

Abstract
Several agents targeting the epidermal growth factor receptor (EGFR) have been FDA-approved to treat cancer patients with varying tumor types including metastatic colorectal cancer. Many patients treated with anti-EGFR therapy however do not respond and those that do initially respond often acquire resistance. Here we show a clear correlation between the efficacy of anti-EGFR inhibitors with their ability to inhibit STAT3 activity in A431 epidermoid carcinoma cells and in a series of wt K-RAS expressing human colon cancer cell lines. Furthermore, the ability of cetuximab to inhibit growth also correlated with its ability to inhibit STAT3 activity in tumor xenograft animal studies. In addition, stable knockdown of the STAT3 phosphatase, protein tyrosine phosphatase receptor delta (PTPRD) resulted in enhanced STAT3 activity and subsequent resistance to cetuximab in DIFI colon carcinoma cells. This resistance could be reversed by STAT3 inhibition. Finally, HN5 cells with acquired resistance to the EGFR tyrosine kinase inhibitor, AG1478 displayed greater STAT3 activity than the HN5 control cell line. These AG1478-refractory HN5 cells were re-sensitized to AG1478, cetuximab and erlotinib when co-treated with a STAT3 inhibitor. Taken together, our current data indicates a key role of STAT3 activity in promoting resistance to anti-EGFR therapy and suggests that anti-EGFR therapy in combination with inhibitors that block STAT3 may provide therapeutic benefit for patients with mCRC and other EGFR driven tumor types.
Keywords: EGFR, STAT3, cetuximab, colon cancer
Introduction
The epidermal growth factor receptor (EGFR) signaling is elicited by ligand binding, initiating the activation of many downstream signaling cascades including: RAS-RAF-ERK1/2, the PTEN regulated phosphatidylinositol 3-kinase (PI3-K)-AKT-mTOR, c-SRC, and signal transducer and activator of transcription (STAT) family members.1 Not surprisingly, genetic alterations leading to EGFR overexpression or mutation are frequently observed in cancer.2-4 These findings led to the development of agents targeting the EGFR, many of which produced promising anti-tumor effects in pre-clinical models.5,6 However, despite the enormous effort and cost, only a very small percentage of tested agents have made it through clinical evaluation to be ultimately approved. The US Food and Drug Administration (FDA) have approved four anti-EGFR agents. These include 2 monoclonal antibodies: cetuximab,7 approved for metastatic colorectal carcinoma (mCRC) and head and neck squamous cell carcinoma (HNSCC), and panitumumab,8 approved for mCRC, and 2 tyrosine kinase inhibitors: gefitinib,9 approved for non-small cell lung carcinoma (NSCLC), and erlotinib,10 approved for NSCLC and advanced pancreatic cancer. Another agent that inhibits both EGFR and HER2 (lapatinib) has also been approved.11 Collectively these strategies of EGFR inhibition have, however, only resulted in small increases in overall survival due to the presence of pre-existing intrinsic resistance mechanisms and the ability of tumors to acquire resistance. Resistance to anti-EGFR agents is common and occurs through several proposed mechanisms.12 Point-mutations in the EGFR catalytic domain predicting response to gefitinib and erlotinib in patients with NSCLC have been recently discovered13,14 and are now routinely screened for in advanced NSCLC patient biopsies.15 While, others have shown that increased activity of alternative receptor tyrosine kinases such as HER2, HER3, c-MET, IGF-1R, and AXL promote resistance to anti-EGFR therapeutics by circumventing their inhibitory effects with compensatory downstream signaling.16-20
Mutations in the Kirsten (K)-RAS gene (present in 30–40% of mCRC), is currently the strongest predictive marker of resistance to EGFR-targeted therapy.12,21-27 Indeed, 90% of mCRC patients harboring K-RAS mutations show no therapeutic benefit to cetuximab or panitumumab. Due to the lack of response, the American Society of Clinical Oncology and the European Medicines Agency have issued guidelines to screen patient biopsies for K-RAS mutations prior to treatment,28,29 and subsequently only administer EGFR-based agents into patients with tumors expressing wild-type (wt) K-RAS.28,30 Disappointingly, however, wt K-RAS expression does not predict successful response.21,22,26,31,32 B-RAF, PTEN, PI3-K, and N-RAS have all been identified as possible biomarkers to predict response to EGFR targeted therapy. Mutational analysis in these signaling molecules have revealed conflicting conclusions with some reports observing significant correlation with response to anti-EGFR treatment while others show no correlation.31-35
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family of cytoplasmic transcription factors that are activated by many cytokine and growth factor receptors including the EGFR.36,37 Phosphorylated STAT3 transmits its signal from the EGFR to the nucleus where it initiates transcription of multiple cancer promoting genes such as SOCS3, SMAD7, and VEGF.36,38,39 Furthermore, STAT3 is constitutively active in many types of tumors including those in which anti-EGFR agents have been clinically approved (mCRC, HNSCC, NSCLC, and pancreatic cancer).38,40,41 Importantly, STAT3 activation was recently identified as a potential predictive marker for resistance to anti-EGFR therapies in patients with mCRC and NSCLC.42,43 Inhibiting STAT3 in combination with anti-EGFR therapeutics have also revealed promising data pre-clinically, emphasizing the potential benefit of targeting of STAT3 to optimize anti-EGFR therapy in the clinic.44-47
Our present study utilizes a STAT3 transcriptional reporter to demonstrate that efficacy of anti-EGFR therapeutics correlates with their ability to inhibit STAT3 activation in culture and in animal xenograft studies. We also identify that reduced expression of the STAT3 phosphatase, protein tyrosine phosphatase delta (PTPRD), which is often reduced in expression in colon cancer, enhances STAT3 activity and subsequent STAT3-mediated resistance to anti-EGFR agents in colon cancer.
