

I. Introduction
With the recognition that lipids are not only key structural elements of membranes, but often function as essential messengers that trigger important metabolic events, there is renewed impetus for understanding the dynamic properties of lipids including lipid transfer processes. The focus of this review will be on spontaneous lipid transfer and/or exchange as occurs between organized lipid assemblies such as bilayers. By spontaneous, I refer to lipid transfer processes that occur in the absence of lipid transfer or binding proteins by mechanism(s) not involving complete fusion of donor and acceptor structures.
Although the distinction between lipid transfer and exchange is often ignored, it is important to realize that exchange is an equilibrium process between donor and acceptor structures of equivalent chemical potential due to their similar lipid compositions. In contrast, transfer involves the redistribution of lipid between donor and acceptor structures of non-equivalent chemical potential due to their differing lipid compositions. Thus, lipid transfer normally proceeds ‘down’ the chemical concentration gradient as the system attempts to achieve chemical equilibrium. Net transfer can occur if two lipids transfer at markedly different rates down their chemical concentration gradients causing the original donor and acceptor lipid assemblies to reorganize their structures at some point during the process to accomodate the net free energy changes within the system. If such reorganization cannot occur, then an external energy source is necessary to maintain the chemical nonequilibrium situation created by kinetic means.
To date, only one review devoted to spontaneous lipid transfer and/or exchange between membranes has been comprehensive in that many different lipids were covered including diacyl phospholipids, monoacyl phospholipids, sphingolipids, free fatty acids, cholesterol as well as various other lipids (e.g., platelet activating factor, cholesteryl esters, long-chain alkane derivatives). This review included most of the literature published until 1988 [1]. In contrast, other reviews have dealt primarily with the transfer of one or two specific lipid classes such as cholesterol and phospholipids [2–9].
Here, the focus will be on spontaneous lipid transfer/exchange as it occurs between organized lipid assemblies (i.e., bilayers, micelles) with the emphasis being on bilayer membranes. As such, lipid transfer to and from other biological surfaces such as lipoproteins and soluble proteins (e.g., bovine serum albumin) will not be addressed [10–11,155]. First, I will reiterate the value of studying spontanous lipid transfer processes by providing pertinent illustrations and then discuss the molecular environmental parameters known to affect spontaneous lipid transfer events. I will then conclude by describing recent findings that provide exciting new insights into the mechanism(s) by which spontaneous interbilayer lipid transfer/exchange can occur.
To facilitate a clear understanding of the first topics, it is helpful to have some insight into the history of spontaneous lipid transfer processes [1,3]. Prior to 1988, the vast majority of investigators had concluded that spontaneous lipid transfer occurred by the partitioning of lipid monomers from the organized lipid ensemble (e.g., bilayer, micelle) into the aqueous milieu and the subsequent diffusion of the lipid monomer to an ‘acceptor’ structure. The rate limiting step in this process was the initial ‘escape’ of the lipid monomer from the ‘donor’ structure. This ‘lipid monomer diffusion’ model is illustrated in Fig. 1A. Only recently has compelling evidence mounted to warrant serious consideration of other mechanisms [96–98,107–109,157]. Accordingly, a detailed discussion of alternative models for lipid transfer (e.g., Fig. 1B and C) will serve as the finale tor this review (section IV). In the meantime, it is important to realize that the ‘lipid monomer diffusion’ model has dominated thinking in this area for many years.
Fig. 1.

Models for spontaneous lipid transfer between membranes. Transferring lipids are depicted with filled headgroups. (A) In the monomer transfer model, individual lipid monomers desorb from donor surface and then diffuse through the bulk aqueous phase prior to incorporation into an acceptor surface. (B) In the transient collision model, the desorption of individual lipid monomers from the donor surface is enhanced by the apposition of hydrated donor and acceptor surfaces. Monomer diffusion occurs across the hydration barrier (approx. 1.5 nm) between the apposed surfaces. (C) In the ‘hemifusion’ model, direct contact between donor and acceptor surfaces permits mixing of the outer monolayer lipids by lateral diffusion.
II. Why study spontaneous lipid transfer processes?
There are three fundamental reasons for studying the spontaneous lipid transfer process. First, it can be a very effective tool for introducing lipids into biological membranes and then studying their fate or metabolic effect or physical manifestation. Second, it can shed light on the subtle aspects of membrane architecture by revealing differences in interactions with other membrane components. Finally, it is important biologically in a variety of processes.
II-A. Value as an experimental tool
Knowing how fast and under what conditions lipids will spontaneously transfer into cell membranes can make this process an effective experimental tool for studying the metabolic effects and fate of certain lipids in cellular systems. In fact, this approach can be a convenient way for introducing lipids bearing various reporter groups into cell or model membranes and has been used with photoactivatable lipids (e.g., Refs. 12–16), with spin-labeled lipids (e.g., Refs. 17–22). and with lipids bearing fluorescent dyes (e.g., 23–30). While one might argue that inserting lipids into membranes is better achieved using soluble lipid transfer proteins. there are experimental drawbacks to this approach. Of course, certain classes of lipids such as cholesteryl esters, triacylglycerols, and neutral glycosphingolipids and certain molecular species of phospholipids exhibit such slow spontaneous interbilayers transfer rates [1,4,5] that using lipid transfer proteins may be the only reasonable alternative for introducing them into cell membranes. Nevertheless, when feasible, avoiding lipid transfer proteins can be beneficial for the following reasons. First, one must isolate the lipid transfer proteins. If not purified to homogeneity, other proteins are introduced into the system that may exert effects in their own right. Secondly, after using lipid transfer proteins, it is often desirable to eliminate them from the system. To be absolutely sure that they are completely removed or inactivated in a way that avoids artifactual effects on the system under study is not always trivial.
One of the more striking illustrations of how spontaneous lipid transfer can be used to study lipid trafficking and processing within cells has been elegantly demonstrated by Pagano and co-workers (e.g., Refs. 24–25,31–33). These researchers have characterized the kinetics of spontaneous interbilayer transfer of different phospholipids and sphingolipids bearing a fatty acyl chain with a fluorescent reporter group. They subsequently used this information to deliver the fluorescent lipids to the plasma membranes of Chinese hamster ovary cells and then follow the intracellular distribution and processing of the lipids by fluorescence microscopy. Interestingly, although the spontaneous incorporation of the fluorescent lipids into the plasma membrane is rapid, the lipids do not randomly transfer to other intracellular membranes, but are sequentially processed at discrete intracellular locations. Moreover, not all lipids follow the same processing path. Since Pagano's pioneering studies began, other researchers have used the same basic approach to study the sorting of lipids within differentiated or non-differentiated adenocarcinoma cells [34] or within polarized epithelial cells when the fluorescent lipids are introduced by spontaneous transfer to either the basolateral or apical cell surfaces [35–37].
In addition to studying the fate of lipids themselves, spontaneous lipid transfer is also valuable for studying the metabolic effects of certain lipids on cells and tissues. For instance, this approach has been quite effective in studying the metabolic effects of gangliosides and related sphingolipid derivatives. Under appropriate conditions, these lipids can rapidly transfer from their micelles, insert themselves into cellular plasma membranes, and modulate a variety of important metabolic events including the binding of certain pericellular adhesion proteins [38–39], viruses, bacterial cells and their toxins [40] as well as regulate cellular differentiation and proliferation, epidermal growth factor receptor activity [39], and protein kinase C activity [41]. Of course, gangliosides are not the only lipid class whose metabolic effects have become better understood via spontaneous lipid transfer [42–46]. Our understanding of the metabolic effects of various monoacyl phosphatides (e.g., Refs. 47–50) and free fatty acids [51–53] have also benefited because these lipids can be effectively delivered to cell membranes by spontaneous lipid transfer processes.
Aside from elucidating the metabolic effects of certain lipids, spontaneous lipid transfer has also provided an effective means for studying the physical effects of adding lipid to cellular membranes. For many years, it has been known that when erythrocytes are incubated with lysophosphatidylcholine (lyso PC) micelles or phospholipid vesicles containing cholesterol, the lyso PC and cholesterol spontaneously transfer to the plasma membrane and dramatically change the shape of these cells [54–56]. The changes in shape are the result of increasing the cell surface area by 1% and are consistent with a slight differential expansion of the outer surface of the bilayer. Subsequently, Huestis and co-workers extended this approach to study the spontaneous transfer of diacyl PCs [20,57–58]. With diacyl PCs, the degree of cell crenation was proportional to the amount of lipid taken up. After crenation reached a plateau, the cells slowly reverted to discoid form. The shape reversal had a half-time of about 22 h. The rates and extents of crenation. lipid transfer, and shape recovery all diminished as the length of the PC acyl chain increased. Interestingly, in case of diacyl aminophospholipids like PE and PS. the induced shape changes are relatively short-lived (t1/2 < 1 h) because energy-dependent lipid fippases are present in the membrane surface [58–59,147].
Shape changes are not the only physical manifestation of incorporating lipids into cells. Golan et al. [60] have reported that incubation of lyso PC with erythrocytes at supralytic concentrations not only lyses the cells, but causes physical immobilization of proteins and lipids within the erthrocytc ghost membrane surface. Thus, a variety of physical effects of lipids can be studied by taking advantage of the spontaneous lipid transfer process.
