

Abstract
A consistent metabolic hallmark observed in multiple cancers is the increase of cellular phosphocholine (PC) and total choline-containing compounds (tCho), which is closely related to malignant transformation, invasion, and metastasis. Enzymes in choline phospholipid metabolism present attractive targets to exploit for treatment, but require a clear understanding of the mechanisms underlying the altered choline phospholipid metabolism observed in cancer. Choline kinase-α (Chk-α) is an enzyme in the Kennedy pathway that phosphorylates free choline (Cho) to PC, and its upregulation in several cancers is a major contributor to increased PC levels. Similarly, increased expression and activity of phospholipase D1 (PLD1), which converts phosphatidylcholine (PtdCho) to phosphatidic acid (PA) and Cho, has been well documented in gastric, ovarian and breast cancer. Here we report a strong correlation between expression of Chk-α and PLD1 with breast cancer malignancy. Data from patient samples established an association between estrogen receptor (ER) status and Chk-α and PLD1 expression. In addition, these two enzymes were found to be interactive. Downregulation of Chk-α with siRNA increased PLD1 expression, and downregulation of PLD1 increased Chk-α expression. Simultaneous silencing of PLD1 and Chk-α in MDA-MB-231 cells increased apoptosis as detected by the TUNEL assay. These data provide new insights into choline phospholipid metabolism of breast cancer, and support multiple targeting of enzymes in choline phospholipid metabolism as a strategy for treatment.
Keywords: breast cancer, choline kinase-alpha, phospholipase D1, RNA interference, magnetic resonance spectroscopy
Introduction
MRS studies have consistently detected an elevation of phosphocholine (PC) and total choline containing compounds (tCho) in human lung, colon, prostate, breast, ovarian, and brain tumors.1,2 Activated choline phospholipid metabolism is a metabolic hallmark of cancer that has been implicated in oncogenesis and tumor progression.1 Elevated levels of PC together with relatively low glycerophosphocholine (GPC) have been consistently detected in human breast cancer cells as compared with normal breast epithelial cells.3 Levels of PC and tCho increased in a stepwise manner in immortalized, oncogene-transformed, and tumor-derived human mammary epithelial cells (HMECs).3 Both GPC and PC levels are elevated in human breast cancers as compared with normal breast tissue.4 Changes in PC and tCho levels are detected in response to traditional chemotherapy as well as novel molecular therapies, making these suitable surrogate markers of treatment response.5-7 PC, one of the intermediate metabolites of choline phospholipid metabolism, is a precursor in the biosynthesis of phosphatidylcholine (PtdCho), as well as a breakdown product. The biosynthesis and hydrolysis pathways of PtdCho are considered vital for mitogenic signal transduction events in cells.8 These pathways are associated with proliferation, and display complex reciprocal interactions with oncogenic signaling pathways.1 Enzymes in choline phospholipid metabolism therefore present unique targets to exploit for cancer treatment, but a clear understanding of the interactions between these enzymes is necessary to optimize such strategies.
Choline kinase (Chk) is a cytosolic enzyme that catalyzes the phosphorylation of free intracellular choline (Cho) to PC, thereby consuming ATP in the presence of magnesium. In mammalian cells, two separate genes Chk-α and Chk-β encode the three known isoforms of Chk, which are Chk-α1, Chk-α2, and Chk-β. Out of these, Chk-α1 and Chk-α2 are the result of alternative splicing of the Chk-α transcript.9 These enzymes are active as homo- or heterodimers.9 Although Chk-α and Chk-β are members of the same family, they behave differently when overexpressed in cells.10 Chk-α expression and activity is found to play a critical role in oncogenesis, tumor progression, and metastasis of many cancers, making it a good choice for targeting.1,11 Increased levels and activity of Chk-α were observed in human breast, colorectal, lung, and prostate cancer.12–14 In addition, increased Chk-α expression was found to correlate with negative estrogen receptor (ER−) status in breast cancer.12 Chk-α has been associated with worse clinical outcome in non-small cell lung cancer, making it a prognostic factor for this disease.15 Increased Chk-α expression in human breast cancer cells was shown to increase their invasiveness.16 We investigated siRNA-mediated downregulation of Chk-α in human breast cancer cells, and observed a significant reduction of cell proliferation and increased differentiation in highly invasive MDA-MB-231 human breast cancer cells following Chk-α downregulation.17
Two other potential targets in choline phospholipid metabolism are phosphatidylcholine-specific phospholipase C (PC-PLC) and phosphatidylcholine-specific phospholipase D (PC-PLD), subsequently called PLC and PLD, respectively. The gene sequence for PLC has not yet been identified, but may become available in the near future. PLD on the other hand is well characterized, and increased PLD expression has been observed in several tumors.18 PLD is a ubiquitous enzyme and is involved in the hydrolysis of PtdCho to phosphatidic acid (PA) and Cho.19 PA is further converted either to diacylglycerol (DAG) or lysophosphatidic acid (LPA) by phosphatidic acid phosphohydrolase and phospholipase A2, respectively.19 Two mammalian genes, PLD1 and PLD2, each with splice variants, have been identified.20-22 The two splice variants of PLD1, PLD1a and PLD1b, differ in 38 amino acids due to the splicing of an alternate exon.21 PLD1 and PLD2 have been shown to accelerate epidermal growth factor receptor (EGFR) endocytosis by interacting with Dynamin, a critical mediator of membrane fission.23 PLD1 also plays a role in exocytosis. The exact mechanism is not yet known but the current model proposes a biophysical role of PLD1-formed PA that generates a negative curvature to enhance fusion to the plasma membrane.24 The association of PLD2 with Grb2 and Rac2 mediates membrane ruffling.25 The metabolic product of PLD1, PA, is known to activate mTOR signaling pathway by binding directly to mTOR.26 Unlike PLD2, which is constitutively expressed, basal expression of PLD1 is very low, and activates G proteins such as ARF, Rho, and Rac.27 PLD1 is overexpressed in uterine28 and endometrial carcinoma,29 and it may be a critical downstream mediator of H-Ras induced tumors, making it an important molecular target.30 Transformed fibroblasts overexpressing either PLD1 or PLD2 exhibited anchorage independent growth and altered growth properties.31 Elevated PLD1 protein expression has been reported to generate rapamycin resistance in MDA-MB-231 cells and other breast cancer cells.32,33
Here we observed a correlation between the expression of Chk-α and PLD1 with malignancy and, in patient samples, found an association between ER status and Chk-α and PLD1 expression. An interdependency was observed between the two enzymes in terms of protein expression that is characteristic of the adaptability of cancer cells. These data highlight the importance of targeting both Chk-α and PLD1 to eliminate any adaptive compensatory effects that would allow cancer cells to survive.
