mammalian and avian spinal cord have shown that they arise from a germinal zone below the central canal and from there migrate to the ventral and lateral columns where they differentiate and myelinate axons (Miller, 2013; Pringle and Richardson, 1993). The dorsal columns are populated by OPCs that are derived both from this ventral source and also from the dorsal column (Cai et al., 2005; Fogarty et al., 2005; Tripathi et al., 2011; Vallstedt et al., 2005). An earlier study of myelination in the developing cat spinal cord (Windle et al., 1934) reported a spatial pattern of myelination that would match the distribution of OPCs described above in the rodent spinal cord. We have examined the temporal-spatial pattern of myelination in the late-fetal/early postnatal stage of the canine spinal cord, and have shown that it matches the pattern seen in the mouse and rat (Ian Duncan unpublished data). Thus, it would seem that the most severe defect seen in the dorsal spinocerebellar tract matches the most distant point of migration of OPCs. In some affected Weimaraners, a small triangular area of nonmyelination was seen in the sub-pial zone of the fasciculus gracilis, which may also be the furthest migration point of OPCs in the dorsal column, and cells that migrate there may be delayed in their differentiation.
While the primary defect in myelination is discussed above, the question of the mechanism of repair of the defect is also unclear, and whether this delayed myelination is complete. In this regard, the myelin abnormalities seen in the two longer lived Chows are informative. Though not formally proven here, we strongly believe the disorder in this breed is allelic to the Weimaraner mutation as, (1) they share the unique spinal cord myelin defect and (2) breeding of homozygous dogs from each breed produced affected pups. In the Chow, the spinal cord myelination defect is more severe than in the Weimaraner and takes longer to recover as reflected clinically by prolonged tremor. This delay extended into adulthood as seen in the 11-month-old Chow where there were still many nonmyelinated axons in the ventral column of the spinal cord (Fig. 4). Indeed, large areas from this column appeared to be persistently hypomyelinated. However, superimposed on this myelin maturation delay was the presence of numerous scattered myelin vacuoles, in which the axons appeared intact. Such vacuolation is a nonspecific myelin change and is seen in many disorders of myelin, most notably in a recent demyelinating disorder described in the cat (Duncan et al, 2009). In the feline disorder, these vacuoles can lead to demyelination that may indeed be the case in the Chow. These changes were only seen in the areas of delayed myelination and demarcated the spinal cord into two areas of myelin, the deeper tracts which are normally myelinated postnatally in affected dogs, and the superficial tracts that are the target of the mutation. By 2 years of age there was no longer large confluent areas of hypomyelination, though many thinly myelinated axons remained along with occasional myelin vacuoles. As only one animal was available at each time point, caution must be observed in the interpretation of these findings and a detailed quantitative study and statistical analyses of myelin sheath thickness were not feasible. However, our data have similarity to those reported previously. Vandevelde et al. (1981) described two similar aged Chows in which they noted but did not illustrate the presence of vacuoles in areas of delayed myelination, and myelin recovery in the older dog. In contrast, we show that myelin sheath thickness does not fully recover and late-stage myelin breakdown with demyelination and subsequent remyelination occurs. These observations add complexity to the role of FNIP2 in myelin development and preservation beyond the early initiation of myelination as we have discussed above.
The unique nature of this myelination defect raises the possibility that it may be useful to search for FNIP2 mutations in children with developmental defects in myelination whose genetic basis is unknown. Thus the sequencing of FNIP2 in such cases may help identify the defect and, given the temporary nature of the disruption in myelination in the canine defect, help provide a favorable prognosis for affected children.
Acknowledgment
Grant sponsor: AKC Canine Health Foundation (to P.I.P. and I.D.D.), University of Michigan Center for Genetics in Health and Medicine (to T.J.P.), and NIAID R21AI090277 (to D.L.B.).
The authors are grateful to many Weimaraner breeders in the USA for supplying information on this disease and blood for DNA, in particular Amy Fast. The authors thank Sarah Martin for technical assistance and Abigail Radcliff for preparation of the article.