II-B. Value in elucidating the organization of membrane components
Aside from being an effective tool for introducing lipids into membranes, spontaneous lipid transfer studies are also valuable for understanding the thermodynamics of lipid mixing in bilayer membranes and thus, helping to provide a better understanding of the lateral organization of components comprising membranes. For instance, studying a lipid's departure rate from a membrane provides clues about its immediate environment including interactions with other components in the membrane. Thus, useful insights into lipid lateral organization in model membrane systems are often revealed. Two laboratories that first recognized and made use of this potential were those of T.E. Thompson and M.C. Phillips. In 1982, Thompson and coworkers [27] noted that the spontaneous interbilayer transfer rate of glucosylceramide (GlcCer) bearing either fluorescent or radioactive labels was extremely slow (t1/2 > 30 days) despite the fact that the GlcCer represented only a small fraction of lipid in an otherwise liquid-crystalline PC bilayer. Based on such behavior and the thermotropic properties of GlcCer [61], these researchers proposed that the GlcCer must be present as gel-like micro-clusters with the PC matrix. Subsequent interbilayer lipid transfer studies involving different homogeneously N-acylated sphingomyelins provided further support that the lamellar phase state of the transferring lipid in the donor vesicle directly reflected its desorption rate [62]. Examination of multi-glycosylated sphingolipids such as gangliotetraosylceramide (asialo-GM1) and III3-N-acetylneuraminosylgangliotetraosylceramide (GM1) revealed more complex spontaneous transfer kinetics suggesting that the sialic acid residue modulates the in-plane organization of glycolipid within the otherwise liquid-crystalline PC bilayer [63–65]. Interpretation of complex spontaneous transfer kinetics becomes especially difficult if the lipid under study possesses marked heterogeneity in the acyl chain region (e.g., natural cerebrosides) [156].
Meanwhile, M.C. Phillips and colleagues studied the spontaneous interbilayer transfer kinetics of cholesterol in PC bilayers in an effort to clearly distinquish effects due to bilayer physical state from those related to composition [66–67]. By studying the influence of cholesterol/PC ratio of the donor bilayer, these investigators found the rate of cholesterol desorption to be consistent with a model in which cholesterol desorbs from that fraction of the bilayer surface which is covered by an equimolear cluster of cholesterol and PC [67]. In subsequent studies, various laboratories have continued to probe how bilayer composition affects cholesterol exchange rates [68–84]. All reports agree that membrane composition plays a critical role in regulating cholesterol exchange and that sphingomyelin, in particular, slows cholesterol exchange rates. The details of the sphingomyelin/cholesterol interactions as deduced by spontaneous lipid transfer studies have been reviewed previously [1–7,9] and clearly illustrate the information that spontaneous lipid transfer studies can reveal about membrane organization and structure.
II-C. Role in biological processes
Spontaneous lipid transfer plays an essential role in a variety of biological processes. One striking illustration involves the parasitic invasion of erythrocytes by Schistosoma mansoni [60,85]. The surface of these parasites is bound by a syncytium covered with two membranes, an outer membrane nearly devoid of protein and an inner membrane that resembles the plasma membrane of mammalian cells. After infection, the worm adheres to and ultimately lyses human erythrocytes. The data suggest that the mechanism involves release of lyso PC from the outer membrane at supralytic concentrations and that the lyso PC is transferred to adherent erythrocytes. Such a mechanism is consistent with new findings showing that spontaneous transfer of phosphatidylcholines is enhanced by transient collisional contacts between model bilayer membranes (see section IV-B).
Another biological process in which spontaneous lipid transfer is essential is during lipid absorption by the intestinal lumen during digestion. The majority of studies involving lipid uptake in the intestine have focused on fatty acid transfer from bile salt micelles to enterocytes [86–90]. However, the recent discovery that non-micellar phases such as unilamellar vesicles may also be formed during fat digestion may prompt further investigation [91–92]. In any case, Dietschy has argued that bile salt micelles enhance lipid uptake by acting as large capacity vehicles for moving fatty acids and other lipids across the unstirred water layers adjacent to enterocytes [89,90,93]. The micelles, by carrying large amounts of lipid, increase the effective lipid concentration to levels much higher than those achieved by aqueous solubilization of individual lipid monomers in the unstirred water layers. Once positioned next to the enterocyte, uptake is thought to occur by desorption of lipid molecules from the bile salt micelles [89,90,93–95]. More recent insights into how bile salts may greatly facilitate the rapid and complete transfer of phospholipid monomers from mixed micelles to the plasma membranes of enteroeytes in the intestinal muscosa have been reported by Nichols and colleagues [96–98] (see sections IV-A and IV-B for further details).
In addition to events involving cellular lipid uptake, be they invasive or digestive in nature, spontaneous lipid transfer may also play a role in the biogenesis and maintenance of membranes. Close examination of the literature reveals sealtered reports in which lipid transfer between subcellular organelles has been measured under conditions where protein involvement appears unlikely [99–103]. Recently, Wojtczak and colleagues have studied the movement of phosphatide acid (PA) and PC between carefully washed microsomal and mitochondrial membranes [102] and between the outer and inner mitochondrial membranes [103]. Significant levels of PA but not PC transfer were observed regardless of whether thiol-blocking agents were present and under conditions where the number of contact sites between outer and inner mitochondrial membranes was diminished. Based on these and other results involving acidic phospholipid spontaneous transfer between model membranes (e.g., Refs. 104–106), Wojtczak and colleagues proposed that PA transfer is not protein-mediated and occurs by spontaneous diffusion of lipid monomers through the aqueous phase [102–103].
Traditionally, a role for spontaneous lipid transfer in intracellular lipid transport and/or membrane biogenesis has been dismissed for two reasons [4,5]. First, it was argued that various cell membranes are known to have different lipid compositions and that this would not be possible if spontaneous lipid transfer really occurred intracellularly. Second, the spontaneous transfer of long-chain diacyl PCs is relatively slow (e.g., POPC t1/2 = 48 h at 37°C). However, these reasons no longer appear to be sufficient to rule out some kind of role for spontaneous lipid transfer in membrane biogenesis. As was pointed out during my earlier discussion of cholesterol interbilayer exchange, the membrane lipid composition does influence the distribution of individual lipids between membranes rather dramatically. Furthermore, it is now clear that certain membranes contain energy-dependent lipid flippases that are capable of altering the composition of specific lipids within each half of the membrane [19,58–59]. Thus, potential mechanisms for minimizing random intracellular spontaneous lipid transfer do exist. In addition, recent reports indicate that the spontaneous transfer rates of long-chain diacyl PCs between bile salt micelles [96–98] as well as between phospholipid bilayers [107–109] can be increased significantly when micelle or vesicle concentrations are high. The mechanism by which these enhanced spontaneous lipid transfer rates are achieved is discussed in detail in section IV-B and represents a significant new advance in the field.
III. Does a lipid's environment affect its transfer behavior?
Both the physico-chemical environment of the aqueous mileu separating organized lipid aggregates and the nature of the lipid matrix surrounding the transferring lipid species can have dramatic effects on spontaneous lipid transfer events. For the sake of this discussion, I have arbitrarily grouped the inter-related matrix effects into three categories.
III-A. Lamellar physical state
Because biological membranes are largely lamellar in nature, much effort has been devoted to understanding spontaneous lipid transfer to and from bilayer membranes. Nearly concurrent with the discovery that phospholipids undergo spontaneous interbilayer transfer, the importance of the lipid bilayers phase state on the kinetics of this process was recognized [110–116]. The details of these intial pioneering studies have been reviewed thoroughly by Lange [3]. Subsequent reviews [1,4–7] have covered studies carried out in the early and mid 1980s which involved the spontaneous transfer of a variety of different lipids. In all cases, spontaneous interbilayer lipid transfer rates were markedly slower when the donor bilayer was in the gel rather than the liquid-crystalline state. Apparently, the increased packing constraints in gel-phase bilayers increase the activation enthalpy and cause slower lipid desorption rates [116].
Recently, new insights into the effect of the phase state of lipid bilayers on spontaneous lipid transfer have been reported by Bayer et al. [117]. In these experiments, bilayer vesicles comprised of either DMPC or DPPC were constructed by sonication or by detergent dialysis and then were incubated together at various temperatures. The resulting transfer of DMPC and DPPC was monitored by high-sensitivity differential scanning calorimetry and other techniques. When the DMPC and DPPC vesicles were large detergent-dialysis vesicles, fusion rather than lipid monomer transfer predominated. In contrast, when vesicles were small and sonicated, a net transfer of DMPC was observed even when the DPPC vesicles were in the gel state. Only incubation temperatures below the Tc of DMPC prevented transfer completely. The time course of transfer exhibited biexponential kinetics. Based on these results and the details of the calorimetric traces, the investigators proposed that the net transfer of DMPC into the DPPC vesicles resulted in an increased lateral pressure in the outer monolayer of the DPPC vesicles that provided a driving force for increased transbilayer exchange (flip-flop) of lipid between the outer and inner monolayers. This behavior was in marked contrast to that in which the small sonicated vesicles were comprised of DMPC bearing either protonated or deuterated acyl chains. In this case, first-order exchange, but no net transfer of DMPC was observed, and only if all lipids were in the liquid-crystalline state. Also, the net DMPC exchange rate was diminished by about 20%.
The link between interbilayer and transbilayer phospholipid exchange has been examined in further detail by Wimley and Thompson [118–119]. DMPC interbilayer and transbilayer exchange were investigated in large unilamellar vesicles comprised of DMPC, DMPC/DSPC, or DMPC/DMPE. By including a small amount of negatively-charged DMPG (1.75 mol%) to prevent vesicle aggregation, the exchange processes could be studied over a range of temperatures known to produce liquid-crystalline, gel, and mixed-phase lipid matrices. Although the activation energy and interbilayer exchange rates were very similar for liquid-crystalline DMPC SUVs and LUVs, the extent of interbilayer exchange of DMPC was about 90% in LUVs in contrast to SUVs where only the DMPC in the outer monolayer was available for exchange. Thus, the transbilayer movement is substantially faster in liquid-crystalline DMPC LUVs than in SUVs. Interestingly, DMPC desorbed from gel-phase LUVs at a much slower rate than from gel-phase SUVs and with a 3-fold higher activation energy. To explain the results, Wimley and Thompson proposed that SUVs are unable to adopt a completely rigid, planar gel phase because of their high curvature and probably possess localized disordered regions equivalent to liquid-like defects between the gel-phase domains [118]. However, the nearly planar LUV bilayers can form gel-phase without requiring extensive defects.
