

Abstract
Brucella are facultative intracellular bacteria that cause chronic infections by limiting innate immune recognition. It is currently unknown whether Brucella FliC flagellin, the monomeric subunit of flagellar filament, is sensed by the host during infection. Here, we used two mutants of Brucella melitensis, either lacking or overexpressing flagellin to show that FliC hinders bacterial replication in vivo. The use of cells and mice genetically deficient for different components of inflammasomes suggested that FliC was a target of the cytosolic innate immune receptor NLRC4 in vivo but not in macrophages in vitro where the response to FliC was nevertheless dependent on the cytosolic adaptor ASC, therefore suggesting a new pathway of cytosolic flagellin sensing. However, our work also suggested that the lack of TLR5 activity of Brucella flagellin and the regulation of its synthesis and/or delivery into host cells are both part of the stealthy strategy of Brucella towards the innate immune system. Nevertheless, since a flagellin-deficient mutant of B. melitensis was found to cause histologically demonstrable injuries in the spleen of infected mice, we suggested that recognition of FliC plays a role in the immunologic standoff between Brucella and its host, which is characterized by a persistent infection with limited inflammatory pathology.
Introduction
The mammalian innate immune system relies on a limited number of pattern recognition receptors (PRRs) to detect microbial-derived molecules during infection and subsequently trigger an appropriate immune response to the invading pathogen. These microbial features are often referred to as PAMPs for pathogen-associated molecular patterns. The PRRs include toll-like receptors (TLRs), which sense PAMPs on the cell surface or in endosomes (Kawai et al., 2011), and Nuceotide-binding domain and leucine-rich repeat containing (NLRs) proteins, which are cytosolic receptors responding to PAMPs and endogenous danger signals (Lamkanfi et al., 2009, Brodsky et al., 2009a). After stimulus recognition, TLRs initiate multiple signalling pathways involved in the innate inflammatory and antimicrobial responses, as well as in the initiation and control of adaptive immune responses (Kawai et al., 2011). In contrast, upon stimulation, several NLRs, including NLRP1 (also known as NALP1), NLRP3 (NALP3 or cryopyrin), and NLRC4 (Ipaf) assemble inflammasomes, which are multiprotein complexes responsible for activation of the inflammatory cysteine protease caspase-1 (Schroder et al., 2010).
Bacterial flagellin, the monomeric subunit of flagellar filament, is a PAMP for both systems. Extracellular flagellin is detected by TLR5 (Hayashi et al., 2001) that activates the MyD88-dependent signalling pathway, leading to the nuclear translocation of NF-κB, and the activation of mitogen activated protein kinases (MAPK), ultimately inducing the secretion of proinflammatory cytokines and chemokines, such as IL-8 (Gewirtz et al., 2001, Eaves-Pyles et al., 2001, Yu et al., 2003). On the other hand, flagellin injected into the cytoplasm of macrophages through bacterial virulence-associated secretion systems is sensed by NLRC4 in association with NAIP5, another member of the NLR family (Kofoed et al., 2011, Zhao et al., 2011). Activation of caspase-1 within the NLRC4 inflammasome leads to the maturation and release of biologically active proinflammatory cytokines IL-1ß and IL-18 (van de Veerdonk et al., 2011). Moreover, this inflammasome can trigger a proinflammatory form of cell death known as pyroptosis, (Bergsbaken et al., 2009). Finally, it has been shown that NLRC4 plays a role in maintaining a normal endosome-lysosome trafficking of phagocytized bacteria within macrophages (Amer et al., 2006, Akhter et al., 2009). There is evidence that both TLR5 and NLRC4 play a role in controlling in vivo infections caused by pathogenic bacteria including Salmonella enterica serotype Typhimurium (Feuillet et al., 2006, Broz et al., 2010), Legionella pneumophila (Hawn et al., 2003, Amer et al., 2006) and Pseudomonas aeruginosa (Feuillet et al., 2006, Sutterwala et al., 2007, Franchi et al., 2012). However, bacterial countermeasures to avoid flagellin recognition by the innate immune system have also been described. Helicobacter pylori and Campylobacter jejuni escape TLR5 recognition as a result of changes in the amino acid sequence of flagellin (Andersen-Nissen et al., 2005), and it has been suggested that S. Typhimurium downregulates fliC expression during macrophage infection to avoid a deleterious strong activation of NLRC4 inflammasome (Cummings et al., 2006, Miao et al., 2010a).
Brucella spp. are Gram-negative bacteria that cause brucellosis, a zoonosis of worldwide importance. In the natural reservoir hosts, including wild and domestic animals, these intracellular pathogens cause abortion and infertility. Humans are accidental hosts and Brucella melitensis and B. abortus are the most frequent cause of human infection (Corbel, 1997). A key characteristic of Brucella infection is its chronic nature. Indeed, animals can remain infected for years, and Brucella causes a protracted debilitating disease in untreated humans that can result in serious clinical complications (Young, 1995). As a result, brucellosis has an important economic impact on livestock and remains a major public health concern in endemic countries (Pappas et al., 2006).
An important aspect of Brucella virulence is its capacity to survive, replicate and persist within infected cells (Atluri et al., 2011). Persistence of Brucella within cells relies at least in part on its ability to control the intracellular trafficking of its vacuole in order to avoid lysosomal degradation and to gain access to its replicative niche derived from the endoplasmic reticulum (Anderson et al., 1986). Moreover, the success of Brucella lies in its stealthy strategy to cope with the innate immune system. First, the structural features of the Brucella envelope allow it to avoid sustained recognition by PRRs and subsequent strong inflammatory responses at the onset of infection (Barquero-Calvo et al., 2007). For example, Brucella produces a lipopolysaccharide that signals poorly through TLR4, compared to other bacteria (Lapaque et al., 2006, Barquero-Calvo et al., 2007). In addition, Brucella can actively control the inflammatory response by producing a protein that interferes with TLR-dependent signalling pathways (Salcedo et al., 2008, Radhakrishnan et al., 2009, Sengupta et al., 2009). Along with the lack of cytotoxicity of Brucella for highly parasitized host cells, all the above-mentioned features could render it less noticeable by the host innate immune system than other pathogens (Gross et al., 2000, Barquero-Calvo et al., 2007, Salcedo et al., 2008). Nonetheless Brucella spp. have virulence factors such as a VirB type IV secretion system (T4SS) (O'Callaghan et al., 1999), cyclic β-1,2-glucan (Briones et al., 2001, Arellano-Reynoso et al., 2005) and flagellar genes (Fretin et al., 2005) that are required for Brucella to persist within its host. Although our previous studies focused on the flagellum and its role in persistent infection, it is unknown whether Brucella flagellin, FliC, is sensed by the host during infection. Here, we combined host and pathogen genetic approaches to assess the potential of Brucella flagellin to stimulate innate immune responses.
Results
Mice fail to control infection by flagellin-deficient B. melitensis mutants
In a previous study, insertional inactivation of genes located in the three flagellar loci of B. melitensis was reported to result in a marked attenuation of its virulence in mice (Fretin et al., 2005). At that time, it was assumed that, as described in enterobacteriaceae, the fliC gene was not expressed in mutants of genes encoding basal flagellar structures. However, we recently demonstrated that the flagellar expression hierarchy of Brucella is not conventional, since the flagellin subunit is still produced in mutants deficient in the hook or basal body (Ferooz et al., 2011). To evaluate the specific impact of the absence of FliC flagellin on the virulence of B. melitensis, non-polar mutants of fliC (ΔfliC) and flbT (ΔflbT) (Ferooz et al., 2011) were used to infect murine macrophages and BALB/c mice. The FlbT regulator of B. melitensis is specifically required for the production of FliC, most likely by allowing translation of the fliC mRNA (Ferooz et al., 2011). Accordingly, flagellin was detected neither in the ΔfliC nor in the ΔflbT strain harvested at the early exponential phase of growth, whereas the protein is produced by the isogenic wt strain (Fig. 1A).
Fig. 1. Flagellin-deficient B. melitensis mutants infect macrophages in vitro with the same kinetics as wt bacteria but show enhanced persistence in mice.

(A) Western blot analysis of the production of flagellin (FliC, upper panel) by B. melitensis strains harvested at the early log phase and the log phase of growth in 2YT rich medium. Anti-Omp89 detection was used as a loading control (lower panel). Data are representative of two independent experiments. ΔfliC pfliC is the complemented strain. (B) Intracellular replication of B. melitensis 16M wt and ΔfliC strains in RAW264.7 murine macrophages. Error bars represent the standard deviation of triplicates in one representative experiment out of three. (C-D) Infection kinetics in the spleens of wt BALB/c mice (n=5) inoculated intraperitoneally (i.p.) with 4 × 104 CFUs of B. melitensis 16M wt, ΔfliC, complemented ΔfliC pfliC, ΔflbT, or ΔfliF strains. (E) Infection kinetics in the spleens of wt C57BL/6 mice (n=5) inoculated i.p. with 4 × 104 CFUs of B. melitensis 16M wt or ΔfliC strains. Data represent the mean CFUs per organ and error bars represent standard deviation. Results have been analyzed by ANOVA I after testing the homogeneity of variance (Bartlett). ** and *** denote highly significant (p<0.01 and p<0.001 respectively) differences in relation to wt infection. These results are representative of at least two independent experiments.
We first compared the intracellular growth of B. melitensis ΔfliC and ΔflbT to that of wt bacteria in RAW264.7 murine macrophages. No difference in colony forming units (CFUs) was detected over a 48-h time course (Fig. 1B). Similar results were obtained in HeLa cells (data not shown). Consistent with a normal multiplication in endoplasmic reticulum-derived vacuoles, both ΔfliC mutant and its isogenic parental strain were found to replicate within calnexin-positive compartments of HeLa cells at 24h p.i (data not shown).