Results
The efficacy of anti-EGFR agents correlate with STAT3 activity
As STAT3 activity has been shown to correlate with patient response rates to anti-EGFR therapy,42 we set out to test the hypothesis that the efficacy of anti-EGFR agents is dependent on its ability to inhibit STAT3 activity. To do this we stably transfected the STAT3 luciferase reporter construct, pAPRE-luc into 2 tumor cell lines with overexpressed EGFR, HN5, and A431. Parental HN5 and A431 and pAPRE-luc transfected cells displayed similar phospho-EGFR and phospho-STAT3 levels in both basal and EGF-stimulated conditions (Fig. S1A). As expected the HN5-APRE and A431-APRE cells clearly displayed enhanced STAT3 reporter activity compared with control cells when stimulated with EGF (Fig. S1B). We next examined the effect of anti-EGFR therapeutics, cetuximab, and erlotinib on STAT3 transcriptional activity in vitro. Both cetuximab and erlotinib dramatically reduced EGF-mediated STAT3 transcriptional activity in a dose dependant manner (Fig. S1C). We next examined whether similar effects were seen in animal xenograft studies. We found that STAT3 transcriptional activity in A431-APRE xenografts was significantly reduced by a single dose of 0.25 or 0.5 mg 4 h post injection of cetuximab (Fig. 1A and B). This reduced STAT3 activity was still evident 26 h post-treatment of 0.5 mg of cetuximab. STAT3 activity in A431-APRE xenografts treated with 0.25 mg of cetuximab, however, had returned to similar activity levels to those seen in untreated mice after 26 h (Fig. 1A and B). Treatment of A431-APRE xenografts with 0.25 and 0.5 mg of cetuximab three times/weekly for a total of 5 injections resulted in significantly reduced xenograft growth compared with PBS treated mice (Fig. 1C). Interestingly, the 0.5 mg treated mice displayed significantly smaller xenografts compared with the 0.25 mg treated mice indicating that the duration of reduced STAT3 activity may in part play a role in overall tumor growth inhibition.

Figure 1. Efficacy of Cetuximab correlates to STAT3 transcriptional activity in A431 cells. (A) Representative images of A431 cells stably transfected with a STAT3 luciferase reporter construct (pAPRE-luc) (1 × 106/injection) that were injected subcutaneously into BALB/cnu−/nu− mice. Seven days later mice were treated i.p. with vehicle control or 0.25 and 0.5 mg of cetuximab (as indicated) at 0 h and imaged for STAT3 luciferase activity using an IVIS lumina™ at 4 (■) and 26 h (□) post cetuximab injection. Visualization of luciferase activity was achieved using the IVIS lumina™ by injecting mice i.p. with 150 mg/kg of the luciferase substrate, D-luciferin. (B) Analysis of total photon emissions (STAT3 transcriptional activity) was performed using the IVIS lumina™ Image analysis software and presented relative to control treated tumors over time ± SD (*P < 0.05). (C) A431-APRE cells were injected subcutaneously into both flanks of BALB/cnu−/nu− mice (1 × 106/injection) and treated with PBS (◆), 0.25 mg (○), or 0.5 mg (□) of cetuximab 3 times a week for a total of 5 injections/mouse once tumors had reached a mean tumor volume of 100 mm3. Data are expressed as average tumor volume over time (n = 8/group; ± S.E.; *P < 0.05).
The efficacy of anti-EGFR agents correlate with STAT3 activity in human colon cancer cell lines
As cetuximab is approved for the use in mCRC patients, we next assessed whether suppression of STAT3 activity is required for cetuximab-mediated growth inhibition in human colon cancer cell lines. Furthermore, as cetuximab is used to treat mCRC patients with wild-type K-RAS expressing tumors only we excluded the use of any cell lines with mutated K-RAS. Seven mCRC cell lines were infected with our Ad-APRE-luc adenovirus to determine STAT3 transcriptional activity following EGF stimulation and treatment with or without cetuximab and erlotinib. Strikingly, we found that the level of reduced cell viability in vitro mediated by cetuximab (Fig. 2A) and erlotinib (Fig. 2B) directly correlates with their ability to inhibit STAT3 transcriptional activity in all wt K-RAS expressing cell lines tested.
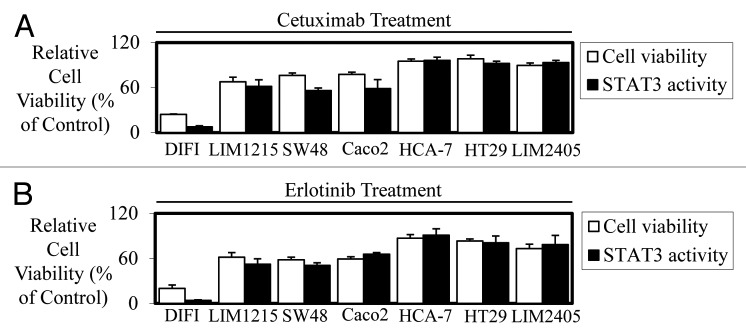
Figure 2. Efficacy of cetuximab and erlotinib correlates to STAT3 transcriptional activity in colon cancer cell lines in vitro. Wild-type K-Ras expressing colon cancer cells were treated in triplicate with (A) cetuximab (± 10 µg/ml) and (B) erlotinib (± 2.5 µM) to assess their effect on cell viability (□) and STAT3 transcriptional activity (■). Cell viability was determined using a commercially available Cell Titer-Glo kit and samples read on a bioluminometer. Data are expressed as % viability compared with untreated control cells ± SD. STAT3 transcriptional activity was determined using a bioluminometer after infection with the Ad-APRE-luc adenovirus and treatment with cetuximab and erlotinib. Data are expressed as percentage STAT3 activity relative to untreated cells ± SD.