To provide additional insight into how packing defects within the lipid matrix affect spontaneous phospholipid transfer, Wimley and Thompson [118] investigated DMPC desorption from DMPC/DSPC LUVs at a variety of temperatures. Although they found no difference in DMPCs desorption rates from liquid-crystalline DMPC LUVs and DMPC/DSPC LUVs, DMPC interbilayer exchange was faster between LUVs comprised of DMPC and DSPC gel-phases than the rates between LUVs comprised only of DMPC gel-phase. To explain the results, Wimley and Thompson proposed that there are packing defects in gel phases composed of both DMPC and DSPC because of the four-carbon difference in the acyl chain length of the two PC species. This explanation seems plausible because similar DMPC desorption rates were observed for LUVs comprised only of gel-phase DMPC and for LUVs comprised of DMPC and DMPE gel-phases, where no chain length mismatch and hence, fewer defects occur [118–119].
An illustration of how molecular packing defects can accelerate spontaneous lipid exchange between biological membranes has been provided recently by Bittman et al. [120]. Based on measurements of cholesterol and PC exchange between Mycoplasma capricolum membranes and lipid vesicles, these investigators found significant increases in lipid exchange as the endogenous phosphatidylglycerol (PG) and diphosphatidylglycerol (DPG) content was changed in the Mycoplasma capricolum plasma membranes. Manipulating the PG/DPG content apparently causes packing defects within the plasma membranes because of the molecular mismatching that occurs in bilayers poised near their lamellar to nonlamellar (hexagonal II) phase transition.
Another way to manipulate the molecular packing within bilayers is to induce curvature. For many years, it has been known that spontaneous cholesterol exchange is faster from small sonicated bilayer vesicles than from larger, planar-like bilayers vesicles [70, 121–122]. Indeed, for the case of cholesterol and zwitterionic diacyl phopholipids [70,121–122], it appears that the characteristics of the donor rather than the acceptor vesicles limit the transfer rate. In fact, the association rate of PC monomers with neutral acceptor vesicles has generally been thought to be diffusion limited by the aqueous mileu rather than by the interfacial characteristics of the acceptor surface [123]. However, in a recent study of the kinetic parameters governing the spontaneous transfer of retinol between bilayer vesicles, Noy and Xu [151–152] reported that retinol's association rate with PC bilayers depends strongly on the composition of the fatty acyl chains of the lipids. In contrast, retinol's dissociation rate from PC bilayers was independent of that composition.
Other recent studies of lipid transfer from micelles to bilayer vesicles also indicate that the nature of the acceptor vesicle can strongly influence the transfer process. Work from this and another lab suggest that the incorporation of negatively-charged ganglioside GM1 from its micelles to PC vesicles is sensitive to the nature of the acceptor vesicle surface [124–125]. Perhaps such findings should not come as a surprise. After all, according to Elamrani and Blume [126]. the rate-limiting step for the spontaneous transfer of lyso PC from its micelles to bilayers is the incorporation of lyso PC monomers into the lipid bilayer. These investigators suggested that the lyso PC incorporation rate is determined by density fluctuations in the lipid bilayer. Clearly, more work is needed to fully understand the subtle features of the lipid transfer process.
III-B. Lamellar compositional effects
In section II-B. a few classic cases were pointed out that illustrate how matrix composition affects lipid desorption rates. Detailed accounts of the comparative effects that PC, PE, gangliosides, and sphingomyelin have on cholesterol interbilayer transfer have also been discussed in previous reviews [1,4–7,9]. Acidic phospholipids also appear to stimulate spontaneous sterol transfer between bilayers as monitored by flourescent sterols [76–79]. A recent summary of this work has been included in the review by Schroeder et al. [9] covering cholesterol's dynamic behavior in membranes. Even so, there are now several newer investigations that provide more insight into the effects of matrix composition on lipid transfer behavior.
Investigations in the laboratories of J. Silvius [30,127] and R. Bittman [80–84] have focused on the role that hydrogen bonding plays in regulating interbilayer phospholipid, sphingolipid and sterol transfer events. Silvius and co-workers [29,30,127] have used a flourescence resonance energy transfer method analogous to that of Nichols and Pagano [28] to examine the partitioning of a variety of exchangeable fluorescent phospho- and sphingolipids between vesicles of various compositions. These investigators have evaluated how the polar head-group structure of a lipid molecule affects its partitioning between different lipid bilayer environments which vary in their surface charges, sterol contents, and/or polar headgroup compositions. Not surprisingly, lipid probes with different charges differed markedly in their equilibrium distributions between neutral and charged lipid vesicles. However, lipid probes with different polar headgroups differed only modestly in their relative affinities for vesicles comprised of ‘hydrogen-bonding’ lipids (PE and PS) compared to ‘non-hydrogen-bonding’ lipids (PC and PG). Probes with different head-groups also showed modest but reproducible differences in their relative affinities for vesicles composed of PC, PG and cholesterol (48: 12:40) compared with PC-PG (80:20) vesicles without cholesterol [30]. In more recent studies, these investigators [127] have compared the partitioning of analogues of different neutral phospholipids between bilayers composed mainly of PC. which can serve as a hydrogen-bond acceptor, and bilayers composed of a quaternary ammonium amphiphile that can neither donate nor accept hydrogen bonds. Neutral phospholipid probes whose headgroups serve as hydrogen-bond donors (e.g., PE) showed an increased affinity for the PC-rich vesicles when compared to probes with lesser or no hydrogen-bond-donating ability (e.g., PC). Careful analysis of the partitioning data revealed that interlipid hydrogen-bonding interactions (in competition with lipid-water interactions) contribute about -300 cal mol−1 to the free energy of a PE molecule in a hydrated liquid-crystalline phospholipid bilayer [127]. As a result, PE probes desorb from POPC vesicles at markedly slower rates than do PC probes with the same acyl chains.
Bittman and co-workers [80–84] have synthesized various sterol, phospholipid, and sphingomyelin derivatives in order to ascertain the relative contribution that intermolecular forces such as van der Waal's interactions and hydrogen-bonding play in determining spontaneous interbilayer lipid transfer behavior. Based on comparison of the exchange behavior of sitosterol and cholesterol between phospholipid vesicles and between lysophospholipid dispersions. Kan and Bittman [80–81] concluded that sitosterol's slower desorption rate is due to additional van der Waal's interactions with nearest neighbors and a lower aqueous phase solubility made possible by the presence of sitosterol's 24-ethyl group. In subsequent studies. Bittman and co-worker's [82–84] examined the kinetics of cholesterol exchange between unilamellar vesicles formed by various N- and S- linked phospholipid derivatives. The results suggest that hydrogen bonding between cholesterol and PC does not play an important role with respect to cholesterol's desorption rate from vesicles. Moreover, cholesterol's much slower desorption rate from sphingomyelin vesicles cannot be adequately explained by interlipid hydrogen bonding between cholesterol's 3-beta-hydroxy group and sphingomyelin's -NH- group. Also, upon systematically modifying the structure of cholesterol, no correlation could be found between the rates of sterol desorption from the lipid-water interface and the relative hydrophobicity of the sterol as estimated by reversed-phase high-performance chromatography [84].
Although the majority of studies involving changes in matrix composition have focused on lipid composition, it is clear that the presence of membrane proteins also modulates spontaneous interbilayer lipid transfer rates. A recent example comes from the work of Nishiva and Chang [128]. Using a PC derivative containing a circular dichroic-active reporter group, these investigators noted that melittin accelerates the transfer of the CD-active PC from gel-phase but not liquid-crystalline vesicles.
III-C. Matrix mesophasic structure
Although biological membranes are predominantly lamellar in nature, other mesophasic forms of lipid assemblies serve important physiological roles. The mesomorphic form of organized lipid assemblies does have dramatic effects on lipid desorption. Illustrations of this behavior are provided by work from several different laboratories. For instance, Nichols and coworkers [96–98] have compared the spontaneous inter-vesicular and intermicellar transfer of various fluorescent PE derivatives. They reported that, when micelle and vesicle concentrations are kept low, PE's dissociation rate from micelles ranges from 200 to 800-fold faster than that from vesicles. These investigators rationalized that the acyl chains of PE within a bile salt micelle find themselves exposed to water more often than PE within a phospholipid vesicle because of the very rapid exchange of bile salts between micelles compared to phospholipid exchange between vesicles. The end result would be an increase in free energy for PE in the micelle. Thus, less activation free energy would be required to transfer phospholipids at the micellar disk's periphery compared to the higher-energy pathway available for phospholipid dissociation from vesicles. In addition, as the acyl chains of the phospholipids become longer, an added geometric factor may cause further destabilization because of the inability of bile salts to shield the entire length of the phospholipid acyl chains from water at the periphery of the micellar disks. As pointed out by Nichols [97], such effects may be very important physiologically in facilitating the rapid and complete transfer of phospholipid monomers from mixed micelles to the plasma membrane of enterocytes in the intestinal mucosa.
In studies involving ganglioside transfer, results from this lab [125] and others [63–64,124] have clearly shown that the spontaneous transfer of ganglioside GM1 from its micelles to bilayer vesicles is much faster than GM1 transfer between bilayer vesicles. A detailed description of these investigations has appeared previously [1].
Of course, micelles are only one type of nonbilayer form that lipids assume. Another complex assembly involved in lipid transport is the lipoprotein. Because of their obvious biological importance, lipoproteins have been studied intensely with respect to spontaneous lipid transfer processes. This work has already been reviewed [6,10,11,155].
III-D. Aqueous mileu
Alterations of the aqueous mileu also have significant effects on the spontaneous interbilayer transfer of lipids. Such effects were first shown for phosphatidylglycerol [110] and later studied in detail for fatty acid interbilayer transfer by Smith, Pownall and co-workers [114–115]. These investigators showed that increasing the ionic strength of the aqueous medium slowed the interbilayer transfer of pyrene-labeled fatty acids. A similar decrease in the transfer rate of long-chain anthroloxy-labeled fatty acids was noted by Storch and Kleinfeld [129] with increasing ionic strength. Such findings were taken a evidence that fatty acid transfer occurs by desorption of monomers from the donor lipid surface followed by diffusion through the aqueous phase.