Despite the absence of an obvious role for Brucella flagellar genes in cellular models of infection, several reports have shown that they are required for the establishment of a persistent infection in vivo (Fretin et al., 2005, Zygmunt et al., 2006). To re-evaluate the role of flagellar proteins in vivo, BALB/c mice were infected via the intraperitoneal route with B. melitensis 16M ΔfliC, ΔflbT and ΔfliF non-polar mutants. None of the mutants was significantly attenuated 5 days p.i., as compared with the parental strain (Fig. 1C and 1D). Moreover, we could confirm that the basal body protein FliF is required for full virulence. Indeed, the ΔfliF mutant was attenuated at 3 and 4 weeks p.i. (Fig. 1D). In contrast, the virulence of the ΔfliC strain was exacerbated when compared to its isogenic parental strain, as ΔfliC-infected mice presented a higher bacterial load in the spleen from 12 days until 60 days p.i. (Fig. 1C). A higher bacterial count was also observed at the same times in the livers of mice infected with the ΔfliC mutant (data not shown). Similarly, an enhanced persistence of the ΔfliC strain in the spleens of the resistant C57BL/6 mice has also been observed (Fig. 1E). The use of a low-copy plasmid carrying fliC gene along with its predicted flanking regulatory sequences, which restores regulated production of flagellin in the ΔfliC strain (Fig. 1A), allowed partial complementation of the phenotype of the newly constructed ΔfliC mutant at 28 days p.i. and full complementation at 60 days p.i. (Fig. 1C). Moreover, we could show that the ΔflbT mutant had similar infection kinetics than the ΔfliC strain in the spleen of BALB/c mice (Fig. 1D). This further supports the fact that the apparent inability of the host to control bacterial infection is specifically due to the lack of flagellin production by Brucella.
Mice infected with B. melitensis ΔfliC mutant exhibit severe splenic pathology.
Brucella is known to induce splenomegaly in infected hosts. During the course of a B. melitensis 16M infection in BALB/c mice, the spleen weight increases and peaks around 0.4 gr (4-fold the spleen weight of an uninfected mice) at 12 days p.i. Afterwards, the spleen weight decreases but remains twice the normal value until the end of the experiment (Fig. 2A). In contrast, we found that the splenomegaly of mice infected with flagellin-deficient mutants, while displaying kinetics similar to those of the wt infection during the first 12 days, continued to increase until 28 days p.i. and reached a plateau of almost 5 or 6 times the normal spleen weight by the end of the experiment (Fig. 2A for ΔfliC, data not shown for ΔflbT). A similar exacerbation of splenomegaly was also observed in C57BL/6 mice at 21 days p.i with the ΔfliC mutant (data not shown). This was in accordance with the enhanced persistence of the flagellin-deficient mutants in mice (Fig. 1C-E).
Fig. 2. Enhanced persistence of B. melitensis ΔfliC in mice is associated with increased pathology.

(A) Kinetics of splenomegaly in wt female BALB/c mice (n=5) injected i.p. with 4 × 104 CFUs of wt or ΔfliC strains of B. melitensis 16M. Data represent the mean spleen weight and error bars represent standard deviation. Results have been analyzed by ANOVA I after testing the homogeneity of variance (Bartlett). *** denotes highly significant (p<0.001) differences in relation to wt infection. (B) Splenic pathology caused by a 28 day-infection was determined using the histopathology scoring system as described in the Material and methods. Data were analysed using a Mann Whitney test, and the mean histopathology scores were significantly different (P=0.009) (C) Representative photomicrographs (x10) of histopathology of spleens from BALB/c mice uninfected or infected for 28 days with B. melitensis wt or ΔfliC strain. WP, white pulp; T, thrombosis; black arrows, granuloma; white arrowhead, neutrophil infiltration. These results are representative of at least two independent experiments.
We further examined the splenic histopathology of BALB/c mice infected for 28 days with wt or ΔfliC B. melitensis strain. At this time, mice infected with the ΔfliC strain showed a markedly exacerbated splenic inflammation characterized by increased vasodilation, thrombosis, neutrophil infiltration and granuloma formation (Fig 2B and 2C). In contrast, mice infected for 28 days with wt B. melitensis had nearly normal splenic morphology, as compared with non-infected mice.
Ectopic production of flagellin attenuates the virulence of B. melitensis in vivo
Mice apparently fail to control infection caused by B. melitensis 16M ΔfliC or ΔflbT at late time points. This observation suggests that production of flagellin by Brucella somehow influences the course of infection. To further test this hypothesis, we engineered a B. melitensis 16M strain, designated BruFliCON, that constitutively expresses a plasmid-encoded copy of fliC from Escherichia coli Plac. Western blot analysis confirmed that, while production of flagellin by wt bacteria is only detectable at the early exponential phase of growth, BruFliCON produced higher levels of flagellin throughout in vitro growth (Fig. 3A). Ectopic production of flagellin did not impair the invasion and replication abilities of Brucella in macrophages in vitro (Fig. 3B). However, we found that the BruFliCON strain was attenuated in vivo compared with wt B. melitensis 16M. While no difference in splenic bacterial load was observed between the two strains at 5 days post infection of BALB/c mice, 0.5 to 1 log fewer CFU of BruFliCON bacteria were recovered at 12, 21 and 28 days p.i. (Fig. 3C). Reduced colonization of BruFliCON was also observed in the liver of infected BALB/c, and similar results were also obtained with C57BL/6 mice (data not shown).
Fig. 3. Constitutive production of flagellin does not impair replication of B. melitensis 16M in macrophages in vitro, but attenuates its virulence in vivo.
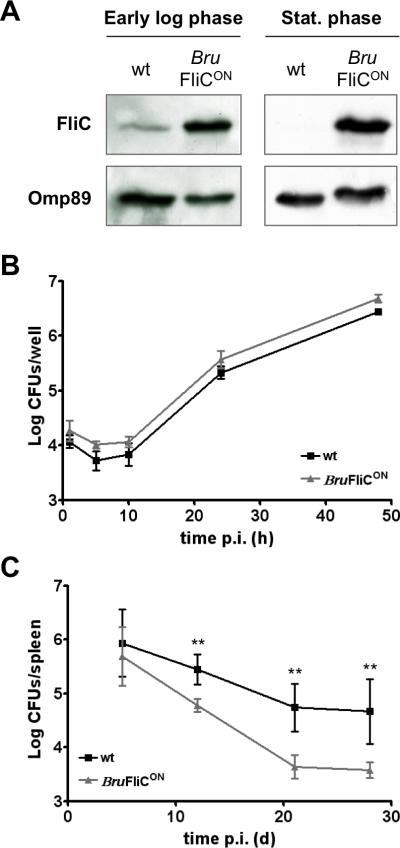
(A) Western blot analysis of flagellin (FliC, upper panel) production in wt and BruFliCON strains during early exponential and stationary phases of growth in 2YT rich medium. Detection of Omp89 was used as a loading control. (B) Intracellular replication of wt and BruFliCON strains in RAW264.7 murine macrophages. Error bars represent the standard deviation of triplicates in one representative experiment out of two. (C) Infection kinetics in the spleens of wt BALB/c mice (n=5) inoculated i.p. with 4 × 104 CFUs of wt or BruFliCON strain. Data represent the mean CFUs per organ and error bars represent standard deviation. Results have been analyzed by ANOVA I after testing the homogeneity of variance (Bartlett). ** and *** denote highly significant (p<0.01 and p<0.001 respectively) differences in relation to wt infection. These results are representative of at least two independent experiments.
Brucella flagellin lacks TLR5 agonist activity
The altered virulence of the ΔfliC and BruFliCON mutants led us to hypothesize that Brucella flagellin is detected by the host in order to mount a protective immune response. To ascertain whether innate immune sensing of flagellin contributes to enhanced control of systemic Brucella infection, we first determined whether Brucella flagellin possesses agonist activity for TLR5. To this end, epitope-tagged FliC flagellins from Brucella (BruFliC-FLAG) or S. enterica serotype Typhimurium (S. Typhimurium; StFliC-FLAG) were expressed in an S. Typhimurium fliCfljB mutant (EHW26) lacking endogenous flagellin expression. Immunoblotting with the anti-FLAG antibody demonstrated that both BruFliC-FLAG and StFliC-FLAG were secreted to the supernatant in similar amounts (Fig. 4A). Addition of the C-terminal FLAG tag to StFliC prevents its assembly into flagellar filaments, thereby allowing for a direct comparison of effects of flagellin monomers in the absence of a confounding effect on motility, since strains expressing either StFliC-FLAG or BruFliC-FLAG were aflagellate and non-motile (data not shown).
Fig. 4. Brucella flagellin lacks TLR5 agonist activity.

(A-C) FLAG-tagged flagellins from S. enterica serotype Typhimurium (StFliC) or Brucella abortus (BruFliC) were expressed in an S. Typhimurium fliCfljB mutant, and culture supernatants containing recombinant flagellins were used to treat cells. (A) Western blot showing production of bacterium-associated flagellins from S. Typhimurium wt (lane 1), S. Typhimurium fliCfljBmutant (lane 2), fliCfljB mutant expressing StFliC-FLAG (lane 3) or fliCfljB mutant expressing BruFliC-FLAG (lane 4). Flagellins were detected both in the pellets (left panel) and in the concentrated supernatants (right panel) of S. Typhimurium strains. 30ng of concentrated supernatant proteins from S. Typhimurium strains expressing recombinant flagellins were used to treat HEK293/hTLR5 cells for 4 or 24h (B) and T84 cells for 8h (C). IL-8 in cell supernatants was measured by ELISA. (D) Activation of p38 and ERK MAPK in T-84 cells by purified recombinant flagellins from Brucella (GST-BruFliC) and S. Typhimurium (GST-StFliC) was measured by Western blot analysis with anti- p38, anti-phosphorylated (P-)p38, anti- ERK, and anti-P-ERK. Detection of tubulin was used as a loading control. Purified flagellins treated with proteinase K (PK) were used as a control. All data shown are from an individual experiment that was repeated at least twice with similar results.