We also demonstrated that a correlation exists between the in vivo efficacy of cetuximab with its ability to inhibit STAT3 activity. In contrast, cetuximab could significantly reduce both the STAT3 transcriptional activity in vivo (Fig. 3A and B) and subcutaneous growth of SW48 tumor xenografts (Fig. 3C). Cetuximab had no significant effect however, on either the STAT3 transcriptional activity in vivo (Fig. 3D and E) or the subcutaneous growth of HCA-7 tumor xenografts (Fig. 3F), suggesting that successful cetuximab-mediated inhibition requires reduction of STAT3 activity in vivo.

Figure 3. Efficacy of cetuximab correlates to STAT3 transcriptional activity in SW48 and HCA-7 xenografts. (A) Representative images of SW48 cells that were infected with the adenoviral STAT3 reporter (Ad-APRE-luc) overnight, were then injected subcutaneously into both flanks of BALB/cnu−/nu− mice (5 × 106/injection). Two days later mice were treated i.p. with cetuximab (± 0.5 mg as indicated) and imaged for STAT3 luciferase activity using an IVIS lumina™ Animal Imager at 0 and 24 h post cetuximab injection. Visualization of STAT3-driven luciferase activity was achieved using the IVIS lumina™ by injecting mice i.p. with 150mg/kg of the luciferase substrate, D-luciferin. (B) Analysis of total photon emissions (STAT3 transcriptional activity) was performed using the IVIS lumina™ Image analysis software and presented relative to PBS-treated tumors after 24 h of treatment ± SD (n = 8/group; *P < 0.05). (C) SW48 cells were injected subcutaneously into both flanks of BALB/cnu−/nu− mice (5 × 106/injection) and treated with PBS or cetuximab (0.5 mg) 3 times a week for 2 wk once tumors had reached a mean tumor volume of 140 mm3. Data are expressed as average tumor volume over time (n = 8/group; ± S.E.; *P < 0.05). (D and E) HCA-7 cells were infected with Ad-APRE-luc, injected into BALB/cnu−/nu− mice, treated with cetuximab, and imaged for STAT3 transcriptional activity as indicated in (A and B). (F) HCA-7 cells were injected into BALB/cnu−/nu− mice and treated with cetuximab as indicated in (C).
Combined blockade of EGFR and STAT3 further enhances inhibition of cell viability
As we demonstrated a correlation between the efficacy of anti-EGFR agents, cetuximab and erlotinib with their ability to inhibit STAT3 activity, we next determined whether dual inhibition of EGFR and STAT3 resulted in enhanced inhibition of cell viability. The JAK-STAT3 inhibitor curcurbitacin produced additive anti-proliferative effects combined with cetuximab (Fig. 4A) and erlotinib (Fig. 4B) in all 7 wt K-RAS colon cancer cell lines tested.

Figure 4. Combined blockade of EGFR and STAT3 further enhances inhibition of cell viability. Wild-type K-Ras expressing colon cancer cells were treated in triplicate with (A) control, cetuximab, curcurbitacin, or a combination of both for 72 h and (B) control, erlotinib, curcurbitacin, or a combination of both for 72 h. Cell viability was determined as outlined above (*P < 0.05; #P < 0.01).
PTPRD confers resistance to anti-EGFR therapy
It was recently discovered that the protein tyrosine phosphatase receptor delta (PTPRD) de-phosphorylates STAT3 and its expression is reduced through either gene deletion or epigenetic silencing in many tumor types including mCRC.48 Oncomine data we extracted from the public colon cancer databases support these reports (Fig. 5). PTPRD gene expression was reduced in colorectal cancer tissue compared with normal tissue (Fig. 5A and B),49,50 higher grade tumor tissue compared with lower grade tumor tissue (Fig. 5C)51 and metastatic tumor compared with primary colorectal tumor tissue (Fig. 5D).52
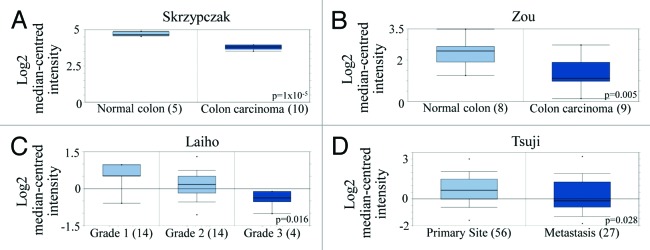
Figure 5. PTPRD expression is reduced in colon cancer and colon cancer metastases. (A and B) PTPRD gene expression in normal tissue vs. cancerous tissue from patient samples. Box-and-whiskers plot with dots representing the high/low values and error bars represent the 90%/10% values. Top and bottom of each box represents the 75%/25% values, and the bar in the middle of the box is the median. The y-axis is normalized mRNA gene expression values that have been log2 transformed. Data was obtained from mRNA studies from (A) Skrzypczak (P = 1 × 10−5) and (B) Zou (P = 0.005) published in Oncomine databases. (C) PTPRD expression in different grades of colorectal adenocarcinoma patient samples from mRNA studies published in Oncomine databases. P = 0.016. (D) Comparison of PTPRD expression from colorectal carcinoma patient samples in both primary (56 patients) and metastasic sites (27 patients). P = 0.028.