The spontaneous interbilayer transfer of phospholipids, be they zwitterionic or charged, is also strongly influenced by the ionic strength of the aqueous medium [105,108,110]. This phenomenon arises from the ‘salting out’ of lipid from the aqueous phase as described by DeCuyper et al. [105] and by Jones and Thompson [108].
Based on the above observations, it should come as no surprise that the interbilayer transfer of certain phospholipids and fatty acids is also sensitive to the pH of the aqueous medium. Indeed, DeCuyper and Joniau [104] demonstrated the marked effect that pH has on the transfer of DMPA between DMPC vesicles. More recently, Silvius and co-workers [30] reported a ten-fold stimulation in the transfer of a fluorescent PA derivative, but not the corresponding PE, PS or PC derivatives, as the pH was systematically increased from 6.0 to 8.3.
Eastman et al. [144] examined the influence of transmembrane proton gradients on the intervesicular exchange of oleic acid and stearylamine. Oleic acid and stearylamine can be induced to transfer from one vesicle population to another by maintaining either a basic or acidic interior pH in the acceptor vesicles, respectively. These pH gradients promote transmembrane asymmetries that concentrate the oleic acid and stearylamine in the inner monolayers of the acceptor vesicles [144–145].
In another study of fatty acid distribution between egg PC vesicles, Gomez-Fernandez and co-workers [146] noted that both the ionized and protonated forms of stearic and oleic acids achieved similar equilibrium distributions between egg PC vesicles of different size. However, vesicle charge strongly influenced the outcome since both free fatty acids in their ionized forms associated preferentially with PC vesicles compared to PG vesicles.
Other manipulations of the aqueous mileu known to affect spontaneous interbilayer lipid transfer involve addition of chaotropic salts [69,72] or poly(ethylene)glycol [130–131]. In such cases, it is difficult to distinquish whether the primary effect is restructuring of the bulk aqueous milieu or the highly polarized hydration zone associated with the surface of organized lipid assemblies. Both effects may be important.
IV. What is the mechanism of spontaneous lipid transfer?
IV-A. Monomer desorption and diffusion through aqueous phase between membranes
The mechanism by which spontaneous lipid transfer occurs between membranes has been investigated in many independent laboratories [23,26,57,66,110–114,132–135]. In the vast majority of cases, it has been concluded that the transfer event is a first-order process in which the rate-limiting step is desorption of lipid molecules from the donor membrane surface followed by rapid diffusion through the aqueous phase to an acceptor membrane (Fig. 1A). Generally, this conclusion has been based on the experimental observation that the lipid transfer rate is not affected by varying the acceptor membrane concentration while the donor membrane concentration is held constant. As has been pointed out by Nichols and Pagano [24,28], the validity of this experimental approach must be confirmed by additional means if donor and acceptor membranes are of differing composition and/or size.
The thermodynamic parameters associated with the kinetics of lipid monomer desorption from a phospholipid vesicle surface have been studied in detail [116,123]. Thermodynamic analysis of the activated state and of fatty acid partitioning indicates that the rate-limiting step is formation of an aqueous intermediate in the interfacial region [114]. Moreover, fatty acid transfer kinetics are not dictated by only the partition equilibirum, but are also influenced by the solvation properties of the interfacial water associated with the vesicles [114]. Comparison of the fatty acid transfer rates and activation energies with those of corresponding methyl ester, alcohol, and alkane derivatives has yielded further insights [115]. For any given derivative, the transfer rate increases as the temperature rises and decreases as the hydrocarbon chain lengthens. Not surprisingly, the activation energy for transfer rises as the chain length increases, but at a given chain length, the activation energy increases in the following order: ester (pH 2.8) < acid (pH 9.0) < alcohol < acid (pH 2.8) < ester (pH 7.4). The transfer rate, however, generally decreases in the same order. Thus, the overall transfer rate is related to both the hydrophobicity (chain length) and the hydrophilicity (polarity of functional groups) of the transferred species [115].
The thermodynamic parameters associated with the kinetics of diacyl PC, lyso PC, and cholesterol desorption from a vesicle surface has also been investigated in detail [116,123]. McLean and Phillips [116] used radiolabeled derivatives of DMPC. DPPC, POPC and cholesterol to determine their exchange rates at various temperatures. For DMPC, t1/2 = 2.0 h at 37°C; t1/2 = 6.5 h at 24.5°C; t1/2 = 82.6 h below 24°C and the activation energy (27–45°C) is 70 kJ mol−1. Using the exchange rates to calculate the activation free energies for exchange of the various lipids, the activation free energies and free energies of transfer from self-aggregates to water were found to increase by 2.2 and 2.1 kJ mol−1 per methylene unit, respectively. Thus, the free energy of transfer is a good predictor of the relative exchange rates of lipid molecules. However, the activation free energies are 30 ± 1 kJ mol−1 greater than the free energies of transfer. This excess free energy is proposed to be associated with restriction of the lipid molecule to the vesicle surface in a transition-state complex [116].
Using fluorescence approaches, Nichols [123] has provided a complete description of the transfer of long-chain PC monomers from membranes into solution based on earlier studies of amphiphile micelle behavior [132–133]. Determining the dependence of acyl chain length and temperature on PC's kinetic rate and equilibrium partition into water permitted conduction of a free energy diagram for monomer-vesicle interactions (Fig. 2). The conclusions of this analysis are: (1) enthalpic energy dominates the creation of the high energy transition state that accompanies fluorscent PC association with and dissociation from the bilayer. (2) Increasing acyl chain length results in more positive values for the activation energy of monomer-vesicle dissociation and more negative values for the free energy of PC transfer from water to vesicles. (3) Enthalpic differences between membrane and water phases dominate the free energy of PC transfer from water to vesicles. (4) Increasing acyl chain length raises the activation enthalpy for association, which is larger than expected for an aqueous diffusion-limited process in bulk water.
Fig. 2.
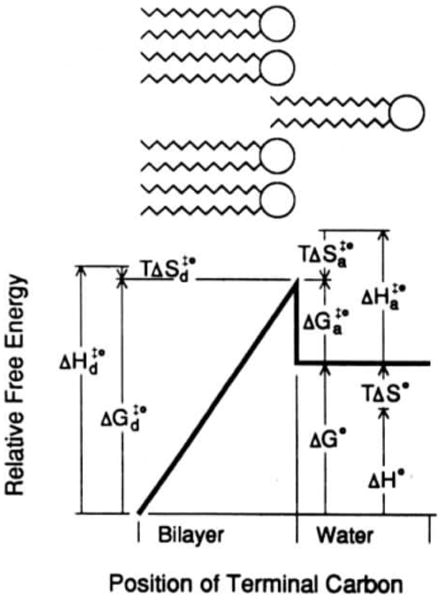
Free energy diagram of fluorescent PC partitioning between liquid-crystalline PC small unilamellar vesicles and water. The upper schematic represents a phospholipid molecule desorbing from a phospholipid bilayer. In the energy diagram below, the length and direction of the arrows represent the absolute magnitudes of the energy components (reprinted with permission from Nichols [123], copyright 1985, American Chemical Society).
An associated cavity model of solvation provides a useful illustration of the nature of the activated complex (Fig. 2). As described by Nichols [123], the peak of the transition state occurs when the solute phospholipid has moved normal to the bilayer plane such that only the terminal carbon remains inserted. This is a high energy state because the terminal carbon does not allow the surrounding phospholipids to collapse to fill the cavity created by an exiting molecule. The transition state requires the creation of two cavities simultaneously, one in the bilayer and one in the water phase. Creation of a water-phase cavity presumably requires the energy to separate hydrogen-bonded water molecules whereas the energy opposing cavity creation in the bilayer arises from van der Waals interactions between closely opposed phospholipid acyl chains and electrostatic interactions between headgroups. Once the exiting molecules leave the bilayer, the surrounding phospholipids collapse to till the cavity resulting in a lower energy state.
IV-B. Transient apposition of hydrated membrane surfaces
As early as 1960, Gurd [136] proposed a model for cholesterol transfer involving the formation of a collisional complex between membranes resulting in the transient diffusion of lipid within the complex. However, as was pointed out above, the majority of experimental data reported prior to 1988 argued against such a mechanism of lipid transfer (e.g., Refs. 23,26,57,66,110–114,132–135). Then, two different laboratories both reported increased spontaneous lipid transfer rates attributable to transient collisions between donor and acceptor surfaces [96–98,107–109]. Although similar claims had been reported previously [22,113], it was difficult to rule out fusion between vesicles [113] and protein-mediated lipid transfer [22,137] in these earlier reports.
In 1988, Nichols [96] reported that the spontaneous transfer rate of a fluorescent PE increased markedly if the donor and acceptor surfaces are mixed PC-taurocholate micelles rather than PC bilayer vesicles. A kinetic analysis of the initial transfer rate data was used to determine a mechanistic model that best described the data. Accordingly, Nichols [96] found that intermicellar PE transfer is faster than intervesicular PE transfer because of (i) exchange between micelles mediated by transient collisions that is not observed between vesicles and (ii) an increased rate of PE monomer desorption from the micelles compared to the vesicles. Collision-dependent transfer dominates at high micelle concentrations: whereas, monomer desorption dominates at low micelle concentrations. Nichols [96] also reported that, although increasing acyl chain length decreases the PE transfer rate associated with monomer desorption, the bi- and termolecular rate constants for PE transfer associated with micellar collisions are relatively independent of acyl chain length. To explain this latter finding, Nichols [96] proposed the formation of transient fusion complexes resulting in an accompanying ‘hydrocarbon continuum’ that requires no exposure of PE's hydrocarbon acyl chains to water during transfer. As pointed out by Nichols [96], a ‘hydrocarbon continuum’ model has been postulated by Patton [138] to explain the intestinal absorption of very hydrophobic lipid molecules. This mechanistic model is very reminiscent of one suggested by Gurd [136] to explain the spontaneous transfer of lipids between membranes.