Culture supernatants of S. Typhimurium fliCfljB expressing recombinant flagellins were used to treat two TLR5-expressing cell lines: HEK293/hTLR5 and the colonic epithelial cell line T84 (Fig. 4B and 4C). Both cell lines secreted interleukin 8 (IL-8) on infection with strains expressing native or FLAG-tagged StFliC, demonstrating that addition of the epitope tag to the C terminus of flagellin did not affect its TLR5 agonist activity. Stimulation of IL-8 secretion was dependent on flagellin in both cell lines, since culture supernatants from the fliCfljB mutant elicited little (Fig. 4C) or no (Fig. 4B) IL-8. In contrast to StFliC-FLAG, expression of BruFliC-FLAG did not elicit IL-8 secretion above the level of the fliCfljB mutant. Similar results were obtained when T84 or HEK-293/hTLR5 cells were infected with S. Typhimurium strains expressing recombinant flagellins (data not shown). The response to BruFliC did not appear to be delayed, since extending the time of the assay to 24h did not allow detection of a response comparable to that elicited by StFliC-FLAG (Fig. 4B). As a second readout for TLR5 signaling, we assayed activation of mitogen-activated protein kinases (MAPK) p38 and ERK by treatment with purified, GST-tagged flagellins. Phosphorylation of both p38 and ERK was induced to a greater extent by GST-StFliC than by GST-BruFliC, and notably no increase in phosphorylation of ERK could be detected after treatment with GST-BruFliC (Fig. 4D). Taken together, these results demonstrate that compared to S. Typhimurium flagellin, the ability of Brucella flagellin to stimulate TLR5 signaling is greatly reduced.
Cytosolic sensing pathways detect Brucella flagellin during infection of macrophages
In addition to TLR5, flagellin that enters the cytosol of host macrophages can be sensed by the NLRC4/NAIP5 pathway (Kofoed et al., 2011, Zhao et al., 2011). To determine whether cytosolic pathways could detect flagellin during Brucella infection, we first used the TEM-1 δ-lactamase assay to detect translocation of flagellin into the cytosol of B. abortus-infected J774 macrophage-like cells. For these experiments, J774 cells were infected with B. abortus 2308 expressing either a C-terminally tagged copy of Brucella flagellin or an irrelevant protein (GST), from a multi-copy plasmid (pFLAG-TEM1; Sun et al, 2007). While cells infected with B. abortus expressing GST::Flag-TEM-1 showed no cytosolic β-lactamase activity (no β-lactamase-positive cells in 4 experiments), 0.94% (range: 0.3-2.1%) of cells infected with B. abortus expressing the flagellin fusion protein were β-lactamase positive, suggesting potential access of low amounts of flagellin to the cytosol of Brucella-infected cells. Next, we determined whether, in primary macrophages, cytosolic flagellin could stimulate innate immune responses. To this end, we compared the ability of B. melitensis and its isogenic ΔfliC mutant to elicit IL-1β secretion from primary bone marrow-derived macrophages (BMDM). Compared to B. melitensis wt, the ΔfliC mutant elicited significantly reduced IL-1β secretion (Fig. 5A). This reduction was not the result of differing numbers of intracellular bacteria of the ΔfliC mutant, since both the ΔfliC mutant and wt B. melitensis were present in the same numbers (data not shown). This partial reduction in IL-1β secretion suggests that recognition of flagellin contributes to activation of the caspase-1 inflammasome. The mechanism of cytosolic flagellin sensing in the context of intracellular infection was further investigated using the B. melitensis FliCON strain, which expresses flagellin constitutively. This strain, as well as a control carrying the empty plasmid pBBR1MCS, was used to infect immortalized BMDM from mice deficient in NLRC4 (Fig. 5B). Constitutive expression of FliC did not affect the ability of B. melitensis to survive intracellularly (Fig. 3B and data not shown). Since in LPS-primed BMDM the amount of IL-1β was maximal at 6h after infection with Brucella (Fig. 5A), we looked at the IL-1β response only at this time point. The BruFliCON strain elicited significantly more IL-1β secretion than the control strain (Fig. 5B), confirming data shown in Fig 5A. While these results suggested that under conditions of flagellin expression, flagellin can be sensed by cytosolic PRRs that lead to activation of caspase-1 and secretion of IL-1β, NLRC4 was not required for flagellin-dependent stimulation of IL-1β secretion by BMDM in vitro (Fig. 5B).
Fig. 5. B. abortus flagellin induces IL-1β in an NLRC4-independent manner.

(A) Primary bone marrow-derived macrophages from C57BL/6 mice were primed with LPS and inoculated with B. melitensis 16M wt or the ΔfliC mutant and IL-1β was measured in the culture supernatants by ELISA. Results are shown as the mean ± standard deviation of data from an individual experiment that was repeated 4 times with similar results. (B) Immortalized, LPS-primed C57BL/6 or Nlrc4-/- bone marrow-derived macrophages were inoculated with B. melitensis 16M wt or the BruFliCONstrain. IL-1β in the supernatant was measured at 6h after inoculation. Data shown are combined from three independent experiments with triplicate samples, and represent the mean ± standard deviation of all data.
Brucella flagellin elicits IL-1β secretion by a mechanism that is distinct from the NLRC4/NAIP5 pathway
Since B. melitensis is known to inhibit innate immune signalling (Salcedo et al., 2008, Radhakrishnan et al., 2009, Sengupta et al., 2009) we determined whether purified flagellin, in the absence of other Brucella factors, would signal similarly to flagellin expressed during cellular infection. For this purpose, purified GST-BruFliC and GST-StFliC were introduced into the cytosol of BMDM using the cationic lipid DOTAP (Franchi et al., 2006). Both BruFliC and StFliC elicited dose-dependent secretion of IL-1β from BMDM from C57BL/6 mice when introduced into the cytosol using DOTAP (Fig. 6A). Neither GST, DOTAP alone, nor recombinant flagellins in the absence of DOTAP elicited any secretion of IL-1β (Fig. 6A and data not shown). Comparison of IL-1β secreted in response to equal amounts of StFliC or BruFliC suggested that the proinflammatory activity of StFliC was slightly higher than that of BruFliC (Fig. 6A). Secretion of IL-1β in response to S. Typhimurium FliC was dependent on NLRC4 and only partially dependent on the adaptor protein ASC (apoptosis-associated speck-like protein), as reported previously (Broz et al., 2010). In contrast, BruFliC elicited IL-1β secretion that required ASC, but was independent of NLRC4, at least in cultured BMDM (Fig. 6B and Fig. 6C). These results suggested that in BMDM, Brucella flagellin was sensed by a cytosolic mechanism that differs from the NLRC4/NAIP5-dependent response to S. Typhimurium FliC (Kofoed et al., 2011, Zhao et al., 2011).
Fig. 6. Introduction of recombinant Brucella flagellin into the host cell cytosol results in ASC-dependent, but NLRC4-independent secretion of IL-1β.
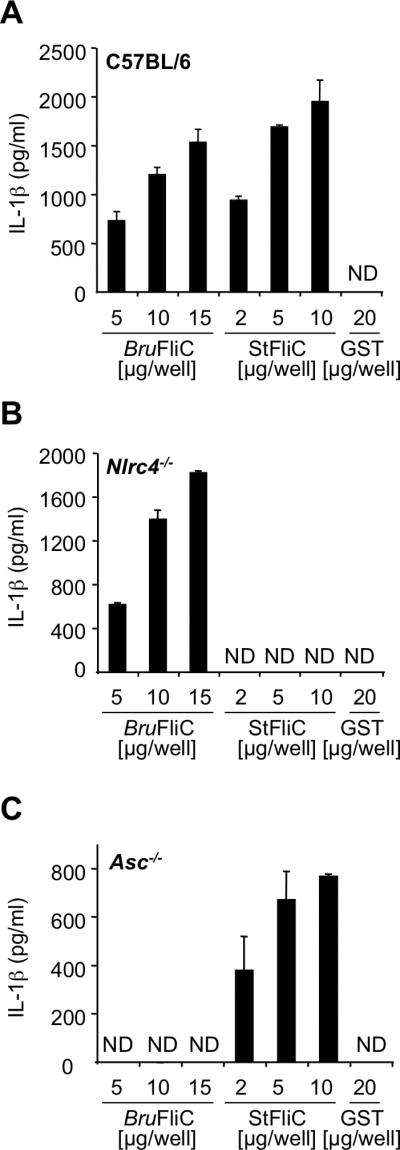
Graded amounts of GST-BruFliC and GST-StFliC fusion proteins were delivered to the cytosol of LPS-primed primary bone marrow-derived macrophages from C57BL/6 (A), Nlrc4-/- (B) or Asc-/- (C) mice, using the cationic lipid DOTAP. Treated macrophages were incubated for 3h before measurement of IL-1β in the supernatants by ELISA. Results are expressed as the mean of triplicate samples, with error bars representing the range of the data from one of two independent experiments with the same outcome.
The cytosolic flagellin-detection pathway is implicated in the control of B. melitensis infection in vivo
To evaluate the potential impact of caspase-1 inflammasomes on the control of Brucella infection in vivo, we infected Nlrc4-/- and Casp1-/- C57BL/6 mice with B. melitensis 16M. Splenic bacterial count was examined 21 days p.i., a time at which wt mice manage to effectively control infection caused by flagellin-producing Brucella strains (Fig. 1C-E and 3C). At this time, we observed that NLRC4 (Fig. 7A) and caspase-1 (Fig. 7B) deficiency moderately but significantly affected the resistance of mice to infection. This suggests that the NLRC4-caspase-1 axis is required for the host to control B. melitensis 16M infection, possibly through recognition of cytosolic flagellin. To further test this hypothesis, the BruFliCON strain was used to infect Nlrc4-/- and Casp1-/- mice. As shown previously, virulence of this strain is attenuated compared to wt B. melitensis 16M, as the spleen of BruFliCON-infected C57BL/6 mice contained less CFUs than these infected by the wt strain (Fig. 7). Interestingly, this virulence defect was rescued in mice deficient for the cytosolic flagellin sensor NLRC4 (Fig. 7A) or the downstream caspase-1 (Fig. 7B). These data indicate that, in contrast to what has been observed in vitro (Fig. 5B and 6B), Brucella flagellin can activate the NLRC4 inflammasome in vivo. Nevertheless, we found that the BruFliCON strain was still attenuated (a significant 0.5 log decreased CFUs in the spleen) compared to B. melitensis 16M wt in Nlrc4-/- and Casp1-/- mice 21 days p.i. (Fig. 7). This suggests that both inflammasome-dependent and inflammasome-independent control of infection operates downstream detection of Brucella flagellin in vivo. This hypothesis is further supported by the finding that although Nlrc4-/- and Casp1-/- mice infected with wt B. melitensis 16M had significantly higher splenic bacterial counts than those of wt mice, it remained significantly lower than those of mice infected with the ΔfliC mutant (Fig. 7).