References
- Aicardi J. The inherited leukodystrophies: A clinical overview. J Inherit Metab Dis. 1993;16:733–743. doi: 10.1007/BF00711905. [DOI] [PubMed] [Google Scholar]
- Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, Esposito D, Gillette WK, Hopkins RF, III, Hartley JL, Furihata M, Oishi S, Zhen W, Burke TR, Jr, Linehan WM, Schmidt LS, Zbar B. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, Smith A. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell. 2013;153:335–347. doi: 10.1016/j.cell.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourikas D, Mir A, Walmsley AR. LINGO-1-mediated inhibition of oligodendrocyte differentiation does not require the leucine-rich repeats and is reversed by p75(NTR) antagonists. Mol Cell Neurosci. 2010;45:363–369. doi: 10.1016/j.mcn.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Qi YC, Hu XM, Tan M, Liu ZJ, Zhang JS, Li Q, Sander M, Qiu M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron. 2005;45:41–53. doi: 10.1016/j.neuron.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Comont PSV, Palmer AC, Williams AE. Weakness associated with spinal subpial myelopathy in a weimaraner puppy. JSmall Anim Pract. 1988;29:367–372. [Google Scholar]
- Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: Classification, diagnosis, and treatment. Neurologist. 2009;15:319–328. doi: 10.1097/NRL.0b013e3181b287c8. [DOI] [PubMed] [Google Scholar]
- Duncan ID, Hoffman RL. Schwann cell invasion of the central nervous system of the myelin mutants. J Anat. 1997;190:35–49. doi: 10.1046/j.1469-7580.1997.19010035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan ID, Kondo Y, Zhang SC. The myelin mutants as models to study myelin repair in the leukodystrophies. Neurotherapeutics. 2011;8:607–624. doi: 10.1007/s13311-011-0080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty M, Richardson WD, Kessaris N. A subset of oligodendrocytes generated from radial glia in the dorsal spinal cord. Development. 2005;132:1951–1959. doi: 10.1242/dev.01777. [DOI] [PubMed] [Google Scholar]
- Gokey NG, Srinivasan R, Lopez-Anido C, Krueger C, Svaren J. Developmental regulation of microRNA expression in Schwann cells. Mol Cell Biol. 2012;32:558–568. doi: 10.1128/MCB.06270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths IR. Myelin mutants: Model systems for the study of normal and abnormal myelination. Bioessays. 1996;18:789–797. doi: 10.1002/bies.950181005. [DOI] [PubMed] [Google Scholar]
- Hobson GM, Garbern JY. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol. 2012;32:62–67. doi: 10.1055/s-0032-1306388. [DOI] [PubMed] [Google Scholar]
- Jones EA, Lopez-Anido C, Srinivasan R, Krueger C, Chang LW, Nagarajan R, Svaren J. Regulation of the PMP22 Gene through an Intronic Enhancer. J Neurosci. 2011;31:4242–4250. doi: 10.1523/JNEUROSCI.5893-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Chung AY, Kim D, Kim YS, Kim HS, Kwon HW, Huh TL, Park HC. Tcf3 function is required for the inhibition of oligodendroglial fate specification in the spinal cord of zebrafish embryos. Mol Cells. 2011;32:383–388. doi: 10.1007/s10059-011-0152-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornegay JN, Goodwin MA, Spyridakis LK. Hypomyelination in Weimaraner dogs. Acta Neuropathol. 1987;72:394–401. doi: 10.1007/BF00687272. [DOI] [PubMed] [Google Scholar]
- Li H, Lu Y, Smith HK, Richardson WD. Olig1 and Sox10 interact synergistically to drive myelin basic protein transcription in oligodendrocytes. J Neurosci. 2007;27:14375–14382. doi: 10.1523/JNEUROSCI.4456-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Louis JC, Magal E, Muir D, Manthorpe M, Varon S. CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J Neurosci Res. 1992;31:193–204. doi: 10.1002/jnr.490310125. [DOI] [PubMed] [Google Scholar]