Results
Chk-α and PLD1 expression levels in patient-derived breast cancer samples and breast cancer cell lines
The expression of Chk-α and PLD1 was determined by qRT-PCR in patient-derived breast cancer samples. As shown in Figure 1A, we observed a significant positive correlation between the mRNA expression levels of Chk-α and PLD1 (Pearson r = 0.67, P = 0.002) in these human breast cancers. Breast cancers that were ER− and contained high PC/GPC had high Chk-α and PLD1 levels, and breast cancers that were ER+ and had low PC/GPC contained relatively low Chk-α and PLD1 levels. Protein levels of Chk-α and PLD1 in nonmalignant MCF-12A cells, non-metastatic MCF-7 breast cancer cells, and highly metastatic MDA-MB-231 breast cancer cells were analyzed by immunoblotting. As shown in Figure 1B, Chk-α expression was highest in ER− MDA-MB-231 cells, followed by ER+ MCF-7 cells, and lowest in MCF-12A cells. Similarly PLD1 was highly expressed in ER− MDA-MB-231 cells, followed by ER+ MCF-7 cells, and lowest in MCF-12A cells as shown in Figure 1C. Data from breast cancer cell lines were consistent with the pattern observed in patient samples, showing that increased expression of Chk-α and PLD1 occurred in ER−, high PC/GPC human breast cancer samples and in ER− MDA-MB-231 cells compared with ER+, low PC/GPC human breast cancer samples and in ER+ MCF-7 breast cancer cells.

Figure 1. (A) Relative fold change in PLD1 mRNA and Chk-α mRNA in patient-derived tumor samples that are either ER+ (n = 11) or ER− (n = 8). (B) Immunoblots representing the protein expression of Chk-α in nonmalignant MCF-12A cells, non-metastatic ER+ MCF-7 cells and highly metastatic ER− MDA-MB-231 cells. (C) Immunoblots representing the protein expression of PLD1 in MCF-12A, MCF-7, and MDA-MB-231 cells. GAPDH was used as a loading control.
mRNA levels after downregulating Chk-α and PLD1 in MDA-MB-231 cells
MDA-MB-231 cells were transfected with Chk-α-siRNA and PLD1-siRNA using Dfect as a transfection reagent (Fig. 2). Dfect alone and 100 nM of control siRNA were used as negative controls. As shown in Figure 2, a significant decrease of PLD1 was seen following transfection with 25 nM and 100 nM PLD1-siRNA compared with untreated cells. Following PLD1 downregulation, a slight increase in Chk-α mRNA levels (~130%) was observed. Significant downregulation of Chk-α mRNA was observed following treatment with 75 nM Chk-α-siRNA and 100 nM Chk-α-siRNA. In summary, effective downregulation at the mRNA level was seen in PLD1 and Chk-α when targeted with 25 nM and 75 nM siRNA.
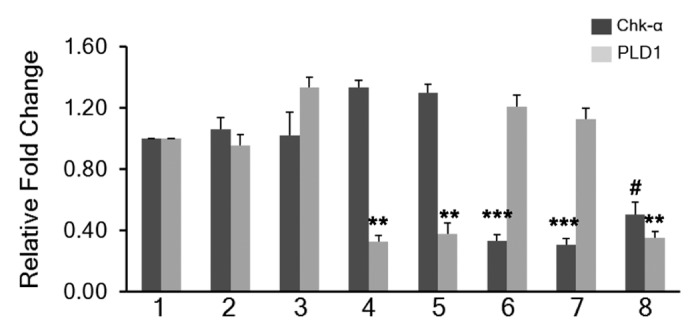
Figure 2. Relative fold change of Chk-α and PLD1 mRNA in MDA-MB-231 cells (n = 7 per treatment group): untreated (1), treated with 25 nM PLD1-siRNA (4), treated with 100 nM PLD1-siRNA (5), treated with 75 nM Chk-α-siRNA (6), treated with 100 nM Chk-α-siRNA (7), and treated with 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (8). MDA-MB-231 cells were also treated with Dfect alone (2), and 100 nM control siRNA (3) (n = 5) as negative controls. Values represent mean + SE, #P < 0.09, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, compared with untreated control cells using ΔCt values.
Protein expression levels after downregulating Chk-α and PLD1 in MDA-MB-231 cells
As shown in Figure 3A, an increase in Chk-α expression was observed when cells were transfected with PLD1. Control siRNA (100 nM) was used as a negative control. These observations were quantified by densitometry as shown in Figure 3B. The increase of Chk-α is evident from the significant increase of this protein observed when cells were treated with combined 25 nM PLD1 siRNA and 75 nM Chk-α-siRNA compared with cells treated with only 75 nM or with 100 nM Chk-α-siRNA (Fig. 3B). Similarly, an increase of Chk-α was also observed following treatment of cells with only PLD1 siRNA, compared with control siRNA (Fig. 3B).