Additional studies of the temperature and rate dependence reveal the thermodynamic parameters associated with the micellar collisional process [98]. Activation enthalpy is the major barrier to transfer and arises from the energy needed to dehydrate the interacting surfaces and overcome the electrostatic, hydrogen-bonding, and van der Waals interactions between bile salts and phospholipids that oppose the close apposition and fusion of the two micelles [98]. However, the increased rate of collision-dependent transfer between mixed micelles comprised of more hydrophobic bile salts (e.g., chenodeoxycaolate) results primarily from increased activation entropy. Presumably, their surfaces tend to structure the surrounding water molecules more than mixed micellar surfaces containing the less hydrophobic taurocholate molecules. Increased entropy favors formation of the fused complex to reduce the surface area of exposed hydrophobic lipids. Since there is no detectable growth in average micelle size, the fused complex must be unstable and short-lived.
Meanwhile, Thompson and co-workers [107–109] observed that, under appropriate conditions. POPC spontaneous transfer between bilayer vesicles is characterized by two rate processes. One is first order and the other second order with respect to vesicle concentration. These two processes reflect the desorption of POPC from the donor surface and transfer due to donor-acceptor collisions, respectively. As vesicle concentration is increased, the transfer flux arising from the concentration-dependent process dominates. For POPC transfer, the critical vesicle concentration is about 2 mM at 37°C. At acceptor lipid concentrations above 2 mM, a second term, proportional to acceptor concentration, must be added to the first-order, vesicle concentration-independent process to describe adequately the kinetics of POPC transfer. Almost all studies in the literature are in the low vesicle concentration regime where the second-order term is neglible. This explains the observation of apparent first-order kinetics in many of these earlier reports.
In subsequent studies, the mechanism by which vesicle-vesicle interactions enhance lipid transfer rates has been investigated as a function of lipid and medium composition as well as temperature and vesicle size [108–109]. The results provide insights into the vesicle-vesicle mediated transfer process. Comparison of the first-and second-order rate constants for POPC and DMPC transfer from POPC vesicles at 30°C shows that both rate constants are about two-orders of magnitude larger for DMPC than for POPC, but that the second-order rate constant is roughly proportional to the first-order constant. This finding precludes a model in which the second-order process reflects lateral diffusion of lipid molecules within a collisionally-induced, transient fusion complex between two vesicles, as suggested by Gurd [136]. If this were the case, the second-order rate constant should be equal for POPC and DMPC since all phospholipids in liquid-crystalline vesicles have very similar lateral diffusion coefficients [139). Another piece of evidence arguing against Gurd's model is the failure to observe significant transfer of cholesterol oleate. a nonexchangeable marker used to monitor fusional processes.
Interestingly, in addition to the observed proportionality between the second- and first-order rate constants. the second-order rate constant has essentially the same temperature dependence as the first-order desorption constant and thus the same activation parameters. These results are consistent with a model in which the second-order process also occurs via monomer transfer between vesicles, but that the interacting vesicles do not enter into direct physical contact. Otherwise, a very large activation energy would be incurred in order to dehydrate the polarized water molecules near the vesicles' surfaces [140–142]. Rather, as depicted in Fig. 3 and proposed by Jones and Thompson [108], monomer desorption from one vesicle is enhanced by close apposition to another vesicle (Fig. 1B). The two vesicles remain separated by a water-filled gap with a dimension dictated by the minimum in the interaction energy function of the two vesicles [108]. Based on calculations by Lis et al. [140], there exists a region in which vesicles experience a net attractive van der Waals interaction once they are separated by a distance greater than that at which short-range repulsive forces dominate the interaction. Upon reaching this favorable separation distance, the formation of the activated state of the desorbing phospholipid monomer is enhanced by the attractive force between the apposing vesicle and desorbing monomer. For PC vesicles, this separation distance is assumed to be about 1.5 nm, the distance at which repulsive and attractive forces are equal [140–142].
Fig. 3.

Energy diagram showing the effect of acceptor vesicle interaction on lipid monomer activation energy. In A, the free energy associated with lipid monomer desorption from the phospholipid bilayer into the aqueous phase is depicted. In B. the decrease in activation energy upon apposition of an acceptor vesicle is depicted. The free energy of interaction is the attractive energy imparted to the monomer by the acceptor vesicle. The vesicle-vesicle separation distance is assumed to be approximately that at which repulsive and attractive forces are equal (approx. 1.5 nm) (reprinted with permission from Jones and Thompson [108], copyright 1990, Amercian Chemical Society).
Estimates of the decrease in activation energy for monomer desorption by apposition with acceptor vesicles reveal that a difference in activation energy of about 1 kcal/mol is sufficient to produce a 5-fold difference in off-rate. This appears within the range of expected excess energy of interaction between a vesicle and a lipid monomer at distances between 0.1 and 1.5 nm. However, calculations of the efficiency of phospholipid transfer mediated by vesicle collisions (50 mM) reveal that it is a highly inefficient process in which 4.4 (10) collisions are required to desorb one phospholipid molecule. Thus, significant impact of the second-order process is only seen at high vesicle concentrations [108].
Interestingly, the relative efficiency of the second-order transfer process appears to be highly dependent on lipid monomer properties, as is illustrated by cholesterol transfer. In contrast to the PC transfer results, Jones and Thompson [108] found that the kinetics of radiolabeled cholesterol transfer between POPC vesicles showed no dependence on vesicle concentration over the same range. These investigators viewed this result as evidence that monomer-vesicle head-group interactions may contribute significantly to the attractive force and that the molecular geometry of a lipid's polar and nonpolar regions, such as hydrophobic length, are important discriminators of second-order transfer efficiency.
In any case, if the model proposed by Jones and Thompson [108] is correct, factors such as surface hydration or charge should have significant effects on the efficiency of the concentration-dependent exchange process. Wimley and Thompson [109] tested these ideas by examining the effect of phosphatidyl-ethanolamine (PE) and phosphatidylglycerol (PG) on second-order PC transfer events. These investigators found that incorporation of 30 mol% PE into the PC vesicles enhanced the efficiency of collisionally-mediated DMPC exchange by two to three orders of magnitude. This PE-induced enhancement disappears if a small amount of charged phospholipid (1.75 mol% PG) is included in the vesicles. These findings are consistent with the mechanistic model proposed by Jones and Thompson [117–108] to explain the second-order transfer process. Bilayer vesicles containing PE tend to be less hydrated than PC and as a result have a smaller equilibrium bilayer separation distance and a deeper energy interaction ‘well’ [140–142]. Thus, it is not surprising that more efficient concentration-dependent PC exchange occurs between vesicles containing significant amounts of PE. Moreover, electrostatic repulsions produced in bilayer surfaces containing charged phospholipids will not only diminish the frequency of inter-vesicle collisions via long-range electrostatic interactions but will also increase the equilibrium separation distance in an intervesicular collisional complex and decrease the depth of the interaction energy minimum [109].
Other evidence suppporting the idea that changes in the dimensions and properties of the aqueous gap between apposed lipid vesicles can regulate spontaneous intervesicular lipid transfer rates comes from studies of poly(ethylene glycoD-induced lipid transfer between PC vesicles. Based on their results, Lentz and co-workers [130–131] proposed that poly(ethylene glycol) enhances the rate of intervesicular lipid transfer by dehydrating and aggregating the PC vesicles thereby altering the properties of the aqueous gap between the membrane surfaces.
In any case, the discovery of the concentration dependent mechanism of spontaneous phospholipid transfer between bilayers raises interesting possibilities for some yet unrecognized role in membrane biogenesis. As pointed out in earlier discussions, the spontaneous transfer rate of diacyl phospholipids has been thought to be too slow to be of consequence in membrane biogenesis. Indeed, this does appear to be the case based on diacyl PC monomer desorption rates from bilayers. However, the recognition that spontaneous PC transfer can be enhanced substantially by collisions between bilayers and that the efficiency of the collisionally-dependent process can be improved two or three orders of magnitude by manipulating membrane composition raises exciting new possibilities for an in vivo role for spontaneous PC transfer. It is interesting to speculate that targeted vesicular trafficking within the cell might bring membranes together and once in close apposition, certain lipids could be exchanged without wholesale fusion of the vesicle to the membrane. Indeed, there are many ways that cells might facilitate and extend the lifetime of a close apposition between membrane surfaces. Silvius and co-workers [143] have carefully studied the aggregation between cationic lipid vesicles and negatively-charged phospholipid vesicles. These investigators noted that the extent of aggregation is markedly enhanced when the vesicles contain high proportions of PE relative to PC. This extended apposition of surfaces results in substantial lipid mixing on the time scale of a few minutes by a process of ‘hemifusion’, in which the outer monolayers of the two apposed bilayers can intermix without simultaneous coalescence of their inner monolayers and aqueous compartments (Fig. 1C). In any case, more work is needed to sort out these exciting possibilities.
V. Implications
Over the past decade, tremendous strides have been made in our understanding of spontaneous lipid transfer processes. There is no longer any doubt about the value of studying such dynamic processes. Clearly, although much has been accomplished, much remains to be done. If recent studies are any indicator, then more and more future studies will be devoted to the transfer behavior of lipid compounds that are of nutritional [148–152] and pharmacologic interest [153–154].
Acknowledgments
I wish to extend special thanks to T.E. Thompson for the many stimulating and helpful discussions that led to my interest in spontaneous lipid transfer processes and to the Hormel Foundation and United States Public Health Service Grant GM45928 for their support.