As PTPRD expression is reduced in mCRC, it is hypothesized that PTPRD plays a tumor suppressor role by regulating STAT3 activity similarly to the role PTEN plays in regulating PI3-K activity. However, reduced expression of PTPRD (and subsequent elevation of STAT3 activity) in mCRC has not been implicated in promoting resistance to anti-EGFR therapy. Therefore we next explored whether reduced PTPRD expression can decrease the efficacy of anti-EGFR agents. Enhanced STAT3 phosphorylation and STAT3 transcriptional activity was seen in DIFI colon cancer cells stably transfected with PTPRD shRNA (DIFI-PTPRD-sh1 and DIFI-PTPRD-sh2) compared with parental DIFI cells (Fig. 6A and B). DIFI-PTPRD-sh1 and DIFI-PTPRD-sh2 cells displayed greater resistance to the inhibitory effects of cetuximab at both 1 and 10 µg/ml in cell viability assays (Fig. 6C). Furthermore, the JAK-STAT3 inhibitor, curcurbitacin could re-sensitize these resistant cells to cetuximab (Fig. 6D), suggesting a role of STAT3 activity in mediating resistance to anti-EGFR therapy.
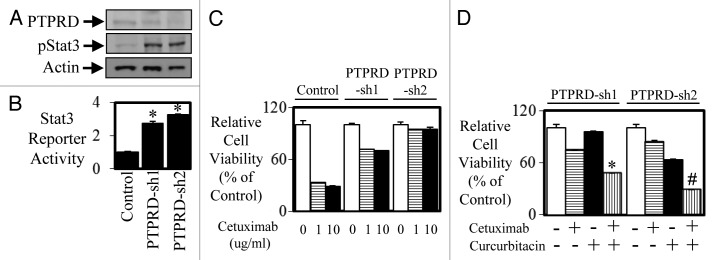
Figure 6. Knockdown of PTPRD leads to enhanced STAT3 activity and enhanced resistance to anti-EGFR therapy. DIFI cells were stably transfected with control or PTPRD shRNA (PTPRD-sh1 and PTPRD-sh2) and then assessed for (A) PTPRD and phospho-STAT3 expression by western blot and (B) STAT3 transcriptional activity was determined using a bioluminometer, 24 h after infection with the Ad-APRE-luc adenovirus. Data are expressed as relative luciferase activity relative to DIFI control cells (*P < 0.05). DIFI control, DIFI-PTPRD-sh1, and DIFI-PTPRD-sh2 were treated with (C) cetuximab (0, 1, or 10 µg/ml) for 72 h and cell viability was determined using a commercially available Cell Titer-Glo kit and samples read on a bioluminometer. Data are expressed as % viability compared with untreated control cells ± SD. DIFI-PTPRD-sh1 and DIFI-PTPRD-sh2 cells were treated (in triplicate) with (D) control, cetuximab, curcurbitacin, or a combination of both for 72 h. Cell viability was determined as outlined above (*P < 0.05; #P < 0.01).
STAT3 activity confers resistance to anti-EGFR therapy in an acquired resistance model
Finally, to test whether STAT3 is elevated in cells that acquire resistance to anti-EGFR treatment, we developed a HN5 cell line that was resistant to the inhibitory effects of AG1478 by long-term culture (>6 mo) of HN5 cells in the presence of AG1478 (Fig. 7A). Both HN5 parental and HN5-AG cells displayed similar basal and EGF-mediated STAT3 activity (Fig. 7B). HN5-AG cells however retained significantly greater levels of STAT3 activity when treated with 0.1 and 1 µM of AG1478 compared with HN5 parental cells. Importantly, HN5-AG cells, which were also resistant to cetuximab and erlotinib, could be re-sensitized to the inhibitory effects of cetuximab (Fig. 7C), erlotinib (Fig. 7D), and AG1478 (Fig. 7E) when co-treated with the JAK-STAT3 inhibitor curcurbitacin.

Figure 7. Acquired resistance in HN5 cells is reversed with STAT3 blockade. (A) HN5 parental (□) and HN5-AG (■) cells were treated with AG1478 (± 100 nM) for 72 h and cell viability was determined as outlined above (#P < 0.01). (B) HN5 parental (□) and HN5-AG (■) cells were infected with Ad-pAPRE-luc then treated with EGF and AG1478 for 24 h as indicated, lysed, and assessed for luciferase activity (STAT3 transcriptional activity). Data are expressed as relative luciferase activity (fold change ± S.D.; *P < 0.05). (C) HN5-AG cells were treated with control, cetuximab, curcurbitacin, or a combination or (D) control, erlotinib, curcurbitacin, or a combination or (E) control, AG1478, curcurbitacin, or a combination for 72 h. Cell viability was determined as outlined above. (*P < 0.05; #P < 0.01).
Discussion
Tumor resistance to molecular targeted therapy is well recognized as a major challenge to successful clinical outcome. The significance of discovering critical mediators of resistance has been highlighted by the identification that K-RAS mutations in mCRC negatively predicts response to EGFR-based therapy in over 90% of cases.21,26,31 Based on these findings, anti-EGFR agents are now excluded as a treatment strategy for mCRC patients with tumors harboring K-RAS mutations, thus allowing for possible successful treatment with other therapeutics. However, 60–80% of patients that do express wt K-RAS and are therefore still considered suitable for anti-EGFR treatment do not respond,21,22,26,31,32,52 suggesting that other mediators of resistance are present.