Figure 3. (A) Representative immunoblots of Chk-α and PLD1 in MDA-MB-231 cells: untreated (1), treated with 25 nM PLD1-siRNA (3), 100 nM PLD1-siRNA (4), 75 nM Chk-α-siRNA (5), 100 nM Chk-α-siRNA (6), and 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (7). Cells treated with 100 nM control siRNA (2) were used as negative controls. (B) Relative band intensity for Chk-α and PLD1 as compared with GAPDH was calculated after the treatment of MDA-MB-231 cells (n = 3–6 per group) with 25 nM PLD1-siRNA (4), 100 nM PLD1-siRNA (5), 75 nM Chk-α-siRNA (6), 100 nM Chk-α-siRNA (7), and 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (8). Cells treated with 100 nM control siRNA (3) were used for comparison. Values represent mean + SE, #P < 0.06, *P ≤ 0.05, **P ≤ 0.01 (one-tailed unpaired t test), compared with control siRNA treated cells (2).
TUNEL assay
A TUNEL assay was performed in MDA-MB-231 cells at 48 h post-transfection with siRNA. As shown in Figure 4, MDA-MB-231 cells treated with DNase I (positive control) showed bright green fluorescence due to fluorescein-12-dUTP-label indicating double strand breaks resulting from apoptosis. Conversely, both untreated and control siRNA transfected MDA-MB-231 cells showed no fluorescence. Increased fluorescence was detected with 100 nM Chk-α-siRNA compared with the control siRNA transfected cells. The most increase in fluorescence intensity/pixel was observed when Chk-α-siRNA and PLD1-siRNA were transfected simultaneously. The observed increases in fluorescence intensity/pixel were significant as compared with control siRNA as well as compared with PLD1-siRNA or Chk-α-siRNA transfection alone. Images obtained from the TUNEL assay were quantified and intensity/pixel values were calculated as shown in Figure 5. The highest increase in fluorescence intensity/pixel was observed in simultaneous Chk-α-siRNA and PLD1-siRNA transfected cells.

Figure 4. Representative images of TUNEL assay performed in MDA-MB-231 cells (n = 5 per group) after transfection with: 25 nM PLD1-siRNA (4), 100 nM PLD1-siRNA (5), 75 nM Chk-α-siRNA (6), 100 nM Chk-α-siRNA (7), and 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (8). MDA-MB-231 cells were treated with DNase I (1) as a positive control while untreated (2) and 100 nM control siRNA (3) treated samples served as negative controls. Panels from left to right represent a bright field view of the cells taken with a 40× objective, propidium iodide staining (red), incorporation of fluorescein-12-dUTP (green), and a merged image of propidium iodide and fluorescein-12-UTP incorporation. Propidium iodide staining represents the total number of cells in the panel while the intensity of fluorescein is indicative of the number of double strand breaks in DNA. Multiple images with the same acquisition parameters were taken from each sample to represent the entire sample.
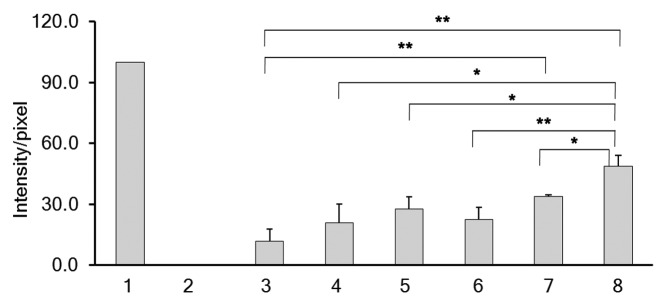
Figure 5. Images obtained from the TUNEL assay were quantified using MATLAB software as described in Materials and Methods and intensity/pixel values were calculated. The intensity/pixel were obtained for samples (n = 5) following transfection with 25 nM PLD1-siRNA (4), 100 nM PLD1-siRNA (5), 75 nM Chk-α-siRNA (6), 100 nM Chk-α-siRNA (7), and 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (8) in MDA-MB-231 cells. MDA-MB-231 cells were treated with DNase I (1) as a positive control while untreated (2) and 100 nM control siRNA (3) treated samples were considered as negative controls. Values represent mean + SE, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Cell viability/proliferation assay after siRNA transfection of MDA-MB-231 cells
MDA-MB-231 cells showed a significant reduction of viability/proliferation following transfection using 75 nM Chk-α-siRNA and 100 nM Chk-α-siRNA compared with control siRNA (Fig. 6). There was a greater reduction of viability/proliferation following simultaneous Chk-α-siRNA and PLD1-siRNA transfection (Fig. 6). In contrast there was no statistically significant reduction of viability/proliferation after PLD1-siRNA transfection compared with 100 nM control siRNA transfection (Fig. 6). There was a significant difference between 25 nM PLD1-siRNA and 25 nM PLD1-siRNA plus 75 nM Chk-α-siRNA treated cells and between 100 nM PLD1-siRNA treated cells and 25 nM PLD1-siRNA plus 75 nM Chk-α-siRNA treated cells (Fig. 6).
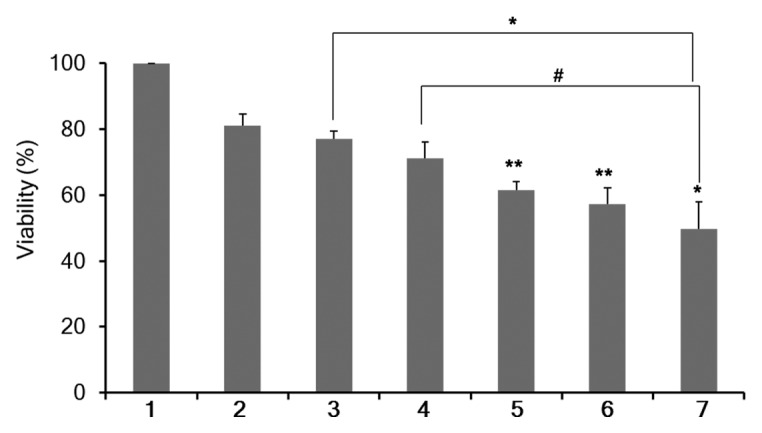
Figure 6. Percent cell viability/proliferation (%) as determined by MTS assay in MDA-MB-231 cells (n = 4 per group). MTS assays were performed 3 d after 48 h of treatment with Dfect (1), 100 nM control siRNA (2), 25 nM PLD1-siRNA (3), 100 nM PLD1-siRNA (4), 75 nM Chk-α-siRNA (5), 100 nM Chk-α-siRNA (6), and 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (7). Values represent mean + SE, #P < 0.075, *P ≤ 0.05, **P ≤ 0.01, compared with control siRNA treated cells unless otherwise indicated.