Abbreviations
- lyso PC
l-O-hexadecanoyl-sn-glycero-3-phosphocholine
- PC
1.2-diacyl-sn-glycero-3-phosphocholine
- PE
1.2-diacyl-sn-glycero-3-phosphoethanolamine
- GlcCer
glucosylceramide
- asialo-GM1
gangliotetraosylceramide
- GM1
II3-N-acetylneuraminosyl-gangliotetraosylceramide
- PA
1.2-diacyl-sn-glycero-3-phosphate
- POPC
l-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DMPC
dimyristoyl-sn-glycero-3-phosphocholine
- DPPC
dipalmitoyl-sn-glycero-3-phosphocholine
- DMPE
dimyristoyl-sn-glycero-3-phosphoethanolamine
- DMPG
dimyristoyl-sn-glycero-3-phospho-1′-sn-glycerol
- SUV
small unilamellar vesicle
- LUV
large unilamellar vesicle
- DSPC
distearoyl-sn-glycero-3-phosphocholine
- PG
1.2-diacyl-sn-glycero-3-phospho-1′-sn-glycerol
- DPG
diphosphatidyl-glycerol
- PS
l.2-diacyl-sn-glycero-3-phosphoserine
- CD
circular dichroism
References
- 1.Brown RE. In: Subcellular Biochemistry. Hilderson HJ, editor. Vol. 16. Plenum Press; New York: 1990. pp. 333–363. [Google Scholar]
- 2.Bruckdorfer KR, Graham JM. In: Biological Membranes. Chapman D, Wallach D, editors. Vol. 13. Academic Press; London: 1976. pp. 103–152. [Google Scholar]
- 3.Lange Y. In: Handbook of Lipid Research: The Physical Chemistry of Lipids. Small DM, editor. Vol. 4. Plenum Press; New York: 1986. pp. 538–549. [Google Scholar]
- 4.Sleight RG. Annu Rev Physiol. 1987;49:193–208. doi: 10.1146/annurev.ph.49.030187.001205. [DOI] [PubMed] [Google Scholar]
- 5.Dawidowicz EA. Curr Top Membr Transp. 1987;29:175–202. [Google Scholar]
- 6.Phillips MC, Johnson WJ, Rothblat GH. Biochim Biophys Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 7.Bittman R. In: Biology of Cholesterol. Yeagle PL, editor. CRC Press; Boca Raton: 1988. [Google Scholar]
- 8.Thompson TE, Brown RE. New Trends in Ganglioside Research: Neurochemical and Neuroregenerative Aspects. In: Ledeen RW, Hogan EL, Tettamanti G, Yates AJ, Yu RK, editors. Fidia Res Ser. Vol. 14. Liviana Press; Padova: 1988. pp. 65–78. [Google Scholar]
- 9.Schroeder F, Jefferson JR, Kier AB, Knittel J, Seallen TJ, Wood WG, Mapala I. Proc Soc Exp Biol Med. 1991;196:235–252. doi: 10.3181/00379727-196-43185. [DOI] [PubMed] [Google Scholar]
- 10.Pownall HJ. Adv Exp Med Biol. 1988;243:173–177. doi: 10.1007/978-1-4613-0733-4_21. [DOI] [PubMed] [Google Scholar]
- 11.Pownall HJ, Smith LC. Chem Phys Lipids. 1989;50:191–211. doi: 10.1016/0009-3084(89)90050-9. [DOI] [PubMed] [Google Scholar]
- 12.Brunner J, Spiess M, Aggeler R, Huber P, Semenza G. Biochemistry. 1983;22:3812–3820. doi: 10.1021/bi00285a016. [DOI] [PubMed] [Google Scholar]
- 13.Schroit AJ, Madsen J, Ruoho AE. Biochemistry. 1987;26:1812–1819. doi: 10.1021/bi00381a004. [DOI] [PubMed] [Google Scholar]
- 14.Zachowski A, Fellmann P, Hervè, Devaux P. FEBS Lett. 1987;223:315–320. doi: 10.1016/0014-5793(87)80311-3. [DOI] [PubMed] [Google Scholar]
- 15.Sonnino S, Chignorno V, Acquotti D, Pitto M, Kirschner G, Tettamanti G. Biochemistry. 1989;28:77–84. doi: 10.1021/bi00427a012. [DOI] [PubMed] [Google Scholar]
- 16.Chigorno V, Valsecchi M, Acquotti D, Sonnino S, Tettamanti G. FEBS Lett. 1990:263, 329–331. doi: 10.1016/0014-5793(90)81406-e. [DOI] [PubMed] [Google Scholar]
- 17.Kanda S, Inoue K, Nojima S, Utsumi H, Wiegandt H. J Biochem. 1982;91:1707–1718. doi: 10.1093/oxfordjournals.jbchem.a133862. [DOI] [PubMed] [Google Scholar]
- 18.Kanda S, Inoue K, Nojima S, Utsumi H, Wiegandt H. J Biochem. 1982;91:2095–2098. doi: 10.1093/oxfordjournals.jbchem.a133904. [DOI] [PubMed] [Google Scholar]
- 19.Seigneuret M, Devaux P. Proc Natl Acad Sci USA. 1984;81:3751–3755. doi: 10.1073/pnas.81.12.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrell JE, Lee KJ, Huestis WH. Biochemistry. 1985;24:2849–2857. doi: 10.1021/bi00333a006. [DOI] [PubMed] [Google Scholar]
- 21.Tamura A, Yoshikawa K, Fujii T, Ohki K, Nozawa Y, Sumida Y. Biochim Biophys Acta. 1986;855:250–256. doi: 10.1016/0005-2736(86)90171-9. [DOI] [PubMed] [Google Scholar]
- 22.Mutsch B, Gains N, Hauser H. Biochemistry. 1986;25:2134–2140. doi: 10.1021/bi00356a043. [DOI] [PubMed] [Google Scholar]
- 23.Roseman MA, Thompson TE. Biochemistry. 1980;19:439–444. doi: 10.1021/bi00544a006. [DOI] [PubMed] [Google Scholar]
- 24.Struck DK, Pagano RE. J Biol Chem. 1980;255:5404–5410. [PubMed] [Google Scholar]
- 25.Pagano RE, Martin OC, Schroit AJ, Struck DK. Biochemistry. 1981;20:4920–4927. doi: 10.1021/bi00520a018. [DOI] [PubMed] [Google Scholar]
- 26.Nichols JW, Pagano RE. Biochemistry. 1981;20:2783–2789. doi: 10.1021/bi00513a012. [DOI] [PubMed] [Google Scholar]
- 27.Correa-Freire MC, Barenholz Y, Thompson TE. Biochemistry. 1982;21:1244–1248. doi: 10.1021/bi00535a021. [DOI] [PubMed] [Google Scholar]
- 28.Nichols JW, Pagano RE. Biochemistry. 1982;21:1720–1726. doi: 10.1021/bi00537a003. [DOI] [PubMed] [Google Scholar]
- 29.Silvius JR, Leventis R, Brown PM, Zuckermann M. Biochemistry. 1987;26:4279–4287. doi: 10.1021/bi00388a015. [DOI] [PubMed] [Google Scholar]
- 30.Gardam MA, Itovitch JJ, Silvius JR. Biochemistry. 1989;28:884–893. doi: 10.1021/bi00428a072. [DOI] [PubMed] [Google Scholar]
- 31.Pagano RE, Sleight RG. Science. 1985;229:1051–1057. doi: 10.1126/science.4035344. [DOI] [PubMed] [Google Scholar]
- 32.Sleight RG, Abanto MN. J Cell Sci. 1989;93:363–374. doi: 10.1242/jcs.93.2.363. [DOI] [PubMed] [Google Scholar]
- 33.Pagano RE. Biochem Soc Trans. 1990;18:361–366. doi: 10.1042/bst0180361. [DOI] [PubMed] [Google Scholar]
- 34.Kok JW, Babia T, Hoekstra D. J Cell Biol. 1991;114:231–239. doi: 10.1083/jcb.114.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simons K, Van Meer G. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- 36.Van Meer G. Annu Rev Cell Biol. 1989;5:247–275. doi: 10.1146/annurev.cb.05.110189.001335. [DOI] [PubMed] [Google Scholar]
- 37.Schwarzmann G, Sandhoff K. Biochemistry. 1990;29:10865–10871. doi: 10.1021/bi00501a001. [DOI] [PubMed] [Google Scholar]
- 38.Roberts DD, Ginsburg V. Arch Biochem Biophys. 1988;267:405–415. doi: 10.1016/0003-9861(88)90046-x. [DOI] [PubMed] [Google Scholar]
- 39.Hakomori S. J Biol Chem. 1990;265:18713–18716. [PubMed] [Google Scholar]
- 40.Karlsson KA. Annu Rev Biochem. 1989;58:309–350. doi: 10.1146/annurev.bi.58.070189.001521. [DOI] [PubMed] [Google Scholar]
- 41.Hannun YA, Bell RM. Science. 1989;243:500–507. doi: 10.1126/science.2643164. [DOI] [PubMed] [Google Scholar]
- 42.Ogura K, Handa S. J Biochem. Vol. 104. Tokyo: 1988. pp. 87–92. [DOI] [PubMed] [Google Scholar]
- 43.Ghidoni R, Riboni L, Tettamanti G. J Neurochem. 1989;53:1567–1574. doi: 10.1111/j.1471-4159.1989.tb08553.x. [DOI] [PubMed] [Google Scholar]
- 44.Van Echten G, Sandhoff K. J Neuroehem. 1989;52:207–214. doi: 10.1111/j.1471-4159.1989.tb10918.x. [DOI] [PubMed] [Google Scholar]
- 45.Kivatinitz SC, Miglio A, Ghidoni R. Biochem, J. 1991:274, 581–585. doi: 10.1042/bj2740581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riboni L, Tettamanti G. J Neuroehem. 1991;57:1931–1939. doi: 10.1111/j.1471-4159.1991.tb06406.x. [DOI] [PubMed] [Google Scholar]