Dobi and colleagues recently demonstrated for the first time that positive expression of STAT3 phosphorylation in mCRC correlated negatively to objective response rates of anti-EGFR therapeutics.42 These results further support the rationale to develop STAT3 inhibitors for the treatment of cancer patients as either single agents or to potentiate anti-EGFR inhibitors. Indeed, pre-clinical data evaluating the inhibition of STAT3 in conjunction with EGFR blockade has produced encouraging outcomes in several tumor types.44,46,47,53
Our current data supports these observations, and provides a luciferase-based platform to thoroughly evaluate anti-STAT3 therapeutics in vivo. Currently, testing the efficacy of anti-EGFR agents (and other therapeutics) in animal models is based on their capability to inhibit tumor growth. Therefore, optimizing therapeutic efficacy is problematic as evaluation of the activation status of EGFR downstream substrates such as STAT3 can only be measured retrospectively after tumors have been resected. Our in vivo model described here, overcomes many of these limitations. The stable incorporation or the adenoviral infection of the STAT3 reporter (APRE-luc) allows for direct assessment of the capability and sustainability of EGFR and STAT3 therapeutic agents to inhibit STAT3 transcriptional activity in real-time in vivo. These systems will also benefit in optimizing dose and scheduling regimens to maximize therapeutic benefit. In addition, our studies offer a proof-of-principle example for testing the efficacy of any future or current agent or combination of agents to inhibit critical downstream signaling molecules throughout the duration of treatment.
In an attempt to model acquired resistance seen in patients, several groups including ours have cultured human cancer cell lines in the continuous and long-term presence of EGFR inhibitors, generating cells that are refractory to the anti-EGFR agent used.17,20,53-60 These laboratory studies have identified similar molecular changes in refractory cells to that seen in retrospective clinical analysis of patient biopsies. Thus these models represent a rationale and valuable method for evaluating acquired resistance observed in the clinic. However, despite identifying increased activity in upstream molecules known to regulate STAT3 activation (EGFR, c-MET, IGF-1R, and c-SRC) and increased activity in molecules known to be regulated by STAT3 (cyclin D1, COX-2, and VEGF), only one very recent report examines modifications to STAT3 activation in anti-EGFR resistant cells. Li and colleagues generated erlotinib-acquired resistant HCC827/ER cells, from the erlotinib-sensitive HCC827 human lung cancer cell line.53 These HCC827/ER cells displayed greater basal levels of phosphorylated STAT3 compared with the parental cell line and their resistance to erlotinib could be reversed when treated with either a STAT3 inhibitor or when STAT3 expression was stably knocked down. Interestingly, our cell line (HN5-AG) with acquired resistance did not have enhanced basal or EGF-stimulated STAT3 activity compared with its control but displayed more STAT3 activity after AG1478 treatment suggesting a possible alternate or compensatory pathway that maintains STAT3 activity when EGFR is de-activated. Others have shown that alternative receptor tyrosine kinases such as HER3, c-MET, IGF-1R, and AXL have increased activity in the absence of EGFR signaling thereby compensating for EGFR blockade and circumventing the inhibitory effects of anti-EGFR therapeutics.16-18,20 Importantly we demonstrate that the blockade of STAT3 activity in HN5-AG cells and DIFI cells with reduced PTPRD expression led to a re-sensitization to cetuximab, which represents a potential new model of resistance. Our mining of PTPRD expression data from the publicly available Oncomine databases suggest PTPRD is reduced in mCRC and could play a key role in both colon cancer development and progression. Our results indicate that resistance to anti-EGFR therapy may occur, at least in part, from altered PTPRD expression and increased STAT3 activity in mCRC.
However, other phosphatases have been implicated in regulating STAT3 phosphorylation61 and thus may also play roles in circumventing the blockade of EGFR and other receptors that drive STAT3 activity. Most recently, Lui and colleagues found that a large cohort of patient HNSCC tumors harbored mutations to several PTPR family members including PTPRD, and this correlated with enhanced STAT3 activity.62 Whether these patients were treated with anti-EGFR therapy and whether they were more resistant to this therapy was not discussed. Interestingly, Li and colleagues found reduced levels of another STAT3 phosphatase, PTPMeg263 in their HCC827/ER refractory lung cancer compared with HCC827 parental cells.53 Likewise, similar findings have been observed for reduced PTEN expression and resistance to anti-EGFR therapy through enhanced PI3-K-AKT-mTOR signaling.33,64,65
In summary, the present study characterizes an in vivo model for detecting suitable dose and scheduling of therapeutic agents targeting the EGFR-STAT3 signaling network. Our current data indicates that the efficacy of anti-EGFR therapy correlates with the ability of these agents to block STAT3 activity. We also identify a potential new role for decreased PTPRD phosphatase expression in mediating resistance to anti-EGFR therapy in colon cancer through enhancing STAT3 activity. These data further enhances the rationale treatment strategy of combining anti-EGFR therapeutics with anti-STAT3 agents in mCRC and other EGFR targeted refractory tumors.
Materials and Methods
Antibodies and reagents
Rabbit polyclonal antibody directed against STAT3 and the goat polyclonal antibody directed against PTPRD were obtained from Santa Cruz Biotechnology. The phospho-STAT3 rabbit polyclonal antibody was from Cell Signaling Technology, while the mouse phospho-tyrosine monoclonal antibody (4G10) was from Upstate Biotechnology. The anti-mouse Actin antibody was purchased from Sigma. The anti-EGFR monoclonal antibody cetuximab and the tyrosine kinase inhibitors erlotinib and AG1478 were all provided by the Ludwig Institute for Cancer Research (Melbourne Branch). Recombinant mouse EGF was from Invitrogen and curcurbitacin I was purchased from Calbiochem/Millipore (EMD Millipore Corporation).
Cells and cell culture
The epidermoid carcinoma cell line A431 and the head and neck carcinoma cell line HN5, have been previously described.66 HN5-APRE and A431-APRE cells were generated by co-transfecting with the firefly luciferase STAT3 transcriptional activity reporter construct, pAPRE-luc67 and pcDNA3 using FuGENE HD and selected for using geneticin. HN5-AG cells were generated by continuously culturing HN5 cells in AG1478 for 6 mo as a result of which this selected sub-population of HN5 cells were resistant to AG1478.
