

Abstract
Aims: S-nitrosylation and S-glutathionylation, redox-based modifications of protein thiols, are recently emerging as important signaling mechanisms. In this study, we assessed S-nitrosylation-based regulation of Janus-activated kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathway that plays critical roles in immune/inflammatory responses and tumorigenesis. Results: Our studies show that STAT3 in stimulated microglia underwent two distinct redox-dependent modifications, S-nitrosylation and S-glutathionylation. STAT3 S-nitrosylation was associated with inducible nitric oxide synthase (iNOS)-produced nitric oxide (NO) and S-nitrosoglutathione (GSNO), whereas S-glutathionylation of STAT3 was associated with cellular oxidative stress. NO produced by iNOS or treatment of microglia with exogenous GSNO inhibited STAT3 activation via inhibiting STAT3 phosphorylation (Tyr705). Consequently, the interleukin-6 (IL-6)-induced microglial proliferation and associated gene expressions were also reduced. In cell-free kinase assay using purified JAK2 and STAT3, STAT3 phosphorylation was inhibited by its selective preincubation with GSNO, but not by preincubation of JAK2 with GSNO, indicating that GSNO-mediated mechanisms inhibit STAT3 phosphorylation through S-nitrosylation of STAT3 rather than JAK2. In this study, we identified that Cys259 was the target Cys residue of GSNO-mediated S-nitrosylation of STAT3. The replacement of Cys259 residue with Ala abolished the inhibitory role of GSNO in IL-6-induced STAT3 phosphorylation and transactivation, suggesting the role of Cys259 S-nitrosylation in STAT3 phosphorylation. Innovation: Microglial proliferation is regulated by NO via S-nitrosylation of STAT3 (Cys259) and inhibition of STAT3 (Tyr705) phosphorylation. Conclusion: Our results indicate the regulation of STAT3 by NO-based post-translational modification (S-nitrosylation). These findings have important implications for the development of new therapeutics targeting STAT3 for treating diseases associated with inflammatory/immune responses and abnormal cell proliferation, including cancer. Antioxid. Redox Signal. 20, 2514–2527.
Introduction
Microglia serve as the first and main form of active immune defense in related CNS diseases. Under the disease conditions, insults to the nervous system trigger a multistage activation of microglia that leads to proliferation, migration to the site of injury, increased expression of immunomodulators, and transformation into phagocytes that are capable of clearing damaged cells and debris (3). Microglial activation involves multiple signaling cascades, including NF-κB, Janus-activated kinase (JAK)–signal transducer and activator of transcription (STAT), and stress-activated protein kinase pathways (25, 28, 49), among which JAK-STAT signaling plays a major role in the regulation of cell cycle progression and proliferation of microglia as well as many other cell types (5). STAT proteins are a family of latent cytoplasmic transcription factors that become phosphorylated by JAK in response to various cytokines and growth factors. Among the seven members of mammalian STAT family identified (STAT1–4, STAT5a, STAT5b, and STAT6), STAT3 is the most pleotropic member and most strongly implicated not only in inflammatory/immune signaling pathways (38) but also in number of pathways important in tumorigenesis and metastasis (5).
Innovation.
Signal transducer and activator of transcription 3 (STAT3) plays critical roles in immune and inflammatory responses as well as tumorigenesis. S-nitrosylation has been recently recognized as an important nitric oxide (NO)-dependent signal transduction mechanism for cell cycle, cell survival, and cell death. However, the regulation of STAT3 by NO or S-nitrosylation remains unclear. The present study for the first time demonstrates that phosphorylation of STAT3 is regulated by NO-mediated S-transnitrosylation of STAT3. Consequently, NO regulates microglial proliferation by modulating downstream target of STAT3, thereby suggesting that STAT3 regulation by redox-based NO signaling might be a potential target for diseases associated with inflammation/immune responses and abnormal cell proliferation.
STAT3 is activated by the interleukin-6 (IL-6) family of cytokines and growth factors. Binding of IL-6 to its receptor gp80 (subunit α) induces homodimerization of gp130 (subunit β) and phosphorylation of the gp130-associated JAK2. JAK2 phosphorylates the Tyr residues on cytoplasmic region of gp130 that serve as docking sites for STAT3. STAT3 binds to the respective tyrosine residues on gp130 through its Src homology 2 (SH2) domain and is subsequently phosphorylated on Tyr705 at the carboxyl terminus by the JAK2 (21). STAT3 phosphorylation induces its dimerization via reciprocal interactions between the SH2 domain and the phosphorylated Tyr705 and then, in turn, translocates into the nucleus where it regulates the expression of many acute-phase protein genes (21). The Tyr705 phosphorylation of STAT3 by JAK2 is dephosphorylated by protein tyrosine phosphatases (PTP), such as SH2 domain-containing PTPs (SHP-1 and SHP-2) (21) and nuclear isoform of T-cell PTP (TC-PTP) (17). In addition to C-terminal tyrosine phosphorylation, transcriptional activity of STAT3 is also regulated by mitogen-activated protein kinase (MAPK)-mediated phosphorylation at Ser727 in response to growth factors (9). In addition, STAT3 activity is also regulated by other types of post-translational modifications, such as S-glutathionylation (51), acetylation (53), and methylation (52). These reports document that STAT3 transactivity is regulated by multiple cellular signaling mechanisms. In addition, here we report that nitric oxide (NO)-mediated secondary modification (S-nitrosylation) of STAT3 also inhibits its phosphorylation as well as its transactivity.