- Lunn KF, Fanarraga ML, Duncan ID. Myelin mutants: New models and new observations. Microsc Res Tech. 1995;32:183–203. doi: 10.1002/jemt.1070320303. [DOI] [PubMed] [Google Scholar]
- Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012;32:5017–5023. doi: 10.1523/JNEUROSCI.5384-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan Y, Mascort J, Blanco A, Costa C, Masian D, Guil-Luna S, Pumarola M, Martin de Las Mulas J. Hypomyelination in three Weimaraner dogs. J Small Anim Pract. 2010;51:594–598. doi: 10.1111/j.1748-5827.2010.00997.x. [DOI] [PubMed] [Google Scholar]
- Miller RH. Development of oligodendrocytes in the vertebrate CNS. In: Duncan ID, Franlin RJM, editors. Myelin repair and neuroprotection in multiple sclerosis. Springer; New York: 2013. pp. 1–21. [Google Scholar]
- O’Connor LT, Goetz BD, Couve E, Song J, Duncan ID. Intracellular distribution of myelin protein gene products is altered in oligodendrocytes of the taiep rat. Mol Cell Neurosci. 2000;16:396–407. doi: 10.1006/mcne.2000.0889. [DOI] [PubMed] [Google Scholar]
- Perlman SJ, Mar S. Leukodystrophies. In: Ahmad SI, editor. Neurode-generative diseases. Landes Bioscience; Austin, TX: 2012. p. xxxi. [Google Scholar]
- Pringle NP, Richardson WD. A singularity of PDGF alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development. 1993;117:525–533. doi: 10.1242/dev.117.2.525. [DOI] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, Krols M, Wu LM, Grove M, Nave KA, Gangloff YG, Brophy PJ. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J Neurosci. 2012;32:1817–1825. doi: 10.1523/JNEUROSCI.4814-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Wegner M. SoxE function in vertebrate nervous system development. Int J Biochem Cell Biol. 2010;42:437–440. doi: 10.1016/j.biocel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Tripathi RB, Clarke LE, Burzomato V, Kessaris N, Anderson PN, Attwell D, Richardson WD. Dorsally and ventrally derived oligodendrocytes have similar electrical properties but myelinate preferred tracts. J Neurosci. 2011;31:6809–6819. doi: 10.1523/JNEUROSCI.6474-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WA, Gangoli N, Gokina P, Kim HA, Covey M, Levison SW, Wood TL. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29:6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallstedt A, Klos JM, Ericson J. Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron. 2005;45:55–67. doi: 10.1016/j.neuron.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Vandevelde M, Braund KG. Dysmyelination in Chow Chow dogs: further studies in older dogs. Acta Neuropathol. 1981;55:81–87. doi: 10.1007/BF00699232. [DOI] [PubMed] [Google Scholar]
- Vandevelde M, Braund KG, Walker TL, Kornegay JN. Dysmyelination of the central nervous system in the Chow-chow dog. Acta Neuropathol. 1978;42:211–217. doi: 10.1007/BF00690359. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner H, Jung M, Klugmann M, Sereda M, Griffiths IR, Nave KA. Mouse models of myelin diseases. Brain Pathol. 1998;8:771–793. doi: 10.1111/j.1750-3639.1998.tb00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windle WF, Fish MW, O’Donnell JE. Myelogeny of the cat as related to development of fiber tracts and prenatal behavior patterns. J Comp Neurol. 1934;59:139–165. [Google Scholar]
- Wood TL, Bercury KK, Cifelli SE, Mursch LE, Min J, Dai J, Macklin WB. mTOR: A link from the extracellular milieu to transcriptional regulation of oligodendrocyte development. ASN Neuro. 2013;5:e00108. doi: 10.1042/AN20120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang J, Wang T, Esteban MA, Pei D. Esrrb activates Oct4 transcription and sustains self-renewal and pluripotency in embryonic stem cells. J Biol Chem. 2008;283:35825–35833. doi: 10.1074/jbc.M803481200. [DOI] [PubMed] [Google Scholar]