- 47.Oishi K, Raynor RL, Charp PA, Kuo JF. J Biol Chem. 1988;263:6865–6871. [PubMed] [Google Scholar]
- 48.Quinn MT, Parthasarathy S, Steinberg D. Proc Natl Acad Sci USA. 1988;85:2805–2809. doi: 10.1073/pnas.85.8.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metz SA. Biochim Biophys Acta. 1988;968:239–252. doi: 10.1016/0167-4889(88)90013-4. [DOI] [PubMed] [Google Scholar]
- 50.Besterman JM, Domanico PL. Biochemistry. 1992;31:2046–2056. doi: 10.1021/bi00122a022. [DOI] [PubMed] [Google Scholar]
- 51.Noy N, Donnelly TM, Zakim D. Biochemistry. 1986;22:2013–2021. doi: 10.1021/bi00356a027. [DOI] [PubMed] [Google Scholar]
- 52.Cooper R, Noy N, Zakim D. Biochemistry. 1987;26:5890–58. doi: 10.1021/bi00392a047. [DOI] [PubMed] [Google Scholar]
- 53.Cooper R, Noy N, Zakim D. J Lipid Res. 1989;30:1719–1726. [PubMed] [Google Scholar]
- 54.Weltzien HU. Biochim Biophys Acta. 1979;559:259–287. doi: 10.1016/0304-4157(79)90004-2. [DOI] [PubMed] [Google Scholar]
- 55.Mohandas N, Greenquist AC, Shohet SB. J Supramol Struct. 1978;9:453–458. doi: 10.1002/jss.400090315. [DOI] [PubMed] [Google Scholar]
- 56.Lange Y, Slayton JM. J Lipid Res. 1982;23:1121–1127. [PubMed] [Google Scholar]
- 57.Ferrell JE, Lee KJ, Huestis WH. Biochemistry. 1985;24:2857–2864. doi: 10.1021/bi00333a007. [DOI] [PubMed] [Google Scholar]
- 58.Daleke DL, Huestis WH. Biochemistry. 1985;24:5406–5416. doi: 10.1021/bi00341a019. [DOI] [PubMed] [Google Scholar]
- 59.Devaux PF. Biochemistry. 1991;30:1163–1173. doi: 10.1021/bi00219a001. [DOI] [PubMed] [Google Scholar]
- 60.Golan DE, Furlong ST, Brown CS, Caulfield JP. Biochemistry. 1988;27:2661–2667. doi: 10.1021/bi00408a005. [DOI] [PubMed] [Google Scholar]
- 61.Correa-Freire MC, Freire E, Barenholz Y, Biltonen RL, Thompson TE. Biochemistry. 1979;18:442–445. doi: 10.1021/bi00570a008. [DOI] [PubMed] [Google Scholar]
- 62.Frank A, Barenholz Y, Lichtenberg D, Thompson TE. Biochemistry. 1983;22:5647–5651. doi: 10.1021/bi00276a023. [DOI] [PubMed] [Google Scholar]
- 63.Brown RE, Sùgar IP, Thompson TE. Biochemistry. 1985;24:4082–4091. doi: 10.1021/bi00336a042. [DOI] [PubMed] [Google Scholar]
- 64.Brown RE, Thompson TE. Biochemistry. 1987;26:5454–5460. doi: 10.1021/bi00391a036. [DOI] [PubMed] [Google Scholar]
- 65.Thompson TE, Barenholz Y, Brown RE, Correa-Freire M, Young WW, Tillack TW. In: Enzymes of Lipid Metabolism II. Freysz L, Dreyfus H, Massarelli R, Gatt S, editors. Plenum; New York: 1986. pp. 387–396. [Google Scholar]
- 66.McLean LR, Phillips M. Biochemistry. 1981;20:2893–2900. doi: 10.1021/bi00513a028. [DOI] [PubMed] [Google Scholar]
- 67.McLean LR, Phillips M. Biochemistry. 1982;21:4053–4059. doi: 10.1021/bi00260a022. [DOI] [PubMed] [Google Scholar]
- 68.Nakagawa Y, Inoue K, Nojima S. Biochim Biophys Acta. 1979;553:307–319. doi: 10.1016/0005-2736(79)90234-7. [DOI] [PubMed] [Google Scholar]
- 69.Clejan S, Bittman R. J Biol Chem. 1984;259:10823–10826. [PubMed] [Google Scholar]
- 70.Fugler L, Clejan S, Bittman R. J Biol Chem. 1985;260:4098–4102. [PubMed] [Google Scholar]
- 71.Bar L, Barenholz Y, Thompson TE. Biochemistry. 1986;25:6701–6705. doi: 10.1021/bi00369a056. [DOI] [PubMed] [Google Scholar]
- 72.Yeagle PL, Young JE. J Biol Chem. 1986;261:8175–8181. [PubMed] [Google Scholar]
- 73.Bar L, Barenholz Y, Thompson TE. Biochemistry. 1987;26:5460–5465. doi: 10.1021/bi00391a037. [DOI] [PubMed] [Google Scholar]
- 74.Lund-Katz S, Laboda HM, McLean LR, Phillips MC. Biochemistry. 1988;27:3416–3423. doi: 10.1021/bi00409a044. [DOI] [PubMed] [Google Scholar]
- 75.Thomas PD, Poznansky MJ. Biochem, J. 1988;251:55–61. doi: 10.1042/bj2510055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nemecz G, Schroeder F. Biochemistry. 1988;27:7740–7749. doi: 10.1021/bi00420a024. [DOI] [PubMed] [Google Scholar]
- 77.Nemecz G, Fontaine RN, Schroeder F. Biochim Biophys Acta. 1988;943:511–521. doi: 10.1016/0005-2736(88)90384-7. [DOI] [PubMed] [Google Scholar]
- 78.Schroeder F, Nemecz G. Biochemistry. 1989;28:5992–6000. doi: 10.1021/bi00440a041. [DOI] [PubMed] [Google Scholar]
- 79.Hapala I, Butko P, Schroeder F. Chem Phys Lipids. 1990;56:37–47. doi: 10.1016/0009-3084(90)90086-7. [DOI] [PubMed] [Google Scholar]
- 80.Kan CC, Bittman R. J Am Chem Soc. 1990;112:884–886. [Google Scholar]
- 81.Kan CC, Bittman R. J Am Chem Sue. 1991;113:6680–6686. [Google Scholar]
- 82.Kan CC, Ruan Z, Bittman R. Biochemistry. 1991;30:7759–7766. doi: 10.1021/bi00245a013. [DOI] [PubMed] [Google Scholar]
- 83.Kan CC, Bittman R, Hajdu J. Biochim Biophys Acta. 1991;1066:95–101. doi: 10.1016/0005-2736(91)90256-8. [DOI] [PubMed] [Google Scholar]
- 84.Kan CC, Yan J, Bittman R. Biochemistry. 1992;31:1866–1874. doi: 10.1021/bi00121a040. [DOI] [PubMed] [Google Scholar]
- 85.Golan DE, Brown CS, Cianci CM, Furlong ST, Caulfield JP. J Cell Biol. 1986;103:819–828. doi: 10.1083/jcb.103.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carey MC, Small DM, Bliss CM. Annu Rev Physiol. 1983;45:651–677. doi: 10.1146/annurev.ph.45.030183.003251. [DOI] [PubMed] [Google Scholar]
- 87.Borgstrom B, Barrowman JA, Lindslrom M. In: Sterols and Bile Acids. Danielson H, Sjovall J, editors. Vol. 12. Elsevier; New York: 1985. pp. 405–425. [Google Scholar]
- 88.Tso P. Adv Lipid Res. 1985;21:143–186. doi: 10.1016/b978-0-12-024921-3.50011-3. [DOI] [PubMed] [Google Scholar]
- 89.Wilson FA, Sallee VL, Dietschy JM. Science. 1971;174:1031–1033. doi: 10.1126/science.174.4013.1031. [DOI] [PubMed] [Google Scholar]
- 90.Westergaard H, Dietschy JM. J Clin Invest. 1976;58:97–108. doi: 10.1172/JCI108465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Staggers JE, Hernell O, Stafford RJ, Carey MC. Biochemistry. 1990;29:2028–2040. doi: 10.1021/bi00460a011. [DOI] [PubMed] [Google Scholar]
- 92.Hernell O, Staggers JE, Carey MC. Biochemistry. 1990;29:2041–2056. doi: 10.1021/bi00460a012. [DOI] [PubMed] [Google Scholar]
- 93.Thompson ABR, Dietschy JM. In: Physiology of the Gastrointestinal Tract. Johnson LR, editor. Raven Press; New York: 1981. pp. 1147–1220. [Google Scholar]
- 94.Hoffman NE. Biochim Biophys Acta. 1970;196:193–203. doi: 10.1016/0005-2736(70)90006-4. [DOI] [PubMed] [Google Scholar]
- 95.Hoffman NE, Yeoh VJ. Biochim Biophys Acta. 1971;233:49–52. doi: 10.1016/0005-2736(71)90356-7. [DOI] [PubMed] [Google Scholar]
- 96.Nichols JW. Biochemistry. 1988;27:3925–3931. doi: 10.1021/bi00411a006. [DOI] [PubMed] [Google Scholar]
- 97.Nichols JW. Hepatology. 1990;12:83S–87S. [PubMed] [Google Scholar]
- 98.Fullington D, Shoemaker DG, Nichols JW. Biochemistry. 1990;29:879–886. doi: 10.1021/bi00456a005. [DOI] [PubMed] [Google Scholar]
- 99.Stuhne-Sekalec L, Stanacev NZ. Can J Biochem. 1978;56:407–413. doi: 10.1139/o78-064. [DOI] [PubMed] [Google Scholar]
- 100.Stuhne-Sekalec L, Stanacev NZ. Can J Biochem. 1980;58:1082–1090. doi: 10.1139/o80-146. [DOI] [PubMed] [Google Scholar]
- 101.Baran`ska J, Wojtczak L. Biochim Biophys Acta. 1984;773:23–31. doi: 10.1016/0005-2736(84)90546-7. [DOI] [PubMed] [Google Scholar]