Fig. 7. NLRC4 inflammasome is implicated in the control of B. melitensis infection in vivo.

Wild type, Nlrc4-/- (A) and Casp1-/- (B) C57BL/6 mice (n=5) were injected i.p. with 4 × 104 CFUs of B. melitensis wt, BruFliCON or ΔfliC strain, as indicated in the figure. Mice were sacrificed 21 days post-infection and CFUs per spleen were determined. These results are representative of at least two independent experiments. Data have been analysed by ANOVA I after testing the homogeneity of variance (Bartlett). * ,** and *** denote significant (p<0.05, p<0.01 and p<0.001 respectively) differences in relation to C57BL/6 wt infection by wt bacteria. # and ## denote significant (p<0.05 and p<0.01 respectively) differences in relation to knock-out mice infection by wt bacteria.
B. melitensis ΔfliC mutant fails to elicit early granuloma formation in the spleen of infected mice
Chronic granulomatous inflammation in the spleen of natural hosts, humans and mice is the hallmark of Brucella infection (Spink et al., 1949, Enright et al., 1990). Recently, we revealed the pivotal role of early splenic granuloma formation in the ability of mice to control bacterial dissemination (Copin et al., 2012). Here, we used a rabbit polyclonal serum raised against B. melitensis (anti-Bru) with the aim to compare the distribution of putative infected cells in the spleen of BALB/c mice inoculated with B. melitensis 16M wt or ΔfliC strain. 5 days after infection with B. melitensis 16M wt, clusters of cells stained with anti-Bru (Bru-positive cells) were found equally in white pulp and red pulp area of the spleen (Fig. 8). These clusters consisted primarily of CD11b+ cells, suggesting that they corresponded to the granuloma previously described (Copin et al., 2012). Strikingly, at the same time, the number of Bru-positive cells clusters counted in splenic sections of ΔfliC-infected mice was reduced (Fig. 8). This apparent defect in early splenic granuloma formation suggests the importance of flagellin sensing by the host for the orchestration of this typical tissue response to Brucella infection.
Fig. 8. The distribution of Bru-positive cells is different in the spleen of mice infected by the ΔfliC mutant, compared to wt infection.

Localization of Bru+ cells (green) and CD11b+ cells (red) in the spleen of BALB/c mice non-infected or infected with B. melitensis wt or the ΔfliC strain. The graph represents the relative number of clusters of Bru+ cells. Errors bars are the standard deviation calculated on countings of four mice from two independent experiments.
Discussion
Intracellular survival and immune evasion both contribute to persistence of Brucella in the host (Atluri et al., 2011). Recent studies have shown that Brucella uses passive as well as active mechanisms to evade detection by TLRs of the innate immune system (Lapaque et al., 2006, Barquero-Calvo et al., 2007, Salcedo et al., 2008, Radhakrishnan et al., 2009, Sengupta et al., 2009). Accordingly, the inflammatory response induced at the onset of Brucella infection is lower than observed with pyogenic infections such as salmonellosis (Barquero-Calvo et al., 2007). Actually, brucellae are not entirely invisible to the immune system, which can still detect them and shape a Th1 response to control infection (Murphy et al., 2001, Copin et al., 2007). However, the host immune response is not sufficient to eliminate bacteria, resulting in a chronic state of infection characterized by a balance between pathogen virulence and host resistance. The impact of Brucella flagellin on infection had not been reported yet. The data presented here suggest that flagellin plays a crucial role in the interplay between Brucella and its host, as its detection by the innate immune system is required for the control of infection in vivo, although some characteristics of Brucella flagellin would contribute to the stealthy strategy of this pathogen.
The use of two mutants of B. melitensis 16M that either overproduce or lack the FliC flagellin has shown that this protein hinders bacterial replication in vivo. Indeed, a strain engineered to ectopically produce flagellin (BruFliCON) was attenuated in mice, whereas deletion of fliC (ΔfliC) enhanced persistence of B. melitensis 16M in these conditions. Our in vivo data are consistent with studies reporting exacerbated infections caused by a flagellin deficient mutant of Salmonella enterica serovar Typhimurium (Vijay-Kumar et al., 2006), Legionella pneumophila (Molofsky et al., 2006) or Pseudomonas syringae pv. Tabaci (Li et al., 2005), as well as virulence attenuation due to flagellin overproduction by S. Typhimurium (Salazar-Gonzalez et al., 2007, Miao et al., 2010a) and Listeria monocytogenes (Grundling et al., 2004). These findings also suggest that Brucella flagellin is an important immune target during infection, and our work provides first insights into the mechanisms involved.
TLR5 and the NLRC4/NAIP5 complex are the only proteins currently known as innate immune sensors of extracellular and cytoplasmic bacterial flagellin, respectively (Miao et al., 2007).
In agreement with a recent paper quoting that purified Brucella flagellin does not induce expression of interferon-inducible resistance proteins (IRGs) in murine macrophages (Lapaque et al., 2009), the data reported in this paper allow us to conclude that Brucella flagellin is not a TLR5 agonist. This is consistent with its atypical sequence as it lacks the amino acid residues required to stimulate this PRR (Andersen-Nissen et al., 2005). Thus, we propose that Brucella evades TLR5-mediated detection, and that it could be part of its stealthy strategy to avoid activation of the innate immune system during the onset of infection. Cytosolic flagellin activates a complex comprising the NLR family proteins NLRC4 and NAIP5 (Franchi et al., 2006, Miao et al., 2006, Kofoed et al., 2011, Zhao et al., 2011). This complex senses a highly conserved region of the C terminal part of the flagellin critical for flagellum filament assembly (Yonekura et al., 2003), but that is required neither for flagellin translocation into the host cell cytosol nor for TLR5 activation (Lightfield et al., 2008). The C-terminal 35 amino acid residues are conserved in Brucella FliC flagellin, as they share respectively 46% and 40% identity with L. pneumophila FlaA and S. Typhimurium FliC, both known to activate NLRC4 (Franchi et al., 2006, Zamboni et al., 2006) and sharing themselves 60% identity. Recently, it has been proposed that the minimal motif of flagellin sensed by NLRC4 comprises the highly conserved last C-terminal residues VLSLL found in L. pneumophila FlaA and S. Typhimurium FliC (Lightfield et al., 2008, Miao et al., 2010b). This motif is semi-conserved in Brucella flagellin that bears an ILSFR motif.
Our results suggest that, similar to what is seen with L. pneumophila infection (Amer et al., 2006, Case et al., 2009) the NLRC4-caspase-1 axis is involved in the control of B. melitensis 16M in vivo (Fig. 7). However, the absence of NLRC4 or caspase-1 stimulation in mice infected with the flagellin-deficient B. melitensis 16M ΔfliC or ΔflbT mutants cannot by itself account for the inability of the host to control infection. Indeed, the relative difference of virulence between B. melitensis 16M wt and ΔfliC strains were only partially reduced in Nlrc4-/- and Casp1-/- mice (Fig. 7), indicating involvement of both NLRC4/caspase-1-dependent and independent mechanisms in the control of Brucella downstream flagellin recognition. This contrasts with what is observed after intratracheal infection of mice with L. pneumophila. Indeed, in this case, the number of flaA mutants and wt bacteria in the lungs of Nlrc4-/- and Casp1-/- is similar (Amer et al., 2006, Case et al., 2009). Therefore, it suggests that Brucella flagellin is an immune target not only for the cytosolic sensor NLRC4 in vivo. The slight attenuation of the BruFliCON strain compared to B. melitensis 16M wt in Nlrc4-/-and Casp1-/- mice 21 days p.i. (Fig. 7) is consistent with the hypothesis that Brucella flagellin stimulates another immune pathway in addition to the NLRC4/caspase-1 axis. The ASC-dependent signalling suggested by our ex-vivo data (Fig. 6) could be this additional pathway. Brucella FliC is the first flagellin found to induce IL-1ß secretion from macrophages in vitro in an NLRC4-independent manner. Whether other poor agonists of TLR5 such as flagellins from β-Proteobacteria activate this uncommon pathway remains to be determined.
Activation of innate immune pathways by flagellin would play a role in limiting replication of Brucella in vivo. Interestingly, we found that the lack of flagellin affected the control of B. melitensis infection by both susceptible BALB/c and resistant C57BL/6 mice (Fig. 1C-1E). BALB/c mice are known for their intrinsic reduced capacity to mount a Th1 immune response and are subsequently less able to control Brucella infections than C57BL/6 mice, having notably an increased bacterial load in the spleens during the plateau phase (Fernandes et al., 1996, Sathiyaseelan et al., 2006, Copin et al., 2007, Vitry et al., 2012). The hypervirulence of the ΔfliC mutant in BALB/c and C57BL/6 mice suggests that immune detection of flagellin in vivo activates one or several immune effector mechanisms that are shared by both mouse species and that are critical for the control of Brucella infection. The immune effector mechanisms triggered by flagellin detection during Brucella infection remain to be uncovered. Processing of the proinflammatory cytokines pro-IL-1ß and pro-IL-18 (Raupach et al., 2006, Dinarello, 2009), pyroptosis (Bergsbaken et al., 2009, Miao et al., 2010a) and control of phagosome maturation (Amer et al., 2006, Akhter et al., 2009) that can all result from caspase-1 activation are important processes for innate immunity against bacterial pathogens (Brodsky et al., 2009b).