1H MRS analysis of choline phospholipid metabolite levels after Chk-α and PLD1 silencing
1H MRS analysis was done to assess cellular PC, GPC and tCho levels. As shown in Figure 7A and B, an increase in PC levels was observed in MDA-MB-231 cells transiently transfected with PLD1-siRNA as compared with untreated MDA-MB-231 cells although this was not statistically significant. The increase was dose-dependent and consistent with the increased expression of Chk-α observed in MDA-MB-231 cells transfected with PLD1-siRNA. When MDA-MB-231 cells were transfected with Chk-α-siRNA, we observed a significant decrease in PC and tCho levels compared with control cells. However, PC and tCho levels did not change when both PLD1-siRNA and Chk-α-siRNA were simultaneously transfected. PC and tCho levels of cells transfected with 100 nM Chk-α-siRNA were significantly lower than those of cells simultaneously transfected with PLD1- and Chk-α-siRNA (Fig. 7).

Figure 7. (A) Representative high-resolution 1H MR spectra showing the GPC, PC, Cho region of MDA-MB-231 cell extracts: untreated (1), treated with 25 nM PLD1-siRNA (2), treated with 100 nM PLD1-siRNA (3), treated with 25 nM PLD1-siRNA + 75nM Chk-α-siRNA (4), treated with 75 nM Chk-α-siRNA (5), and treated with100 nM Chk-α-siRNA (6). (B) Histogram showing quantified levels of PC (white box), GPC (gray box), and tCho (black box), in MDA-MB-231 cells (n = 4 per group): untreated (1), treated with Dfect (2), treated with 100 nM control siRNA (3), treated with 25 nM PLD1-siRNA (4), treated with 100 nM PLD1-siRNA (5), treated with 75 nM Chk-α-siRNA (6), treated with 100 nM Chk-α-siRNA (7), and treated with 25 nM PLD1-siRNA + 75 nM Chk-α-siRNA (8). Values represent mean + SE, *P ≤ 0.05, compared with untreated control cells unless otherwise indicated.
Discussion
Abnormal choline phospholipid metabolism is linked to oncogenesis, tumor progression and metastasis.1 Here we observed a strong correlation between malignancy, ER status, and expression of Chk-α and PLD1 in breast cancer cell lines and patient-derived breast tumor samples. Elevated levels of Chk-α and PLD1 were found in both highly metastatic ER− MDA-MB-231 breast cancer cells and in ER− patient-derived breast cancer samples as compared with non-metastatic ER+ MCF-7 breast cancer cells and ER+ patient-derived breast cancer samples, supporting their role in malignancy. The strong positive correlation between elevated Chk-α and PLD1 levels in human breast tumor samples and breast cancer cell lines suggests a functional dependency between these two key regulatory enzymes in choline phospholipid metabolism.
The involvement of PLD1 in tumor progression has been identified in various molecular pathways. PA, the product of PLD1 activity, phosphorylates p70S6K at threonine 389, activating mTOR and promoting cell cycle progression.28 Increased PLD1 activity causes an increase in the Ras-ERK/PI3K-NFκB signaling cascade, which in turn further upregulates PLD1, indicating a positive feedback loop.34 Moreover, PLD1 inhibition decreased matrix metalloproteinase (MMP)2 and MMP9 upregulation, reducing invasion in breast cancer cells.34 A decrease in invasion was also reported when PLD1 siRNA significantly abolished PDGF-induced upregulation of MMP2 and MMP9.35 PLD1 was also shown to drive a positive feedback loop for Wnt/β catenin signaling, and its inhibition has been suggested as a possible disruption strategy of the pathway.36 Given the critical role of PLD1 in tumor progression and metastasis, we failed to observe any apoptosis and reduction of cell viability/proliferation when we transfected PLD1-siRNA in highly malignant MDA-MB-231 cells in spite of observing an effective downregulation of PLD1. However, these results were consistent with previously published data where abrogation of PLD activity only rendered cells sensitive to the apoptotic insult of serum withdrawal, but did not induce apoptosis on its own.37 The increase of Chk-α observed following PLD1 silencing may have compensated for the critical role played by PLD1 in cell survival.
Chk-α, which converts Cho to PC, has a well-established role in tumor progression.1 Chk-α mediates proliferation of primary HMECs by stimulating DNA synthesis and promoting G1 → S transition of the cell cycle.38 Chk-α inhibition decreased the phosphorylation of ERK1/2 to p-ERK1/2 on T202/Y204, and the phosphorylation of AKT to p-AKT on S473, indicating a possible role of Chk-α in the regulation of MAPK and PI3K/Akt signaling.39 Moreover, Chk-α is phosphorylated by c-Src and forms a complex with EGFR, which contributes to the regulation of cell proliferation and tumorigenesis.40 In addition to tumorigenesis, an increase in invasiveness and drug resistance has also been observed with Chk-α overexpression in breast cancer cells.16
Downregulation of Chk-α using Chk-α-siRNA induced apoptosis and reduced cell viability/proliferation in MDA-MB-231 cells. These observations are consistent with previously reported data where Chk-α downregulation caused apoptosis.41,42 However, following PLD1 downregulation, we observed an increase in Chk-α protein and PC levels. Whether the increase in the Chk-α protein levels was due to an increase in Chk-α mRNA levels remains unclear as the increase in Chk-α mRNA levels, although observed, was statistically insignificant. The increase of Chk-α protein may have generated a survival signal to compensate for the downregulation of PLD1. To assess the effect of Chk-α and PLD1 downregulation on cell survival, we performed TUNEL and MTS assays. We observed increased double strand breaks in MDA-MB-231 cells transfected with 25 nM PLD1-siRNA and 75 nM Chk-α-siRNA as compared with cells transfected with PLD1- and Chk-α-siRNA alone, indicating a synergistic apoptotic effect of Chk-α-siRNA and PLD1-siRNA. A similar trend was observed in cell viability/proliferation using an MTS assay. Observations that decreased PLD activity can only induce apoptosis in case of serum withdrawal37 can be explained by the fact that serum treatment of cells induces Chk activity.43
In conclusion, we have shown for the first time increased expression of Chk-α following PLD1 downregulation, which is possibly mediating survival. These results provide new insights into choline phospholipid metabolism in breast cancer cells. With simultaneous downregulation of Chk-α and PLD1, we were able to attenuate this survival. Further studies are required to understand the mechanisms by which PLD1 downregulation increases Chk-α expression.