Human colon cancer cell line DIFI, LIM1215, SW48, Caco2, HCA-7, HT29, and LIM2405 were all provided by the Ludwig Institute for Cancer Research (Melbourne Branch). DIFI-PTPRDsh1 and DIFI-PTPRDsh2 cells were generated by transfecting PTPRD shRNA48 into DIFI parental cells using FuGENE HD and selected for using geneticin. All cells were maintained in Dulbecco’s modified Eagle’s medium contained 10% fetal bovine serum (FBS) (DKSH), 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin (Invitrogen). Cells were incubated in a humidified atmosphere of 10% CO2 at 37 °C.
Generation of the Ad-APRE-luc adenovirus
The pAPRE-luc DNA construct was digested with SacI and Bgl-II yielding an APRE-luc insert that was subsequently cloned into an in-house pENTR 1A-CAGA construct.68 LR recombination was then performed with the pAd/PL-DEST destination vector (Invitrogen) to generate the pAPRE-luc Adenoviral Expression plasmid. The Expression plasmid was digested with Pac I to expose the ITRs and then transfected into 293A cell line using Lipofectamine LTX transfection reagent (Invitrogen). Cells were harvested approximately 2 wk after transfection when lysis was observed in the majority of cells. The adenovirus was amplified and used to detect STAT3 transcriptional activity in cultured cell lines and in mice.
Luciferase assays
HN5-APRE and A431-APRE cells were seeded into a 96-well plate and allowed to adhere overnight. Alternatively, the human colon cancer cell lines were infected with the Ad-APRE-luc adenovirus and allowed to adhere overnight. After 24 h, cells were washed with PBS and cultured with EGF (50 ng/ml) ± anti-EGFR inhibitor where indicated for a further 24 h. Cells were then lysed and assessed for luciferase activity using the Luciferase Reporter Assay Kit (Promega) following the manufacturer’s instructions.
Cell viability assays
Cells were plated in 96-well plates and allowed to adhere overnight. Triplicate wells were treated with varying concentrations of inhibitors where indicated for 72 h. Cell were then lysed and cell viability relative to vehicle control was determined using a commercially available Cell Titer-Glo kit (Promega) following manufacturer’s instructions. Samples were read on a bioluminometer.
Western blot analysis
Cells were lysed with lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton-X-100, 50 mM NaF, 2 mM MgCl2, 1 mM Na3VO4, and protease inhibitor cocktail [Roche]) and clarified by centrifugation (13 000 g for 15 min at 4 °C). Proteins were then separated by SDS-PAGE (Invitrogen), blotted onto nitrocellulose and probed with the indicated primary antibodies. The signal was visualized using the ECL chemilluminescence detection kit (GE Healthcare) following incubation with appropriate secondary antibodies.
Tumor growth and Bioluminescence imaging in vivo
A431-APRE (1 × 106), SW48 (5 × 106), and HCA-7 (1 × 107) cells were inoculated s.c. into both flanks of 4–6 wk old BALB/c nude mice (Animal Research Centre). Tumor volume in mm3 was determined using the formula (length × width2)/2, where length was the longest axis and width the measurement at right angles to the length.69 This research project was approved by the Animal Ethics Committee of the Ludwig Institute for Cancer Research and Department of Surgery, University of Melbourne at the Royal Melbourne Hospital.
For bioluminescence imaging, mice with subcutaneous tumors were injected i.p. with 150 mg/kg D-luciferin (Xenogen Corp.) in PBS, anesthetized with isofluorane and placed under the IVIS camera (Xenogen Corp.) at the indicated times. The bioluminescence images were recorded between 10 to 20 min after each luciferin injection and bioluminescence intensity was quantified as the sum of detected photons per second within the region of interest using the LivingImage software (Xenogen Corp.).
Data mining using Oncomine
Oncomine 4.4.4.3 (www.oncomine.org, Compendia Bioscience™, part of Life Technologies) was utilized for the analysis of PTPRD gene expression. Oncomine is an online tool that contains 715 mRNA and copy number expression data sets from 86 733 cancer and normal tissue samples. These data sets are compiled from publically available cancer microarray data which is then processed using the same criteria before being made available.70 We used the Oncomine compendium to study the profile of PTPRD expression in cancer tissues vs. their normal tissue counterparts. This was achieved by subjecting each data set to threshold criteria for inclusion in the analysis. The threshold criteria that were initially utilized for this study were a P value < 0.05 and an mRNA expression fold change > 1.4. The fold change is classified as a change in the mRNA expression level in the cancer tissue compared with the normal expression level for that tissue specifically for your gene of interest. Based on the threshold criteria, Oncomine will then assign a gene rank percentile for all genes studied within a data set. This figure is the percentage ranking of your gene of interest based on the P value relative to the P values of all the other genes within the same data set. The gene expression data generated through Oncomine is log transformed and standard deviation normalized to one per array studied.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
R.B.L. is a Melbourne Brain Centre Post-Doctoral Research Fellow (Dept of Surgery and Dept of Medicine, Royal Melbourne Hospital, The University of Melbourne) and is supported by the NHMRC Centre for Research Excellence Grant 1001216.
Author Contribution
N.U., T.L.P., S.S.S., I.N., and R.B.L. all contributed to acquisition of data, analysis, and interpretation of results. J.M.M., T.A.C., H.J.Z. and R.B.L. contributed to the conception and design of the study, and R.B.L. was involved in drafting and revising the manuscript.