Besides its impact on the innate immune system, it is known that bacterial flagellin is also a target of the adaptive immune response (Salazar-Gonzalez et al., 2005). However, whether the adaptive immune system responds to MHC class II-presented flagellin peptides during infection by Brucella is currently not known.
While searching for immune effector mechanisms triggered by flagellin detection and involved in the control of Brucella replication in mice, we found that the ΔfliC mutant fails to elicit early granulomatous response in the spleen of mice infected for 5 days, a time at which the mutant is found at a similar level as the wt strain (Fig. 8). Thus, we suggest that detection of flagellin by the host would play a role in early granuloma development during brucellosis. Although the granulomatous response was stronger at 28 days p.i. (Fig. 2B), when the ΔfliC strain colonized spleens at higher extent than wt, an early alteration in this response could contribute to the apparent failure of mice to control infections caused by the flagellin-deficient mutants of B. melitensis 16M. Indeed, granulomatous inflammation is the typical tissue response to Brucella infection in both mice and humans (Spink et al., 1949, Hunt et al., 1967, Enright et al., 1990), and a recent study has demonstrated the crucial role of early formation of splenic granuloma in the control of B. melitensis 16M (Copin et al., 2012). Whether granuloma formation during infection by Brucella depends on ASC, NLRC4 and/or caspase-1 is currently unknown. Up to now, a role for the NLRC4 inflammasome in such a response has never been reported. However, it was recently shown that granuloma formation in chronic M. tuberculosis infection is dependent on ASC, whereas it does not require caspase-1 (McElvania Tekippe et al., 2010).
S. Typhimurium translocates flagellin from its containing-vacuole into the cytosol of infected cells by a SPI1-T3SS-dependent but flagellar secretory apparatus-independent process (Sun et al., 2007). Similarly, a Dot/Icm T4SS-mediated flagellin translocation has been suggested in the case of L. pneumophila (Ren et al., 2006, Molofsky et al., 2006). Here, we show that Brucella flagellin is also translocated into the host cell cytosol. Interestingly, flagellin translocation was not seen when a virB2 mutant was used to infect macrophages (data not shown), suggesting that VirB T4SS may play a role in flagellin translocation. Interestingly, a requirement for the T4SS to elicit splenic microgranuloma formation has been proposed (Rolan et al., 2009). According to our results, it could be envisioned that the VirB T4SS of Brucella elicits a granulomatous response by translocating flagellin. However, since the T4SS is also essential for Brucella to reach its replicative niche (Celli et al., 2003), additional studies would be necessary to determine whether the role of the T4SS in release of flagellin to the host cytosol is direct or indirect. The TEM1 β-lactamase reporter assay has been previously used to demonstrate translocation of S. Typhimurium flagellin into the cytosol of infected macrophages (Sun et al., 2007). We observed that the amount of flagellin translocated into cells by Brucella is far less than by Salmonella. While flagellin could be detected in the cytosol of 77.5% of macrophages infected for 4h with S. Typhimurium (Sun et al., 2007), less than 1% of cells were positive 16h after infection with B. abortus. Therefore, although the intrinsic ability of Brucella and Salmonella flagellin to induce IL-1ß secretion from BMDM appeared to be similar (Fig. 6), Brucella might evade activation of a robust innate immune response from cytosolic PRRs by controlling the production and/or delivery of flagellin into the host cell. Accordingly, we could show that the attenuation of the BruFliCON strain that ectopically produces flagellin is due at least in part to a strong NLRC4 inflammasome activation in vivo (Fig. 7). Thus, we propose that the tight regulation of flagellin synthesis and/or delivery during infection is part of its stealthy strategy. This has also been suggested for S. Typhimurium, which downregulates the expression of fliC during macrophage infection (Cummings et al., 2006).
In conclusion, we propose that flagellin is an important molecular actor of the interplay between Brucella and its host. Although flagellin escapes detection by TLR5 and Brucella controls its production and/or delivery to the infected host cell cytosol, its detection by cytosolic PRRs initiates a response that results in an immunologic standoff between Brucella and its host, leading to a persistent infection with limited inflammatory pathology. The increased bacterial tissue loads and destructive pathology, seen with the flagellin-deficient mutant demonstrates that innate and possibly also adaptive, recognition of flagellin is a process that is important to the chronic and stealthy nature of Brucella infection. As such, flagellin could be considered as a “host protective factor” (Shames et al., 2010) in the context of brucellosis.
Experimental procedures
Bacteria and growth conditions
Bacterial strains and plasmids are listed in Table 1. Cultures of Brucella strains were freshly inoculated from frozen stock onto 2YT medium (10% yeast extract, 10 g liter-1 tryptone, 5 g liter-1 NaCl) plates before subculturing aerobically at 37°C in 2YT broth supplemented with appropriate antibiotics. LB broth was used for Escherichia coli and Salmonella enterica serotype Typhimurium (S. Typhimurium) cultures. Antibiotics were used at the following concentrations: carbenicillin, 100 mg/liter; chloramphenicol, 30 mg/liter; kanamycin, 60 mg/liter; or nalidixic acid, 50 mg/liter.
Table 1.
Bacterial strains and plasmids used in this study.
| Designation | Genotype and/or Phenotype | Source or Reference |
|---|---|---|
| Strains | ||
| Brucella melitensis strains | ||
| 16M | wild type isolate | |
| Δ fliC | ΔfliC::Kan | (Ferooz et al., 2011) |
| Δ flbT | ΔflbT::Kan | (Ferooz et al., 2011) |
| Δ fliF | ΔfliF::Kan | (Ferooz et al., 2011) |
| BruFliCON | pBBR1-fliC | This work |
| Brucella abortus strain | ||
| 2308 | wild type isolate | |
| Salmonella enterica serovar Typhimurium strains | ||
| 14028 | ATCC 14028 Wild-Type | ATCC |
| IR715 | 14028 Spontaneous NalR | (Stojiljkovic et al., 1995) |
| LT2 | LT2 Wild-Type | (Lilleengen, 1948) |
| EHW26 | IR715 fliC::Tn10 fljB::MudJ (fliCfljB) | (Raffatellu et al., 2005) |
| Escherichia coli strains | ||
| CC118 λpir | araD139 Δ(ara, leu)7697 ΔlacX74 phoAΔ20 galE galK thi rpsE rpoB argEam recA1 λpir | (Simon et al., 1983) |
| DH10B | F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara, leu)7697 galU galK rpsL(StrR) endA1 nupG | Invitrogen |
| DH5 | F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacΔZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 phoA supE44 λ- thi-1 gyrA96 relA1 | (Woodcock et al., 1989) |
| S17-1 λpir | recA thi pro rK– mK+ RP4:2-Tc:MuKm Tn7 λpir | (Simon et al., 1983) |
| Plasmids | ||
| pCR2.1 | TOPO cloning vector | Invitrogen |
| pUC-KIXX | pUC4::Tn5 KanR | (Beck et al., 1982) |
| pBBR1MCS | mob RK2, lacZα, CmR | (Kovach et al., 1994) |
| pRH001 fliC | pMR10 (CmR, B. melitensis 16M PfliC-fliC-tfliC) | This work |
| pBBR1-fliC | pBBR1MCS(CmR, B. melitensis 16M fliC) | This work |
| pBBRFlag | pBBR1MCS::3xFLAG | This work |
| pYHS1116 | pBBRFlag::StFliC | This work |
| pYHS1073 | pBBRFlag::BaFliC | This work |
| pWSK29 | CarbR, pSC101 ori | (Wang et al., 1991) |
Molecular techniques
DNA manipulations were performed according to standard techniques (Ausubel et al., 1991). Primers used are listed in Table 2.
Table 2.
Primers used in this work
| Primer | Sequence | Restriction site | Application |
|---|---|---|---|
| BaFliC-F | ACCATATGGCTAGCATTCTTACAAACTCGTCG | NdeI | FLAG-tagged BaFliC and BaFliC::FT fusion protein |
| BaFliC-R | ACTGCAGTTAGCCGCGGAACAGCGACAGGATCGAC | SalI | |
| StFliC-F | ACCATATGGCACAAGTCATTAATACAAACAGC | NdeI | FLAG-tagged StFliC and StFliC::FT fusion protein |
| StFliC-R | AGTCGACTTAACGCAGTAAAGAGAGGACGTTTTGC | SalI | |
| StFliC-F2 | GAATTCATGGCACAAGTCATTAATACAAACAGC | EcoRI | GST-StFliC fusion protein |
| StFliC-STOP-R | ACTCGAGTTAACGCAGTAAAGAGAGGACGTTTTGC | Xhol | |
| BaFliC-F2 | GAATTCATGGCTAGCATTCTTACAAACTCG | EcoRI | GST fusion proteins |
| BaFliC-R2 | ACTCGAGTTAGCCGCGGAACAGCGACAG | Xhol | GST fusion proteins |
| PfliC | CGGGATCCAATGCCCGGGATCATGTTGATGC | BamHI | complementation plasmid |
| tfliC | GCTCTAGATGCCAGACAGGATGTCGGGC | XbaI | |
| Plac | GCTCtagAtagAtagAGCGCAACGCAATTAATGTGAG | XbaI | fliC overexpression plasmid |
| fliC-Plac | GTTTGTAAGAATGCTAGCCATAGCTGTTTCCTGTGTGAAATTG | ||
| BmfliC-F | ATGGCTAGCATTCTTACAAACTCGT | ||
| BmfliC-R | CGGGATCCTTAGCCGCGGAACAGCG | BamHI |
Bold: Extra 5’ DNA; Bold/Underlined: Multiple cloning site; Bold/Underlined/Italicized: Restriction site utilized in cloning; Lower case: Start or stop codon.
Generation of the complementation vector pRH001-fliC
fliC coding sequence (cds) and its predicted upstream and downstream regulatory sequences were amplified by PCR using the PfliC and tfliC primers pair. The PCR product (PfliC-fliC-tfliC) was then cloned into the EcorRV site of pGEM. In a second step, this fragment was excised using BamHI and XbaI, and inserted into the corresponding sites of pMR10cat (R. Roberts, unpublished) in the opposite orientation to the Plac.