Materials and Methods
Chk-α and PLD1 expression in human breast cancers
Tumor biopsies from 19 breast cancer patients were analyzed for expression of Chk-α and PLD1. The patients (mean age 46.5 y, age range from 28.5–68.5) were diagnosed with invasive breast cancer stage IIB–IIIB. ER positive (ER+) tumor was defined as ≥10% positive cells determined by immunohistochemistry. The patients were stratified according to their ER status (n = 11 for ER+, n = 8 for ER−). Total RNA isolation was performed by using a rotor-stator homogenizer and an RNeasy Mini Kit (Qiagen). The RNA integrity number (RIN) determined by Bioanalyzer 2100 (Agilent) was 7.6 ± 1.2, where RIN > 7.0 can generally be accepted as good quality RNA for gene expression analyses. cDNA was synthesized from 1 μg total RNA by using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). Chk-α and PLD1 expression levels were measured by using iCycler real-time PCR detection system (Bio-Rad) and iQ SYBR Green Supermix (Quanta BioSciences). The qRT-PCR reactions were performed in triplicate and the gene expression levels were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT1). Pearson correlation analysis (SPSS 16.0 Inc.) was performed between Chk-α and PLD1. A P value of <0.05 was considered significant.
Cells and cell culture conditions
MDA-MB-231, an ER-, progesterone receptor- and Her2-negative (triple negative) metastatic human breast cancer cell line, was grown in RPMI 1640 (Sigma-Aldrich Co.) supplemented with 10% fetal bovine serum (FBS). MCF-7, an ER+, poorly metastatic human breast cancer cell line, was cultured in Eagle’s MEM (Mediatech Inc.) supplemented with 10% FBS. MCF-12A, a spontaneously immortalized, nonmalignant human mammary epithelial cell line, was cultured in DMEM-Ham’s F12 medium (Mediatech Inc.) supplemented as previously described.44 All cell lines were obtained from the American Type Culture Collection (ATCC) and were maintained in a humidified atmosphere with 5% CO2 in air at 37 °C. The MCF-7 and MDA-MB-231 human breast cancer cell lines were used within 6 mo of purchasing from ATCC. The cell lines were tested and authenticated by ATCC using two independent methods: the ATCC cytochrome C oxidase I PCR assay, and short tandem-repeat profiling using multiplex PCR.
RNA interference experiments
The sequence for siRNA that is specific for human Chk-α (Chk-α-siRNA), 5′-CAUGCUGUUC CAGUGCUCC-3′, was purchased as a duplex (Thermo Fisher Scientific Inc.). Similarly, siRNA specific for PLD1 (PLD1-siRNA) was designed using an algorithm provided by the company (Thermo Fisher Scientific Inc.) with the sequence 5′-GGGAAGAAGG AGACAGAAA-3′. Nontargeting control siRNA was also obtained from Thermo Fisher Scientific Inc. siRNAs were transfected 24 h after seeding cells using DharmaFECT4 (Thermo Fisher Scientific Inc.) following the manufacturer’s protocol.
RNA isolation, cDNA synthesis, and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was isolated from MDA-MB-231 cells 48 h post transfection using QIAshredder and RNeasy Mini kit (Qiagen) as per the manufacturer’s protocol. cDNA was prepared using the iScript cDNA synthesis kit (Bio-Rad). cDNA samples were diluted 1:10 and real-time PCR was performed using IQ SYBR Green supermix and gene specific primers in the iCycler real-time PCR detection system (Quanta Bioscience). All primers were designed using Beacon designer software 7.8 (premier Biosoft, USA) and were obtained from Invitrogen. The expression of target RNA relative to the housekeeping gene HPRT1 was calculated based on the threshold cycle (Ct) as R = 2−Δ(ΔCt), where ΔCt = Ct of target − Ct of HPRT1.
Protein isolation and immunoblot assay
Whole cell lysates from MDA-MB-231 cells, transfected with either Chk-α-siRNA or PLD1-siRNA or both using Dfect or lipofectamine, were prepared in radioimmune precipitation (RIPA) buffer fortified with protease inhibitor cocktail (Sigma-Aldrich), 200 mM dithiothreitol, 100 mM phenylmethanesulfonylfluoride (PMSF), and 200 mM sodium orthovanadate. Whole cell lysates were fractionated by SDS-PAGE (60 µg per lane), transferred to nitrocellulose membrane, and subjected to immunoblot assays using either custom-made rabbit polyclonal antibody against human Chk-α (1:100 dilution), rabbit polyclonal antibody against human PLD1 (1:500 dilution, Abcam) or a mouse monoclonal antibody against human GAPDH (1:50000, Sigma-Aldrich). Horseradish peroxidase-conjugated secondary antibody against mouse or rabbit IgG was used at 1:2500 dilutions, and the signal was visualized using the SuperSignal West Pico Chemiluminescent substrate kit (Pierce Biotechnology) recorded on Blue Bio film (Denville Scientific). The films were scanned and densitometric values relative to the respective GAPDH band intensity were obtained using the Gel-analysis-tool in ImageJ (NIH).