- 102.Bara`ska J, Wojtczak L. Arch Bioehim Biophys. 1988;260:301–308. doi: 10.1016/0003-9861(88)90454-7. [DOI] [PubMed] [Google Scholar]
- 103.Wojtczak L, Bara`ska J, Zborowski J. Biochim Biophys Acta. 1990;1044:284–287. doi: 10.1016/0005-2760(90)90315-o. [DOI] [PubMed] [Google Scholar]
- 104.De Cuyper M, Joniau M. Biochaim Biophys Acta. 1985;814:374–380. [Google Scholar]
- 105.De Cuyper M, Joniau M, Dangreau H. Biochemistry. 1983;22:415–420. doi: 10.1021/bi00271a027. [DOI] [PubMed] [Google Scholar]
- 106.De Cuyper M, Joniau M, Engberts JB, Sudholter EJ. Colloids Surf. 1984;10:313–319. [Google Scholar]
- 107.Jones JD, Thompson TE. Biochemistry. 1989;28:129–134. doi: 10.1021/bi00427a019. [DOI] [PubMed] [Google Scholar]
- 108.Jones JD, Thompson TE. Biochemistry. 1990;29:1593–1600. doi: 10.1021/bi00458a034. [DOI] [PubMed] [Google Scholar]
- 109.Wimley WC, Thompson TE. Biochemistry. 1991;30:4200–4204. doi: 10.1021/bi00231a014. [DOI] [PubMed] [Google Scholar]
- 110.Papahadjopoulos D, Hui S, Vail WJ, Poste G. Biochim Biophys Acta. 1976;448:245–264. [PubMed] [Google Scholar]
- 111.Martin MC, MacDonald RC. Biochemistry. 1976;15:321–327. doi: 10.1021/bi00647a013. [DOI] [PubMed] [Google Scholar]
- 112.Duckwitz-Peterlein G, Eilenberger G, Overath P. Biochim Biophys Acta. 1977;469:311–325. doi: 10.1016/0005-2736(77)90167-5. [DOI] [PubMed] [Google Scholar]
- 113.Kremer JM, Kops-Werkhoven MM, Pathmamanoharan C, Gijzeman OL, Wiersema PH. Biochim Biophys Acta. 1977;471:177–188. doi: 10.1016/0005-2736(77)90248-6. [DOI] [PubMed] [Google Scholar]
- 114.Doody MC, Pownall HJ, Kao YJ, Smith LC. Biochemistry. 1980;19:108–116. doi: 10.1021/bi00542a017. [DOI] [PubMed] [Google Scholar]
- 115.Pownall HJ, Hickson DL, Smith LC. J Am Chem Soc. 1983;105:2440–2445. [Google Scholar]
- 116.McLean LR, Phillips MC. Biochemistry. 1984;23:4624–4630. doi: 10.1021/bi00315a017. [DOI] [PubMed] [Google Scholar]
- 117.Bayer TM, Schmidt CF, Sackmann E. Biochemistry. 1988;27:6078–6085. [Google Scholar]
- 118.Wimley WC, Thompson TE. Biochemistry. 1990;29:1296–1303. doi: 10.1021/bi00457a027. [DOI] [PubMed] [Google Scholar]
- 119.Wimley WC, Thompson TE. Biochemistry. 1991;30:1702–1709. doi: 10.1021/bi00220a036. [DOI] [PubMed] [Google Scholar]
- 120.Bittman R, Clejan S, Hui SW. J Biol Chem. 1990;265:15110–15117. [PubMed] [Google Scholar]
- 121.McLean LR, Phillips MC. Biochim Biophys Acta. 1984;776:21–26. doi: 10.1016/0005-2736(84)90246-3. [DOI] [PubMed] [Google Scholar]
- 122.Thomas PD, Poznansky MJ. Biochem, J. 1988;254:155–160. doi: 10.1042/bj2540155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nichols JW. Biochemistry. 1985;24:6390–6398. doi: 10.1021/bi00344a011. [DOI] [PubMed] [Google Scholar]
- 124.Masserini M, Freire E. Biochemistry. 1987;26:237–242. doi: 10.1021/bi00375a032. [DOI] [PubMed] [Google Scholar]
- 125.Brown RE, Hyland KJ. Biochemistry. 1992 doi: 10.1021/bi00158a024. in press. [DOI] [PubMed] [Google Scholar]
- 126.Elamrani K, Blume A. Biochemistry. 1982;21:521–526. doi: 10.1021/bi00532a017. [DOI] [PubMed] [Google Scholar]
- 127.Shin TB, Leventis R, Silvius JR. Biochemistry. 1991;30:7491–7497. doi: 10.1021/bi00244a018. [DOI] [PubMed] [Google Scholar]
- 128.Nishiya T, Chang TMS. Chem Phys Lipids. 1989;51:83–90. doi: 10.1016/0009-3084(88)90066-7. [DOI] [PubMed] [Google Scholar]
- 129.Storch J, Kleinfeld AM. Biochemistry. 1986;25:1717–1726. doi: 10.1021/bi00355a041. [DOI] [PubMed] [Google Scholar]
- 130.Burgess SW, Massenburg D, Yates J, Lentz BR. Biochemistry. 1991;30:4193–4200. doi: 10.1021/bi00231a013. [DOI] [PubMed] [Google Scholar]
- 131.Wu JR, Lentz BR. Biochemistry. 1991;30:6780–6787. doi: 10.1021/bi00241a022. [DOI] [PubMed] [Google Scholar]
- 132.Nakagawa T. Colloid Polymer Sci. 1974;252:56–64. [Google Scholar]
- 133.Thilo L. Biochim Biophys Acta. 1977;469:326–334. doi: 10.1016/0005-2736(77)90168-7. [DOI] [PubMed] [Google Scholar]
- 134.Duckwitz-Peterlein G, Moraal H. Biophys Struct Mech. 1978;4:315–326. doi: 10.1007/BF00537614. [DOI] [PubMed] [Google Scholar]
- 135.Arvinte T, Hildenbrand K. Biochim Biophys Acta. 1984;775:86–94. doi: 10.1016/0005-2736(84)90238-4. [DOI] [PubMed] [Google Scholar]
- 136.Gurd FRN. In: Lipide Chemistry. Hanahan DJ, editor. Wiley; New York: 1960. pp. 208–259. [Google Scholar]
- 137.Thurnhofer H, Hauser H. Biochemistry. 1990;29:2142–2148. doi: 10.1021/bi00460a026. [DOI] [PubMed] [Google Scholar]
- 138.Patton JS. In: Physiology of the Gastrointestinal Tract. Johnson LR, editor. Raven; New York: 1981. pp. 1123–1146. [Google Scholar]
- 139.Quo AL, Wade CG. Biochemistry. 1979;18:2300–2308. doi: 10.1021/bi00578a026. [DOI] [PubMed] [Google Scholar]
- 140.Lis LJ, McAlister M, Fuller N, Rand RP, Parsegian VA. Biophys J. 1983;37:657–666. [PMC free article] [PubMed] [Google Scholar]
- 141.Mcintosh TJ, Simon SA. Biochemistry. 1986;25:4948–4952. doi: 10.1021/bi00365a034. [DOI] [PubMed] [Google Scholar]
- 142.Rand P, Parsegian VP. Biochim Biophys Acta. 1987;988:351–376. [Google Scholar]
- 143.Stamatatos L, Leventis R, Zuckermann MJ, Silvius JR. Biochemistry. 1988;27:3917–3925. doi: 10.1021/bi00411a005. [DOI] [PubMed] [Google Scholar]
- 144.Eastman SJ, Wilschut J, Cullis PR, Hope MJ. Biochim Biophys Acta. 1989:178–184. doi: 10.1016/0005-2736(89)90026-6. [DOI] [PubMed] [Google Scholar]
- 145.Hope MJ, Cullis PR. J Biol Chem. 1987;262:4360–4366. [PubMed] [Google Scholar]
- 146.Micol V, Ortiz A, Gomez-Fernandez JC. Chem Phys Lipids. 1990;55:245–251. doi: 10.1016/0009-3084(90)90162-k. [DOI] [PubMed] [Google Scholar]
- 147.Schroit AJ, Zwaal RFA. Biochim Biophys Acta. 1991;1071:313–329. doi: 10.1016/0304-4157(91)90019-s. [DOI] [PubMed] [Google Scholar]
- 148.Fex G, Johannesson G. Biochim Biophys Acta. 1988;944:249–255. doi: 10.1016/0005-2736(88)90438-5. [DOI] [PubMed] [Google Scholar]
- 149.Ho MTP, Massey JB, Pownall HJ, Anderson RA, Hollyfield JG. J Biol Chem. 1989;264:928–935. [PubMed] [Google Scholar]
- 150.Ho MTP, Pownall HJ, Hollyfield JG. J Biol Chem. 1989;264:17759–17763. [PubMed] [Google Scholar]
- 151.Noy N, Xu ZJ. Biochemistry. 1990;29:3883–3888. doi: 10.1021/bi00468a013. [DOI] [PubMed] [Google Scholar]
- 152.Noy N, Xu ZJ. Biochemistry. 1990;29:3888–3892. doi: 10.1021/bi00468a014. [DOI] [PubMed] [Google Scholar]
- 153.Vallari DS, Record M, Smith ZL, Snyder F. Biochim Biophys Ada. 1989;1006:250–254. doi: 10.1016/0005-2760(89)90204-x. [DOI] [PubMed] [Google Scholar]
- 154.Van Wijk GMT, Gadella TWJ, Wirtz KWA, Hosteller KY, Van den Bosch H. Biochemistry. 1992;31:5912–5917. doi: 10.1021/bi00140a030. [DOI] [PubMed] [Google Scholar]
- 155.Pownall HJ, Bick DLM, Massey JB. Biochemistry. 1991;30:5696–5700. doi: 10.1021/bi00237a009. [DOI] [PubMed] [Google Scholar]
- 156.Jones JD, Thompson TE. Biochemistry. 1990;29:3892–3897. doi: 10.1021/bi00468a015. [DOI] [PubMed] [Google Scholar]
- 157.Steck TL, Kezdy FJ, Lange Y. J Biol Chem. 1988;263:13023–13031. [PubMed] [Google Scholar]