Generation of the B. melitensis 16M FliCON strain
The fliC overexpression vector pBBR1-fliC was obtained as follows: first, the constitutive promoter of the lac operon Plac was amplified by PCR using the Plac and fliC-Plac primers pair. In the resulting PCR product, Plac is flanked by translation stop codons in all three reading frame in 5’ and by the 21st fliC coding sequence (cds) base pairs in 3’. fliC cds was amplified by PCR using the BmfliC-F and BmfliC-R primers. A third PCR using the Plac and BmfliC-R primers was used to ligate the two PCR products by cohesive ends. Stop codons and close fusion of fliC cds to Plac without any linker ensure the production of FliC flagellin that does not bear additional N-terminal amino acid residues. The PCR product (XbaI-Plac-fliC-BamHI) was then cloned into the EcorRV site of pGEM. In a last step, this fragment was excised using XbaI and BamHI, and inserted into the corresponding sites of pBBR1 MCS-I (Kovach et al., 1994) in the opposite orientation to the endogenous Plac. This gave rise to pBBR1-Plac-fliC. This final construction was transformed into E. coli strain S17-1 (Simon et al., 1983), and introduced into B. melitensis 16M by conjugation.
Generation of C-terminally FLAG-tagged flagellins
A derivative of the broad host range plasmid pBBR1MCS (pBBR1-FLAG) was first generated by ligating a fragment containing “SphI-promoter-NdeI-SalI-3x-Flag-STOP-PstI-SacI” into pBBR1MCS4 treated with SphI and SacI. The S. Typhimurium fliC gene was amplified using primers StFliC-F and StFliC-R, and the resulting amplicon was ligated into NdeI and SalI-digested pBBR1-FLAG to yield plasmid pYHS1116, encoding StFliC-FLAG. The B. abortus fliC gene was amplified using primers BaFliC-F and BaFliC-R and cloned in the same way to generate pYHS1073, encoding BruFliC-FLAG. In both constructs, expression of the recombinant proteins was controlled by a previously described constitutive Brucella promoter, BMEII0193 (Eskra et al., 2001). The constructs were confirmed by DNA sequencing across the junction fragments. Plasmids pYHS1116 (StFliC-FLAG) and pYHS1073 (BruFliC-FLAG) were introduced into a Salmonella fliC fljB mutant (EHW26, (Raffatellu et al., 2005)) by electroporation. The B. abortus and B. melitensis FliC proteins are identical except for a substitution of Ala156 to Thr in B. abortus.
Generation of fusions to TEM-1 ß-lactamase
To express BruFliC fused with TEM1, B. abortus fliC was amplified by using the primer pair BaFliC-F and BaFliC-R. The amplicon was cloned into pCR2.1, then subsequently digested with NdeI and PstI, and ligated pFlagTEM1 (Raffatellu et al., 2005) digested with the same enzymes to yield pBaFliCTEM1. The expression of BruFliC::TEM1 in pBaFliCTEM1 is under the control of inducible Trc promoter. Constructs expressing StFliC::TEM1 were described previously (Sun et al., 2007).
Generation of GST-flagellin fusion proteins
For construction of plasmids expressing GST fused at the N-terminus of flagellins, flagellin genes were amplified to delete predicted N-terminal secretion domains. The fliC gene from S. Typhimurium was amplified without its first 332 nucleotides using primer pair of StFliC-F2 and StFliC-STOP-R. Similarly B. abortus fliC lacking its first 87 nucleotides was amplified using primer pair of BaFliC-F2 and BaFliCR2. Both amplicons were cloned in pCR2.1, excised as BamHI/SalI fragments, and ligated to BamHI/SalI –digested pGEX-4T-1. The cloning junctions were confirmed by DNA sequence analysis, and the resulting constructs, pGEX-StFliC and pGEX-BaFliC, were transformed into E. coli BL-21. Expression of GST::StFliC and GST::BruFliC was induced by IPTG, and the recombinant flagellins were purified using Glutathione-Sepharose 4B (GE Healthcare). Protein concentration was measured with DC protein assay (BioRad).
Preparation of concentrated S. Typhimurium culture supernatant containing recombinant flagellins
S. Typhimurium strains were grown for 4 to 5 hours at 37°C with vigorous shaking by diluting an overnight culture 1 to 100 in 20 ml LB broth plus 1 mM IPTG. Once the OD600 reached 0.8 to 1.2 bacteria were removed by centrifugation at 4000 rpm for 15 min and 12 ml of the resulting supernatant was passed through a 0.45 βm filter and subject to concentration by using an Amicon Ultra-15 with cutoff of 5K (Millipore) followed by a wash with 10 ml PBS. Protein concentration was determined by DC protein assay (BioRad) and SDS-PAGE followed by Coomassie blue stain. The final protein concentration was adjusted to 1 μ/ul.
Generation of rabbit anti-BaFliC serum and Western blot
B. abortus fliC (BaFliC) was amplified using primers BaFliC-F and BaFliC-R and cloned into pET103 in frame with a 6xHis tag. The resulting BaFliC::6xHis fusion protein was produced and purified by using Ni-NTA kit (Qiagen). Rabbit serum against BaFliC was generated by Antagene (Antagene Inc., Calif.). For detection of secreted BaFliC the supernatant from 1 ml culture was precipitated using trichloroacetic acid (TCA) and separated on a 12% SDS-PAGE gel. Proteins were electrotransferred to a polyvinylidene difluoride (PVDF) membrane. BaFliC was detected by using rabbit anti-BaFliC as primary antibody and as goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) as secondary antibody. S. Typhimurium Phase I flagellin (FliC) was detected using Salmonella Hi antiserum (Difco). C-terminal FLAG-tagged S. Typhimurium and B. abortus flagellins were detected using anti-FLAG monoclonal antibody (1:5000, Sigma) and a goat anti-mouse IgG antibody conjugated to HRP. HRP activity was detected with a chemiluminescent substrate (PerkinElmer Life Sciences). Flagellin produced by B. melitensis 16M was detected as described previously (Fretin et al., 2005).
Measurement of TLR5 agonist activity of flagellins
The human colonic epithelial cell line T-84 was cultured in were maintained in Dulbecco's modified Eagle medium (DMEM)-F12 medium (Gibco), containing 1.2 g/liter sodium bicarbonate, 2.5 mM L-glutamine, 15 mM HEPES, and 0.5 mM sodium pyruvate (Gibco), supplemented with 10% fetal calf serum (FCS). The day before assay cells from 1/3 of a 80 to 90% confluent T75 flask were seeded per each 24-well plate containing DMEM-F12 and 2% FCS. HEK293 cells were cultured as previously described (Keestra et al., 2010).
T84 cells in 24-well plates were either infected with 10 μl of bacteria grown as above or treated by adding 30 μl of concentrated bacterial culture supernatant and incubated for 4 hours at 37°C under 5% CO2. For the HEK293 stably transfected with human TLR5, cells were grown in 48-well tissue culture plates and infected for 4-48 h with 10 μl of bacteria grown as described above or treated by adding 10 μl of concentrated bacteria culture supernatant and incubated for 8 hours at 37°C under 5% CO2. Supernatants were aspirated and centrifuged for 10 min at 6,000 rpm to remove residual bacteria and cell debris before measurement of IL-8 concentration by ELISA.
Mitogen-activated protein kinase (MAPK) phosphorylation assay
T84 cells were seeded in six well plates at a density of 4 × 108 cells per well and incubated for 24h in DMEM/F12 + 10% FBS. The following day, cells were rinsed with PBS and the medium replaced with serum-free medium. For analysis of MAP kinase phosphorylation, cells were treated with concentrations of GST-BruFliC or GST-StFliC ranging from 250ng/ml to 1 μg/ml. As a negative control, cells were treated with the highest concentration of flagellin (1μg/ml) that had previously been treated with proteinase K (20mg/ml proteinase K for 1h at 37°C, then for 10 min at 75°C to inactivate the protease). After 30 and 90 min, cells were lysed 0.1 ml in phosphosafe extraction reagent (Novagen) containing 2.5% protease inhibitor (Sigma) according to the instructions of the manufacturer. The protein concentration was determined using the Micro BCA kit (Pierce). Total protein (0.01 mg) was resolved by SDS-PAGE and transferred to a polyvinylidene fluoride membrane. Primary antibodies were purchased from Cell Signalling Technology, including the following phosphorylation-specific antibodies: p-ERK and p-p38 (Thr180/Tyr182). Secondary antibodies (goat anti rabbit conjugated to horseradish peroxidase) were purchased from Jackson Immunoresearch and used according to the recommendations of the manufacturer. Peroxidase activity was visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore). For each primary antibody, a separate membrane was used.
Detection of flagellin in the cytosol of infected macrophages
The β-lactamase translocation assay was performed as previously described (Sun et al., 2007). Briefly J774A.1 mouse macrophages were seeded in 96-well coverglass bottom plates and infected with B. abortus 2308 expressing either a BruFliC::Flag-TEM-1 fusion proteins, or an irrelevant control (Glutathione-S-transferase::Flag-TEM-1) at a multiplicity of infection of 500. Plates were centrifuged at 250 g for 5 min at room temperature to synchronize infection. After incubation for 1 hour at 37 °C in 5% CO2, free bacteria were removed from the cells by three washes with PBS. A volume of 0.2 ml of Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, 1% non-essential amino acids, 1 mM glutamine containing 1 mM IPTG and 100 ug/ml gentamicin was added to each well, and plates were incubated at 37 °C in 5% CO2. After 16 h, cells were washed once with Hank’s balanced salt solution (Invitrogen) and loaded with the fluorescent substrate CCF2/AM (1mM, Invitrogen) for 1.5 h at room temperature using the standard loading protocol recommended by the manufacturer. Fluorescence microscopy analysis was performed using an Axiovert M200 (Carl Zeiss), equipped with a CCF2 filter set (Chroma Technology). Fluorescence micrographs were captured using a Zeiss Axiocam MRC5 and Zeiss AxioVision 4.5 software. Images were imported into Adobe PhotoShop for color adjustment. The number of blue cells containing cleaved CCF2/AM was counted visually and expressed as the percentage of total cells in the well. The experiment was performed four times and the result expressed as geometric mean and range of the four experiments.