TUNEL assay
Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling (TUNEL) assays were performed using the DeadEnd Fluorometric TUNEL System (Promega) as per the manufacturer’s protocol. The TUNEL assay measures the fragmented DNA of apoptotic cells by catalytically incorporating fluorescein-12-dUTP at 3′-OH DNA ends using recombinant Terminal Deoxynucleotidyl Transferase enzyme (rTdT). The intensity per pixel (intensity/pixel) obtained under standardized conditions as outlined below was used as a measure of the fragmented DNA.
Cells were seeded in Chamber Slides (Lab-Tek II, Thermo Fisher Scientific) with an area of approximately 4 cm2/well and coated with poly-d-lysine (Sigma-Aldrich). Twenty-four hours post seeding, cells were transfected with either Chk-α-siRNA or PLD1-siRNA or both. Forty-eight hours post transfection, cells were fixed with 4% paraformaldehyde for 25 min at 4 °C, followed by permeabilization with 0.2% Triton X-100 in PBS for 5 min. Subsequently, equilibration buffer was added followed by rTdT. Labeled cells were counter-stained with propidium iodide to visualize the cell nuclei, and images were captured using a fluorescence microscope (Nikon Inc.) equipped with a digital camera, using a standard fluorescein filter set to view the green fluorescence of fluorescein at 520 ± 20 nm and the red fluorescence of propidium iodide at >620 nm. All experiments were performed at least 5 times and images were captured from five different regions per slide using a 40× objective.
A program using MATLAB 7.4.0 (The MathWorks) was generated to analyze the intensity of green fluorescence of fluorescein-12-dUTP-labeled DNA in each image. The intensity/pixel in the experimental samples was normalized to the negative and positive control samples. Samples treated with DNase I were considered positive controls and the intensity/pixel of these samples were normalized to 100%; the threshold value of the sample intensity was normalized to 0 based on the intensity of the negative untreated control samples.
Proliferation assay
To evaluate the effect of the different siRNA combinations on cell viability/proliferation, approximately 4000 MDA-MB-231 cells were seeded in each well of a 96-well plate and cultured overnight. Twenty-four hours post seeding, cells were transiently transfected for 48 h with either Chk-α-siRNA, PLD1-siRNA, or both. After 48 h treatment, cells were maintained in fresh culture medium for another 3 d. MTS assays (Promega Corp.) were performed as per manufacture’s protocol. Dfect treatment without siRNA and nontargeting siRNA were used as negative controls and values were normalized to the Dfect values.
Dual-phase extraction and high-resolution 1H MRS studies
Cells were cultured for 24 h and then transfected with either Chk-α-siRNA or PLD1-siRNA or both for 48 h. Adherent cells were collected by trypsinization and counted using a hemocytometer. Approximately 107 cells were used for cell extraction. Water-soluble as well as lipid extracts were obtained using the dual-phase extraction method.17,45 Briefly, the pelleted cells were mixed with 4 mL of ice-cold methanol and vigorously vortexed. After keeping samples on ice for 10 min, 4 mL of chloroform were added, vortexed vigorously and kept on ice for 10 min. Finally, 4 mL of water were added and the samples were shaken well. All procedures were performed on ice and samples were stored at 4 °C overnight for phase separation and then centrifuged at 15 000 × g at 4 °C for 30 min. The water/methanol phase containing water-soluble cellular metabolites such as Cho, PC, and GPC were treated with ~100 mg of chelex beads (Sigma-Aldrich) to remove any divalent cations. After removing the beads, methanol was evaporated using a rotary evaporator. The remaining water phase was lyophilized. Cell extracts were re-suspended in 0.6 mL deuterated water (D2O) for MRS analysis. Five microliters of 0.75% (w/w) 3-(trimethylsilyl) propionic 2,2,3,3-d4 acid sodium salt (TSP) in D2O was used as an internal standard. Fully relaxed 1H MR spectra of the water-soluble extracts were acquired on a Bruker Avance 500 spectrometer (Bruker BioSpin Corp.) as previously described.17 Signal integrals of the -N-(CH3)3 resonances of PC at ~3.225 ppm, GPC at ~3.235 ppm, and free Cho at ~3.208 ppm were determined and normalized to cell numbers and compared with the TSP standard. To determine concentrations of cell samples, peak integration (IMET) from 1H spectra for PC, GPC, and Cho were compared with that of the internal standard TSP according to the equation:
Equation 1
In this equation, [MET] represents the intracellular concentration of a given metabolites such as PC, GPC, or Cho expressed as mmol/L. ATSP is the number of moles of TSP in the sample, N is the cell number, and V is the cell volume. Because the number of protons contributing to the signals of PC, GPC, and Cho and the TSP peak is the same, a correction for differences in the number of protons was not required. The resulting PC, GPC, and Cho concentrations were averaged for four independent experiments. The total choline concentration was obtained by adding values for PC, GPC, and Cho.
Statistical analysis
Statistical significance was evaluated using an unpaired two-tailed Student t test unless otherwise stated. P values < 0.05 were considered statistically significant unless otherwise stated.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NIH R01CA136576, R01CA138515, R01CA73850, R01CA82337, P50CA103175, P30CA006973, and R01 CA134695. We thank Ms Flonne Wildes for expert technical assistance and Dr Franca Podo for useful discussions.