Glossary
Abbreviations:
- EGFR
epidermal growth factor receptor
- STAT3
signal transducer and activator of transcription 3
- PTPRD
protein tyrosine phosphatase receptor delta
- PI3-K
phosphoinositide 3-kinase
- ERK
extracellular signal-related kinase
- JAK
Janus kinase
- IGF-1R
insulin-like growth factor-1 receptor
- COX2
cyclooxygenase 2
- VEGF
vascular endothelial growth factor
- mCRC
metastatic colorectal carcinoma
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/28179
References
- 1.Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008;26:263–74. doi: 10.1080/08977190802312844. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18:73–9. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–65. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 4.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-I. [DOI] [PubMed] [Google Scholar]
- 5.Baselga J, Norton L, Masui H, Pandiella A, Coplan K, Miller WH, Jr., Mendelsohn J. Antitumor effects of doxorubicin in combination with anti-epidermal growth factor receptor monoclonal antibodies. J Natl Cancer Inst. 1993;85:1327–33. doi: 10.1093/jnci/85.16.1327. [DOI] [PubMed] [Google Scholar]
- 6.Fan Z, Baselga J, Masui H, Mendelsohn J. Antitumor effect of anti-epidermal growth factor receptor monoclonal antibodies plus cis-diamminedichloroplatinum on well established A431 cell xenografts. Cancer Res. 1993;53:4637–42. [PubMed] [Google Scholar]
- 7.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 8.Giusti RM, Shastri K, Pilaro AM, Fuchs C, Cordoba-Rodriguez R, Koti K, Rothmann M, Men AY, Zhao H, Hughes M, et al. U.S. Food and Drug Administration approval: panitumumab for epidermal growth factor receptor-expressing metastatic colorectal carcinoma with progression following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens. Clin Cancer Res. 2008;14:1296–302. doi: 10.1158/1078-0432.CCR-07-1354. [DOI] [PubMed] [Google Scholar]
- 9.Cohen MH, Williams GA, Sridhara R, Chen G, McGuinn WD, Jr., Morse D, Abraham S, Rahman A, Liang C, Lostritto R, et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10:1212–8. doi: 10.1158/1078-0432.CCR-03-0564. [DOI] [PubMed] [Google Scholar]
- 10.Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, Kris MG, Tran HT, Klein P, Li X, et al. TRIBUTE Investigator Group TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005;23:5892–9. doi: 10.1200/JCO.2005.02.840. [DOI] [PubMed] [Google Scholar]
- 11.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 12.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–7. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 13.Riely GJ, Politi KA, Miller VA, Pao W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin Cancer Res. 2006;12:7232–41. doi: 10.1158/1078-0432.CCR-06-0658. [DOI] [PubMed] [Google Scholar]
- 14.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25:587–95. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 15.Azzoli CG, Baker S, Jr., Temin S, Pao W, Aliff T, Brahmer J, Johnson DH, Laskin JL, Masters G, Milton D, et al. American Society of Clinical Oncology American Society of Clinical Oncology Clinical Practice Guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol. 2009;27:6251–66. doi: 10.1200/JCO.2009.23.5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002;62:200–7. [PubMed] [Google Scholar]
- 17.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 18.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–56. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giles KM, Kalinowski FC, Candy PA, Epis MR, Zhang PM, Redfern AD, Stuart LM, Goodall GJ, Leedman PJ. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther. 2013;12:2541–58. doi: 10.1158/1535-7163.MCT-13-0170. [DOI] [PubMed] [Google Scholar]
- 21.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 22.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 23.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 24.De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–15. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 25.Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–9. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 27.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 28.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 29.Van Cutsem EJ, Oliveira J, ESMO Guidelines Working Group Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2008;19(Suppl 2):ii33–4. doi: 10.1093/annonc/mdn079. [DOI] [PubMed] [Google Scholar]
- 30.Duffy MJ, O’Donovan N, Crown J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat Rev. 2011;37:151–9. doi: 10.1016/j.ctrv.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 31.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 32.Hawkes E, Cunningham D. Relationship between colorectal cancer biomarkers and response to epidermal growth factor receptor monoclonal antibodies. J Clin Oncol. 2010;28:e529–31, author reply e532-3. doi: 10.1200/JCO.2010.29.5626. [DOI] [PubMed] [Google Scholar]
- 33.Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68:1953–61. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15:3184–8. doi: 10.1158/1078-0432.CCR-08-2961. [DOI] [PubMed] [Google Scholar]
- 35.Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, Molinari F, De Dosso S, Saletti P, Martini M, Cipani T, Marrapese G, Mazzucchelli L, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS One. 2009;4:e7287. doi: 10.1371/journal.pone.0007287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/S0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 37.Zhong Z, Wen Z, Darnell JE., Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–8. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 38.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54. [PubMed] [Google Scholar]