Bone-marrow derived Macrophages
Bone marrow-derived macrophages were isolated from C57BL/6, or congenic mutant mice following standard protocols as described previously (Sun et al., 2007).
Macrophage infection
For assaying inflammasome activation, 24-well microtiter plates were seeded with bone marrow-derived macrophages at a concentration of 2 x105 cells/well in 0.5 ml of RPMIsup and incubated over night at 37°C in 5% CO2. For priming of macrophages, cells were treated for 4h before infection with LPS (100 ng/ml), as previously described (Franchi et al., 2006). Inocula of B. melitensis 16M were prepared by growing with shaking in TSB for 24h. Bacteria were treated with a non-agglutinating (1:4,000) dilution of anti-Brucella rabbit serum (Difco) for 1h at 37°, as described (Rolan et al., 2007) then diluted in RPMIsup to a concentration of 4 × 107 CFU/ml. Approximately 2 × 107 bacteria in 0.5 ml of RPMIsup, containing B. melitensis 16M wt or its isogenic fliC mutant, were added to each well of macrophages. Three independent assays were performed with triplicate samples, and each experiment included control (C57BL/6) macrophages together with macrophages from mutant mice. Microtiter plates were centrifuged at 250 × g for 5 min at room temperature in order to synchronize infection. Cells were incubated for 20 min at 37°C in 5% CO2, and free bacteria were removed by three washes with phosphate-buffered saline (PBS). RPMIsup plus 50mg gentamicin per ml was added to the wells, and the cells were incubated at 37°C in 5% CO2. After 1 h, the RPMIsup plus 50μg/ml gentamicin was replaced with medium containing 25μg/ml gentamicin. Wells were sampled after infection by aspirating the medium, lysing the macrophages with 0.5 ml of 0.5% Tween-20 and rinsing each well with 0.5 ml of PBS. Viable bacteria were quantified by dilution in sterile PBS and plating on TSA containing appropriate antibiotics.
Liposome-mediated delivery of flagellins to the macrophage cytosol
Recombinant flagellin proteins were delivered to the macrophage cytosol using the cationic lipid DOTAP (Roche), as described previously (Franchi et al., 2006). Briefly, 50 ml of DOTAP was incubated for 30 min in serum-free media with 2 mg of recombinant flagellins purified as described above. After incubation, 3.5 ml serum-free media was added and 500 ml was used to stimulate 1 × 106 macrophages seeded in 24-well microtiter plates for 3h.
Measurement of cytokines
Mouse IL-1ß was measured in culture supernatants by enzyme-linked immunoabsorbent assay (ELISA) (R&D Systems). Human IL-8 was detected using an ELISA kit from BioLegend.
Mice
Wild type (wt) BALB/c, wt C57BL/6, C57BL/6 Nlrc4-/- (obtained from Dr. VM. Dixit and described in (Mariathasan et al., 2004)) and C57BL/6 Casp1-/- (obtained from Dr. R. Flavell and described in (Kuida et al., 1995)) mice were used in this study. They were bred in the animal facility of the University of Namur (Belgium). The animal handling and procedures of this study were in accordance with the current European legislation (directive 86/609/EEC) and in agreement with the corresponding Belgian law “Arrêté royal relatif à la protection des animaux d'expérience du 6 avril 2010 publié le 14 mai 2010”. The complete protocol was reviewed and approved by the Animal Welfare Committee of the University of Namur, Belgium (Permit Number: 05-558).
Infection of mice
Mice were injected intraperitoneally (i.p.) with 4 × 104 CFUs of B. melitensis 16M in 500μl of PBS. Control animals were injected with the same volume of PBS. Infectious doses were validated by plating serial dilutions of the inocula. At selected time intervals, mice were sacrificed by cervical dislocation. Immediately after being killed, spleen and liver were collected for bacterial counts and histopathologic analyses. For bacterial counts, spleens and livers were homogenized in PBS/0.1% X-100 triton (Sigma). Serial dilutions were plated on 2YT media plates for enumeration of tissue-associated CFU.
Histology
Spleens were fixed for 24h in Bouin's fixative, dehydrated for 24h in methanol, then incubated in toluol and finally in warm paraffin prior to paraffin embedding. Sections (5μm) were rehydrated and stained with hemalun, erythrosin and safran. Blinded histopathology scoring for splenic granuloma formation was performed by a pathologist (MX), according to the following criteria. 0, <5% of splenic parenchyma containing granulomas; 1, 5-20%; 2, 20-40%; 3, 40-40%; 4, >60%.
Immunofluorescence microscopy
Spleens were fixed for 6h at 4°C in 2% paraformaldehyde (pH 7.4), washed in PBS, incubated overnight at 4°C in a 20% PBS-sucrose solution under agitation, and washed again in PBS. Tissues were embedded in the Tissue-Tek OCT compound (Sakura), frozen in liquid nitrogen, and cryostat sections (10μm) were prepared. Tissues sections were rehydrated in PBS, then incubated successively in a PBS solution containing 1% blocking reagent (Boeringer) (PBS-BR 1%) and in PBS-BR 1% containing any of the following mAbs or reagents: DAPI nucleic acid stain, Alexa Fluor 350 phalloidin, M1/70 (anti-CD11b, BD Biosciences), homemade anti-B. melitensis 16M serum (Copin et al., 2012). Slides were mounted in Fluoro-Gel medium (Electron Microscopy Sciences, Hatfield, PA). Labelled tissues sections were visualized under a Zeiss fluorescent inverted microscope (Axiovert 200) equipped with high-resolution monochrome camera (AxioCam HR, Zeiss).
Statistical analysis
ANOVA I was used for infection data analysis after testing the homogeneity of variance (Bartlett test). Average comparisons were performed by pairwise Scheffe's test. A Mann Whitney test was used for analysis of histopathology scoring. Errors bars represent standard deviation.
Acknowledgements
We thank V. Dixit, J. Tschopp and A. Tardivel for providing us with the NLRC4 KO mice. Part of this work has been granted by an ARC Convention from the French community of Belgium (N° 08/13-015). M. Terwagne holds a PhD grant from FNRS (Fond National pour la Recherche Scientifique) and J. Ferooz holds a PhD grant from FRIA (Fonds pour la formation à la Recherche dans l’Industrie et l’Agriculture).
This work was supported by US PHS grants AI50553 and AI097107 to R.M.T. and US PHS grant DK091191 to G.N. V.L.A was supported by T32 IA60555.
References
- Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 2009;5:e1000361. doi: 10.1371/journal.ppat.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A. 2005;102:9247–9252. doi: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson TD, Cheville NF. Ultrastructural morphometric analysis of Brucella abortus-infected trophoblasts in experimental placentitis. Bacterial replication occurs in rough endoplasmic reticulum. Am J Pathol. 1986;124:226–237. [PMC free article] [PubMed] [Google Scholar]
- Arellano-Reynoso B, Lapaque N, Salcedo S, Briones G, Ciocchini AE, Ugalde R, et al. Cyclic beta-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nat Immunol. 2005;6:618–625. doi: 10.1038/ni1202. [DOI] [PubMed] [Google Scholar]
- Atluri VL, Xavier MN, de Jong MF, den Hartigh AB, Tsolis RE. Interactions of the human pathogenic Brucella species with their hosts. Annu Rev Microbiol. 2011;65:523–541. doi: 10.1146/annurev-micro-090110-102905. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, E., M.D., Seidman JG, SMith JA, Struhl K. Current protocols in Molecular Biology. John Wiley & Sons; New-York: 1991. [Google Scholar]
- Barquero-Calvo E, Chaves-Olarte E, Weiss DS, Guzman-Verri C, Chacon-Diaz C, Rucavado A, et al. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS One. 2007;2:e631. doi: 10.1371/journal.pone.0000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck E, Ludwig G, Auerswald EA, Reiss B, Schaller H. Nucleotide sequence and exact localization of the neomycin phosphotransferase gene from transposon Tn5. Gene. 1982;19:327–336. doi: 10.1016/0378-1119(82)90023-3. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones G, Inon de Iannino N, Roset M, Vigliocco A, Paulo PS, Ugalde RA. Brucella abortus cyclic beta-1,2-glucan mutants have reduced virulence in mice and are defective in intracellular replication in HeLa cells. Infect Immun. 2001;69:4528–4535. doi: 10.1128/IAI.69.7.4528-4535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky IE, Medzhitov R. Targeting of immune signalling networks by bacterial pathogens. Nat Cell Biol. 2009a;11:521–526. doi: 10.1038/ncb0509-521. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Monack D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin Immunol. 2009b;21:199–207. doi: 10.1016/j.smim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case CL, Shin S, Roy CR. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun. 2009;77:1981–1991. doi: 10.1128/IAI.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med. 2003;198:545–556. doi: 10.1084/jem.20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copin R, De Baetselier P, Carlier Y, Letesson JJ, Muraille E. MyD88-dependent activation of B220-CD11b+LY-6C+ dendritic cells during Brucella melitensis infection. J Immunol. 2007;178:5182–5191. doi: 10.4049/jimmunol.178.8.5182. [DOI] [PubMed] [Google Scholar]
- Copin R, Vitry MA, Hanot Mambres D, Machelart A, De Trez C, Vanderwinden JM, et al. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog. 2012;8:e1002575. doi: 10.1371/journal.ppat.1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbel MJ. Brucellosis: an overview. Emerg Infect Dis. 1997;3:213–221. doi: 10.3201/eid0302.970219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol Microbiol. 2006;61:795–809. doi: 10.1111/j.1365-2958.2006.05271.x. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- Eaves-Pyles T, Murthy K, Liaudet L, Virag L, Ross G, Soriano FG, et al. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J Immunol. 2001;166:1248–1260. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]
- Enright FM, Araya LN, Elzer PH, Rowe GE, Winter AJ. Comparative histopathology in BALB/c mice infected with virulent and attenuated strains of Brucella abortus. Vet Immunol Immunopathol. 1990;26:171–182. doi: 10.1016/0165-2427(90)90065-z. [DOI] [PubMed] [Google Scholar]