Glossary
Abbreviations:
- Chk-α
choline kinase-alpha
- Cho
choline
- DAG
diacylglycerol
- Dfect
DharmaFECT4
- FBS
fetal bovine serum
- GPC
glycerophosphocholine
- LPA
lysophosphatidic acid
- MMP
matrix metalloproteinase
- MRS
magnetic resonance spectroscopy
- PA
phosphatidic acid
- PC
phosphocholine
- PE
phosphoethanolamine
- PLD1
phospholipase D1
- PtdCho
phosphatidylcholine
- siRNA
small interfering RNA
- tCho
total choline-containing metabolites (GPC+PC+ free choline)
- TSP
3-(trimethylsilyl) propionic-2,2,3,3,-d4 acid
- TUNEL
Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/28165
References
- 1.Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer. 2011;11:835–48. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Podo F. Tumour phospholipid metabolism. NMR Biomed. 1999;12:413–39. doi: 10.1002/(SICI)1099-1492(199911)12:7<413::AID-NBM587>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 3.Aboagye EO, Bhujwalla ZM. Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Res. 1999;59:80–4. [PubMed] [Google Scholar]
- 4.Sitter B, Lundgren S, Bathen TF, Halgunset J, Fjosne HE, Gribbestad IS. Comparison of HR MAS MR spectroscopic profiles of breast cancer tissue with clinical parameters. NMR Biomed. 2006;19:30–40. doi: 10.1002/nbm.992. [DOI] [PubMed] [Google Scholar]
- 5.Eliyahu G, Kreizman T, Degani H. Phosphocholine as a biomarker of breast cancer: molecular and biochemical studies. Int J Cancer. 2007;120:1721–30. doi: 10.1002/ijc.22293. [DOI] [PubMed] [Google Scholar]
- 6.Sharma U, Baek HM, Su MY, Jagannathan NR. In vivo 1H MRS in the assessment of the therapeutic response of breast cancer patients. NMR Biomed. 2011;24:700–11. doi: 10.1002/nbm.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beloueche-Babari M, Arunan V, Troy H, te Poele RH, te Fong AC, Jackson LE, Payne GS, Griffiths JR, Judson IR, Workman P, et al. Histone deacetylase inhibition increases levels of choline kinase α and phosphocholine facilitating noninvasive imaging in human cancers. Cancer Res. 2012;72:990–1000. doi: 10.1158/0008-5472.CAN-11-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai H, Erhardt P, Troppmair J, Diaz-Meco MT, Sithanandam G, Rapp UR, Moscat J, Cooper GM. Hydrolysis of phosphatidylcholine couples Ras to activation of Raf protein kinase during mitogenic signal transduction. Mol Cell Biol. 1993;13:7645–51. doi: 10.1128/mcb.13.12.7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoyama C, Liao H, Ishidate K. Structure and function of choline kinase isoforms in mammalian cells. Prog Lipid Res. 2004;43:266–81. doi: 10.1016/j.plipres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Gallego-Ortega D, Ramirez de Molina A, Ramos MA, Valdes-Mora F, Barderas MG, Sarmentero-Estrada J, Lacal JC. Differential role of human choline kinase alpha and beta enzymes in lipid metabolism: implications in cancer onset and treatment. PLoS One. 2009;4:e7819. doi: 10.1371/journal.pone.0007819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacal JC. Choline kinase: a novel target for antitumor drugs. IDrugs. 2001;4:419–26. [PubMed] [Google Scholar]
- 12.Ramírez de Molina A, Gutiérrez R, Ramos MA, Silva JM, Silva J, Bonilla F, Sánchez JJ, Lacal JC. Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene. 2002;21:4317–22. doi: 10.1038/sj.onc.1205556. [DOI] [PubMed] [Google Scholar]
- 13.Ramírez de Molina A, Rodríguez-González A, Gutiérrez R, Martínez-Piñeiro L, Sánchez J, Bonilla F, Rosell R, Lacal J. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem Biophys Res Commun. 2002;296:580–3. doi: 10.1016/S0006-291X(02)00920-8. [DOI] [PubMed] [Google Scholar]
- 14.Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D’Ascenzo S, Millimaggi D, Pavan A, Dolo V, Canevari S, et al. Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Res. 2005;65:9369–76. doi: 10.1158/0008-5472.CAN-05-1146. [DOI] [PubMed] [Google Scholar]
- 15.Ramírez de Molina A, Sarmentero-Estrada J, Belda-Iniesta C, Tarón M, Ramírez de Molina V, Cejas P, Skrzypski M, Gallego-Ortega D, de Castro J, Casado E, et al. Expression of choline kinase alpha to predict outcome in patients with early-stage non-small-cell lung cancer: a retrospective study. Lancet Oncol. 2007;8:889–97. doi: 10.1016/S1470-2045(07)70279-6. [DOI] [PubMed] [Google Scholar]
- 16.Shah T, Wildes F, Penet MF, Winnard PT, Jr., Glunde K, Artemov D, Ackerstaff E, Gimi B, Kakkad S, Raman V, et al. Choline kinase overexpression increases invasiveness and drug resistance of human breast cancer cells. NMR Biomed. 2010;23:633–42. doi: 10.1002/nbm.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glunde K, Raman V, Mori N, Bhujwalla ZM. RNA interference-mediated choline kinase suppression in breast cancer cells induces differentiation and reduces proliferation. Cancer Res. 2005;65:11034–43. doi: 10.1158/0008-5472.CAN-05-1807. [DOI] [PubMed] [Google Scholar]
- 18.Su W, Chen Q, Frohman MA. Targeting phospholipase D with small-molecule inhibitors as a potential therapeutic approach for cancer metastasis. Future Oncol. 2009;5:1477–86. doi: 10.2217/fon.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol. 2004;82:225–53. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- 20.Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA. Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem. 1995;270:29640–3. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- 21.Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, Morris AJ. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J Biol Chem. 1997;272:3860–8. doi: 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- 22.Steed PM, Clark KL, Boyar WC, Lasala DJ. Characterization of human PLD2 and the analysis of PLD isoform splice variants. FASEB J. 1998;12:1309–17. doi: 10.1096/fasebj.12.13.1309. [DOI] [PubMed] [Google Scholar]