- 39.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–40. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11:595–6. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 41.Lin Q, Lai R, Chirieac LR, Li C, Thomazy VA, Grammatikakis I, Rassidakis GZ, Zhang W, Fujio Y, Kunisada K, et al. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol. 2005;167:969–80. doi: 10.1016/S0002-9440(10)61187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobi E, Monnien F, Kim S, Ivanaj A, N’Guyen T, Demarchi M, Adotevi O, Thierry-Vuillemin A, Jary M, Kantelip B, et al. Impact of STAT3 phosphorylation on the clinical effectiveness of anti-EGFR-based therapy in patients with metastatic colorectal cancer. Clin Colorectal Cancer. 2013;12:28–36. doi: 10.1016/j.clcc.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Haura EB, Sommers E, Song L, Chiappori A, Becker A. A pilot study of preoperative gefitinib for early-stage lung cancer to assess intratumor drug concentration and pathways mediating primary resistance. J Thorac Oncol. 2010;5:1806–14. doi: 10.1097/JTO.0b013e3181f38f70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonner JA, Yang ES, Trummell HQ, Nowsheen S, Willey CD, Raisch KP. Inhibition of STAT-3 results in greater cetuximab sensitivity in head and neck squamous cell carcinoma. Radiother Oncol. 2011;99:339–43. doi: 10.1016/j.radonc.2011.05.070. [DOI] [PubMed] [Google Scholar]
- 45.Kijima T, Niwa H, Steinman RA, Drenning SD, Gooding WE, Wentzel AL, Xi S, Grandis JR. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13:355–62. [PubMed] [Google Scholar]
- 46.Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–89. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 47.Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, Wang L, Gooding WE, Johnson DE, Grandis JR. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res. 2012;18:4986–96. doi: 10.1158/1078-0432.CCR-12-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009;106:9435–40. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, Pachlewski J, Oledzki J, Ostrowski J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010;5:e13091. doi: 10.1371/annotation/8c585739-a354-4fc9-a7d0-d5ae26fa06ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou TT, Selaru FM, Xu Y, Shustova V, Yin J, Mori Y, Shibata D, Sato F, Wang S, Olaru A, et al. Application of cDNA microarrays to generate a molecular taxonomy capable of distinguishing between colon cancer and normal colon. Oncogene. 2002;21:4855–62. doi: 10.1038/sj.onc.1205613. [DOI] [PubMed] [Google Scholar]
- 51.Laiho P, Kokko A, Vanharanta S, Salovaara R, Sammalkorpi H, Järvinen H, Mecklin JP, Karttunen TJ, Tuppurainen K, Davalos V, et al. Serrated carcinomas form a subclass of colorectal cancer with distinct molecular basis. Oncogene. 2007;26:312–20. doi: 10.1038/sj.onc.1209778. [DOI] [PubMed] [Google Scholar]
- 52.Tsuji S, Midorikawa Y, Takahashi T, Yagi K, Takayama T, Yoshida K, Sugiyama Y, Aburatani H. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br J Cancer. 2012;106:126–32. doi: 10.1038/bjc.2011.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL, Ramalingam SS, Curran WJ, Khuri FR, Deng X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol Cancer Ther. 2013;12:2200–12. doi: 10.1158/1535-7163.MCT-13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benavente S, Huang S, Armstrong EA, Chi A, Hsu KT, Wheeler DL, Harari PM. Establishment and characterization of a model of acquired resistance to epidermal growth factor receptor targeting agents in human cancer cells. Clin Cancer Res. 2009;15:1585–92. doi: 10.1158/1078-0432.CCR-08-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ciardiello F, Bianco R, Caputo R, Caputo R, Damiano V, Troiani T, Melisi D, De Vita F, De Placido S, Bianco AR, et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res. 2004;10:784–93. doi: 10.1158/1078-0432.CCR-1100-03. [DOI] [PubMed] [Google Scholar]
- 56.Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–19. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, Mendelsohn J, Fan Z. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67:8240–7. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 58.Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, Rak J, Kerbel RS. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–101. [PubMed] [Google Scholar]
- 59.Wheeler DL, Iida M, Kruser TJ, Nechrebecki MM, Dunn EF, Armstrong EA, Huang S, Harari PM. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009;8:696–703. doi: 10.4161/cbt.8.8.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamasaki F, Johansen MJ, Zhang D, Krishnamurthy S, Felix E, Bartholomeusz C, Aguilar RJ, Kurisu K, Mills GB, Hortobagyi GN, et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007;67:5779–88. doi: 10.1158/0008-5472.CAN-06-3020. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, Polakiewicz RD, Kinzler KW, Vogelstein B, Velculescu VE, et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A. 2007;104:4060–4. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lui VW, Peyser ND, Ng PK, Hritz J, Zeng Y, Lu Y, Li H, Wang L, Gilbert BR, General IJ, et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc Natl Acad Sci U S A. 2014;111:1114–9. doi: 10.1073/pnas.1319551111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su F, Ren F, Rong Y, Wang Y, Geng Y, Wang Y, Feng M, Ju Y, Li Y, Zhao ZJ, et al. Protein tyrosine phosphatase Meg2 dephosphorylates signal transducer and activator of transcription 3 and suppresses tumor growth in breast cancer. Breast Cancer Res. 2012;14:R38. doi: 10.1186/bcr3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, Tsurutani J, Dennis PA, Mills GB, Arteaga CL. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–22. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 65.She QB, Solit D, Basso A, Moasser MM. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res. 2003;9:4340–6. [PubMed] [Google Scholar]
- 66.Luwor RB, Baradaran B, Taylor LE, Iaria J, Nheu TV, Amiry N, Hovens CM, Wang B, Kaye AH, Zhu HJ. Targeting Stat3 and Smad7 to restore TGF-β cytostatic regulation of tumor cells in vitro and in vivo. Oncogene. 2013;32:2433–41. doi: 10.1038/onc.2012.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Robledo O, Kinzie E, Blanchard F, Richards C, Miyajima A, Baumann H. Receptor subunit-specific action of oncostatin M in hepatic cells and its modulation by leukemia inhibitory factor. J Biol Chem. 2000;275:25273–85. doi: 10.1074/jbc.M002296200. [DOI] [PubMed] [Google Scholar]
- 68.Luwor RB, Wang B, Nheu TV, Iaria J, Tsantikos E, Hibbs ML, Sieber OM, Zhu HJ. New reagents for improved in vitro and in vivo examination of TGF-β signalling. Growth Factors. 2011;29:211–8. doi: 10.3109/08977194.2011.615311. [DOI] [PubMed] [Google Scholar]
- 69.Luwor RB, Johns TG, Murone C, Huang HJ, Cavenee WK, Ritter G, Old LJ, Burgess AW, Scott AM. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001;61:5355–61. [PubMed] [Google Scholar]
- 70.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.