- Eskra L, Canavessi A, Carey M, Splitter G. Brucella abortus genes identified following constitutive growth and macrophage infection. Infect Immun. 2001;69:7736–7742. doi: 10.1128/IAI.69.12.7736-7742.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes DM, Jiang X, Jung JH, Baldwin CL. Comparison of T cell cytokines in resistant and susceptible mice infected with virulent Brucella abortus strain 2308. FEMS immunology and medical microbiology. 1996;16:193–203. doi: 10.1111/j.1574-695X.1996.tb00136.x. [DOI] [PubMed] [Google Scholar]
- Ferooz J, Lemaire J, Letesson JJ. Role of FlbT in flagellin production in Brucella melitensis. Microbiology. 2011;157:1253–1262. doi: 10.1099/mic.0.044867-0. [DOI] [PubMed] [Google Scholar]
- Feuillet V, Medjane S, Mondor I, Demaria O, Pagni PP, Galan JE, et al. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc Natl Acad Sci U S A. 2006;103:12487–12492. doi: 10.1073/pnas.0605200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol. 2012;13:449–456. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretin D, Fauconnier A, Kohler S, Halling S, Leonard S, Nijskens C, et al. The sheathed flagellum of Brucella melitensis is involved in persistence in a murine model of infection. Cell Microbiol. 2005;7:687–698. doi: 10.1111/j.1462-5822.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- Gross A, Terraza A, Ouahrani-Bettache S, Liautard JP, Dornand J. In vitro Brucella suis infection prevents the programmed cell death of human monocytic cells. Infect Immun. 2000;68:342–351. doi: 10.1128/iai.68.1.342-351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundling A, Burrack LS, Bouwer HG, Higgins DE. Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. Proc Natl Acad Sci U S A. 2004;101:12318–12323. doi: 10.1073/pnas.0404924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawn TR, Verbon A, Lettinga KD, Zhao LP, Li SS, Laws RJ, et al. A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires' disease. J Exp Med. 2003;198:1563–1572. doi: 10.1084/jem.20031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Hunt AC, Bothwell PW. Histological findings in human brucellosis. Journal of clinical pathology. 1967;20:267–272. doi: 10.1136/jcp.20.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Keestra AM, de Zoete MR, Bouwman LI, van Putten JP. Chicken TLR21 is an innate CpG DNA receptor distinct from mammalian TLR9. J Immunol. 2010;185:460–467. doi: 10.4049/jimmunol.0901921. [DOI] [PubMed] [Google Scholar]
- Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–595. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach ME, Phillips RW, Elzer PH, Roop RM, 2nd, Peterson KM. pBBR1MCS: a broad-host-range cloning vector. BioTechniques. 1994;16:800–802. [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Inflammasomes: guardians of cytosolic sanctity. Immunol Rev. 2009;227:95–105. doi: 10.1111/j.1600-065X.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- Lapaque N, Forquet F, de Chastellier C, Mishal Z, Jolly G, Moreno E, et al. Characterization of Brucella abortus lipopolysaccharide macrodomains as mega rafts. Cell Microbiol. 2006;8:197–206. doi: 10.1111/j.1462-5822.2005.00609.x. [DOI] [PubMed] [Google Scholar]
- Lapaque N, Muller A, Alexopoulou L, Howard JC, Gorvel JP. Brucella abortus induces Irgm3 and Irga6 expression via type-I IFN by a MyD88-dependent pathway, without the requirement of TLR2, TLR4, TLR5 and TLR9. Microb Pathog. 2009;47:299–304. doi: 10.1016/j.micpath.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Li X, Lin H, Zhang W, Zou Y, Zhang J, Tang X, Zhou JM. Flagellin induces innate immunity in nonhost interactions that is suppressed by Pseudomonas syringae effectors. Proc Natl Acad Sci U S A. 2005;102:12990–12995. doi: 10.1073/pnas.0502425102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol. 2008;9:1171–1178. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilleengen K. Typing of Salmonella typhimurium by means of bacteriophage. Acta. Pathol. Microbiol. Scand. Suppl. 1948;77:2–125. [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One. 2010;5:e12320. doi: 10.1371/journal.pone.0012320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 2007;29:275–288. doi: 10.1007/s00281-007-0078-z. [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010a;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010b;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, Swanson MS. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med. 2006;203:1093–1104. doi: 10.1084/jem.20051659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EA, Sathiyaseelan J, Parent MA, Zou B, Baldwin CL. Interferon-gamma is crucial for surviving a Brucella abortus infection in both resistant C57BL/6 and susceptible BALB/c mice. Immunology. 2001;103:511–518. doi: 10.1046/j.1365-2567.2001.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan D, Cazevieille C, Allardet-Servent A, Boschiroli ML, Bourg G, Foulongne V, et al. A homologue of the Agrobacterium tumefaciens VirB and Bordetella pertussis Ptl type IV secretion systems is essential for intracellular survival of Brucella suis. Mol Microbiol. 1999;33:1210–1220. doi: 10.1046/j.1365-2958.1999.01569.x. [DOI] [PubMed] [Google Scholar]
- Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect Dis. 2006;6:91–99. doi: 10.1016/S1473-3099(06)70382-6. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR Domain-containing Protein Mimics Properties of the Toll-like Receptor Adaptor Protein TIRAP. J Biol Chem. 2009;284:9892–9898. doi: 10.1074/jbc.M805458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffatellu M, Chessa D, Wilson RP, Dusold R, Rubino S, Baumler AJ. The Vi capsular antigen of Salmonella enterica serotype Typhi reduces Toll-like receptor-dependent interleukin-8 expression in the intestinal mucosa. Infect Immun. 2005;73:3367–3374. doi: 10.1128/IAI.73.6.3367-3374.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2006;2:e18. doi: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolan HG, Tsolis RM. Mice lacking components of adaptive immunity show increased Brucella abortus virB mutant colonization. Infect Immun. 2007;75:2965–2973. doi: 10.1128/IAI.01896-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolan HG, Xavier MN, Santos RL, Tsolis RM. Natural antibody contributes to host defense against an attenuated Brucella abortus virB mutant. Infect Immun. 2009;77:3004–3013. doi: 10.1128/IAI.01114-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez RM, McSorley SJ. Salmonella flagellin, a microbial target of the innate and adaptive immune system. Immunol Lett. 2005;101:117–122. doi: 10.1016/j.imlet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Salazar-Gonzalez RM, Srinivasan A, Griffin A, Muralimohan G, Ertelt JM, Ravindran R, et al. Salmonella flagellin induces bystander activation of splenic dendritic cells and hinders bacterial replication in vivo. J Immunol. 2007;179:6169–6175. doi: 10.4049/jimmunol.179.9.6169. [DOI] [PubMed] [Google Scholar]
- Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, et al. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008;4:e21. doi: 10.1371/journal.ppat.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathiyaseelan J, Goenka R, Parent M, Benson RM, Murphy EA, Fernandes DM, et al. Treatment of Brucella-susceptible mice with IL-12 increases primary and secondary immunity. Cellular immunology. 2006;243:1–9. doi: 10.1016/j.cellimm.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Sengupta D, Koblansky A, Gaines J, Brown T, West AP, Zhang D, et al. Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter, MAL. J Immunol. 2009;184:956–964. doi: 10.4049/jimmunol.0902008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames SR, Finlay BB. Breaking the stereotype: virulence factor-mediated protection of host cells in bacterial pathogenesis. PLoS Pathog. 2010;6:e1001057. doi: 10.1371/journal.ppat.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology. 1983;1:784–791. [Google Scholar]
- Spink WW, Hoffbauer FW, et al. Histopathology of the liver in human brucellosis. J Lab Clin Med. 1949;34:40–58. [PubMed] [Google Scholar]
- Stojiljkovic I, Baumler AJ, Heffron F. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. Journal of bacteriology. 1995;177:1357–1366. doi: 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YH, Rolan HG, Tsolis RM. Injection of flagellin into the host cell cytosol by Salmonella enterica serotype Typhimurium. J Biol Chem. 2007;282:33897–33901. doi: 10.1074/jbc.C700181200. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. 2011;32:110–116. doi: 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Wu H, Jones R, Grant G, Babbin B, King TP, et al. Flagellin suppresses epithelial apoptosis and limits disease during enteric infection. Am J Pathol. 2006;169:1686–1700. doi: 10.2353/ajpath.2006.060345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitry MA, De Trez C, Goriely S, Dumoutier L, Akira S, Ryffel B, et al. Crucial role of IFN-gamma-producing CD4+ Th1 cells but dispensable function of CD8+ T cell, B cell, Th2 and Th17 responses in the control of Brucella melitensis infection in mice. Infect Immun. 2012 doi: 10.1128/IAI.00761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, et al. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic acids research. 1989;17:3469–3478. doi: 10.1093/nar/17.9.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura K, Maki-Yonekura S, Namba K. Complete atomic model of the bacterial flagellar filament by electron cryomicroscopy. Nature. 2003;424:643–650. doi: 10.1038/nature01830. [DOI] [PubMed] [Google Scholar]
- Young EJ. An overview of human brucellosis. Clin Infect Dis. 1995;21:283–289. doi: 10.1093/clinids/21.2.283. quiz 290. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zeng H, Lyons S, Carlson A, Merlin D, Neish AS, Gewirtz AT. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expression via posttranscriptional mechanism. Am J Physiol Gastrointest Liver Physiol. 2003;285:G282–290. doi: 10.1152/ajpgi.00503.2002. [DOI] [PubMed] [Google Scholar]
- Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol. 2006;7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- Zygmunt MS, Hagius SD, Walker JV, Elzer PH. Identification of Brucella melitensis 16M genes required for bacterial survival in the caprine host. Microbes Infect. 2006;8:2849–2854. doi: 10.1016/j.micinf.2006.09.002. [DOI] [PubMed] [Google Scholar]