- 23.Lee CS, Kim IS, Park JB, Lee MN, Lee HY, Suh PG, Ryu SH. The phox homology domain of phospholipase D activates dynamin GTPase activity and accelerates EGFR endocytosis. Nat Cell Biol. 2006;8:477–84. doi: 10.1038/ncb1401. [DOI] [PubMed] [Google Scholar]
- 24.Bader MF, Vitale N. Phospholipase D in calcium-regulated exocytosis: lessons from chromaffin cells. Biochim Biophys Acta. 2009;1791:936–41. doi: 10.1016/j.bbalip.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 25.Mahankali M, Peng HJ, Cox D, Gomez-Cambronero J. The mechanism of cell membrane ruffling relies on a phospholipase D2 (PLD2), Grb2 and Rac2 association. Cell Signal. 2011;23:1291–8. doi: 10.1016/j.cellsig.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–5. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 27.Cockcroft S. Signalling roles of mammalian phospholipase D1 and D2. Cell Mol Life Sci. 2001;58:1674–87. doi: 10.1007/PL00000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhingra S, Rodriguez ME, Shen Q, Duan X, Stanton ML, Chen L, Zhang R, Brown RE. Constitutive activation with overexpression of the mTORC2-phospholipase D1 pathway in uterine leiomyosarcoma and STUMP: morphoproteomic analysis with therapeutic implications. Int J Clin Exp Pathol. 2011;4:134–46. [PMC free article] [PubMed] [Google Scholar]
- 29.Shen Q, Stanton ML, Feng W, Rodriguez ME, Ramondetta L, Chen L, Brown RE, Duan X. Morphoproteomic analysis reveals an overexpressed and constitutively activated phospholipase D1-mTORC2 pathway in endometrial carcinoma. Int J Clin Exp Pathol. 2010;4:13–21. [PMC free article] [PubMed] [Google Scholar]
- 30.Buchanan FG, McReynolds M, Couvillon A, Kam Y, Holla VR, Dubois RN, Exton JH. Requirement of phospholipase D1 activity in H-RasV12-induced transformation. Proc Natl Acad Sci U S A. 2005;102:1638–42. doi: 10.1073/pnas.0406698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min DS, Kwon TK, Park WS, Chang JS, Park SK, Ahn BH, Ryoo ZY, Lee YH, Lee YS, Rhie DJ, et al. Neoplastic transformation and tumorigenesis associated with overexpression of phospholipase D isozymes in cultured murine fibroblasts. Carcinogenesis. 2001;22:1641–7. doi: 10.1093/carcin/22.10.1641. [DOI] [PubMed] [Google Scholar]
- 32.Gozgit JM, Pentecost BT, Marconi SA, Ricketts-Loriaux RS, Otis CN, Arcaro KF. PLD1 is overexpressed in an ER-negative MCF-7 cell line variant and a subset of phospho-Akt-negative breast carcinomas. Br J Cancer. 2007;97:809–17. doi: 10.1038/sj.bjc.6603926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–42. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 34.Kang DW, Park MH, Lee YJ, Kim HS, Lindsley CW, Alex Brown H, Min S. Autoregulation of phospholipase D activity is coupled to selective induction of phospholipase D1 expression to promote invasion of breast cancer cells. Int J Cancer. 2011;128:805–16. doi: 10.1002/ijc.25402. [DOI] [PubMed] [Google Scholar]
- 35.Kang DW, Min S. Platelet derived growth factor increases phospholipase D1 but not phospholipase D2 expression via NFkappaB signaling pathway and enhances invasion of breast cancer cells. Cancer Lett. 2010;294:125–33. doi: 10.1016/j.canlet.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 36.Kang DW, Lee SH, Yoon JW, Park WS, Choi KY, Min S. Phospholipase D1 drives a positive feedback loop to reinforce the Wnt/beta-catenin/TCF signaling axis. Cancer Res. 2010;70:4233–42. doi: 10.1158/0008-5472.CAN-09-3470. [DOI] [PubMed] [Google Scholar]
- 37.Zhong M, Shen Y, Zheng Y, Joseph T, Jackson D, Foster DA. Phospholipase D prevents apoptosis in v-Src-transformed rat fibroblasts and MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun. 2003;302:615–9. doi: 10.1016/S0006-291X(03)00229-8. [DOI] [PubMed] [Google Scholar]
- 38.Ramírez de Molina A, Báñez-Coronel M, Gutiérrez R, Rodríguez-González A, Olmeda D, Megías D, Lacal JC. Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 2004;64:6732–9. doi: 10.1158/0008-5472.CAN-04-0489. [DOI] [PubMed] [Google Scholar]
- 39.Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, Trent JO, Chesney J. A novel small molecule antagonist of choline kinase-α that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene. 2011;30:3370–80. doi: 10.1038/onc.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyake T, Parsons SJ. Functional interactions between Choline kinase α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene. 2012;31:1431–41. doi: 10.1038/onc.2011.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bañez-Coronel M, Ramírez de Molina A, Rodríguez-González A, Sarmentero J, Ramos MA, García-Cabezas MA, García-Oroz L, Lacal JC. Choline kinase alpha depletion selectively kills tumoral cells. Curr Cancer Drug Targets. 2008;8:709–19. doi: 10.2174/156800908786733432. [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez-González A, Ramírez de Molina A, Bañez-Coronel M, Megias D, Lacal JC. Inhibition of choline kinase renders a highly selective cytotoxic effect in tumour cells through a mitochondrial independent mechanism. Int J Oncol. 2005;26:999–1008. [PubMed] [Google Scholar]
- 43.Warden CH, Friedkin M. Regulation of choline kinase activity and phosphatidylcholine biosynthesis by mitogenic growth factors in 3T3 fibroblasts. J Biol Chem. 1985;260:6006–11. [PubMed] [Google Scholar]
- 44.Paine TM, Soule HD, Pauley RJ, Dawson PJ. Characterization of epithelial phenotypes in mortal and immortal human breast cells. Int J Cancer. 1992;50:463–73. doi: 10.1002/ijc.2910500323. [DOI] [PubMed] [Google Scholar]
- 45.Tyagi RK, Azrad A, Degani H, Salomon Y. Simultaneous extraction of cellular lipids and water-soluble metabolites: evaluation by NMR spectroscopy. Magn Reson Med. 1996;35:194–200. doi: 10.1002/mrm.1910350210. [DOI] [PubMed] [Google Scholar]