Microglial proliferation is regulated by NO generated by endogenous inducible NO synthase (iNOS) under the inflammatory condition (26). NO is a signaling molecule derived from L-Arg in a reaction catalyzed by different isoforms of NOS (i.e., iNOS, eNOS, and nNOS) (34). NO is known to exert its effects through two pathways: one that relies on the activation of soluble guanylyl cyclase (sGC) thus increased cGMP and a second one that is independent of cGMP (11). Recently, S-nitrosylation, a redox-based post-translational modification of proteins by NO, is recognized to regulate the activities of an increasing number of target proteins, including metabolic, structural, cytoskeletal, and signaling proteins (44, 45). In addition, Protein S-nitrosylation is emerging as a major post-translational modification regulating many cellular activities, including phosphorylation/dephosphorylation (kinases and phosphatases), acetylation (histone deacetylase), palmitoylation, ubiquitinylation, metalloprotease (MMP9), and caspase (caspase 3) (2, 15, 23, 24, 44, 54). The protein S-nitrosylation can be mediated through a direct reaction between NO and protein thiol in the presence of electron acceptors or by formation of transition metal adduct (19). However, recent studies have shown that protein S-nitrosylation in in vivo conditions is mainly mediated through the formation of low molecular mass S-nitrosothiols (RSNO) (8). S-nitroso-L-cysteine and S-nitrosoglutathione (GSNO) are the most characterized and abundant endogenous RSNOs synthesized by reaction between NO and cysteine or NO and glutathione (GSH). Recently, these low molecular mass RSNOs have been implicated as a potent anti-inflammatory and antioxidant mediators, vasodilators, and inhibitors of platelet aggregation (6, 12, 27, 32, 39). However, the mechanism(s) underlying the role of NO and RSNO in the regulation of microglial proliferation is not understood well at present.
In this study, we document that endogenous NO generated by iNOS or exogenous GSNO treatment induces microglial cell cycle arrest and inhibited cell proliferation via redox-based post-translational modification (S-nitrosylation) of STAT3. S-nitrosylation of STAT3 inhibits its target DNA interaction and thus downstream gene expressions for microglial proliferation and survival. These studies also report that Cys259 in STAT3 was specifically S-nitrosylated by GSNO and its mutation to Ala (C259A) abolished GSNO-induced STAT3 S-nitrosylation as well as inhibition of STAT3 activation. Taken together, these data describe, for the first time, S-nitrosylation-mediated regulation of STAT3. These findings are important for STAT3-mediated inflammatory mechanisms in cancer and neurodegenerative diseases and thus related therapeutics.
Results
NO produced by iNOS inhibits microglial proliferation by inhibiting STAT3 activity
We previously observed that GSNO treatment inhibited the phosphorylation of STAT3 in T cells (32). Based on the established role of STAT3 in cell cycle progression and proliferation and the reported role of endogenous NO produced by iNOS in cell cycle arrest and inhibition of microglial proliferation (26), we investigated the role of endogenous NO in microglial STAT3 regulation (phosphorylation) and cell proliferation. Figure 1A shows that lipopolysaccharide (LPS) treatment (0.1 μg/ml) of BV2 microglia increased iNOS protein expression (Fig. 1A-i) and NO production (Fig. 1A-iv) significantly within 6 h of treatment and that continued to increase over time. To assess the role of endogenous NO on the regulation of STAT3 activity, the cells were treated with LPS for 1, 3, 6, 12, and 24 h and then treated with IL-6 (30 ng/m) for 0.5 h and the phosphorylated STAT3 (Tyr705) levels were analyzed by Western analysis. Figure 1A-ii and iv show that IL-6-induced increase in STAT3 phosphorylation was decreased by LPS pretreatment and that this inhibition paralleled with treatment time-dependent iNOS expression and NO production (Fig. 1A-i, iv). In addition, the decreased STAT3 phosphorylation (Fig. 1A-ii, iv) correlated with increase in S-nitrosylation of STAT3 (Fig. 1A-iii, iv) up to 12 h after LPS treatment, indicating an inverse relationship between S-nitrosylation and phosphorylation of STAT3. At 24 h after LPS treatment, STAT3 phosphorylation continued to decline with some decrease in STAT3 S-nitrosylation, suggesting the S-nitrosylation-dependent mechanism, at least in part, in the inhibition of STAT3 phosphorylation at this time point.
FIG. 1.

NO produced by iNOS inhibits cell proliferation by inhibiting STAT3 activity. (A) Cultured BV2 murine microglial cells were treated with LPS (0.1 μg/ml) for different time periods, and the production of NO and expression of iNOS protein were analyzed (A-i, iv). To assess the role of endogenous NO produced by LPS treatment on phosphorylation of STAT3 (Tyr705), the cells were pretreated with LPS for different time periods (0–24 h), then treated with IL-6 (30 ng/ml) for 0.5 h, and cellular phosphorylated STAT3 (pSTAT3) levels were analyzed (A-ii, iv). Under the same experimental conditions, the effect of LPS on STAT3 S-nitrosylation (sno-STAT3 levels) was analyzed by biotin switch assay as described under the Materials and Methods section (A-iii, iv). The NO production by LPS treatment and its effect on IL-6-induced STAT3 phosphorylation and STAT3 S-nitrosylation were quantified by densitometry (A-iv). (B). The cells were treated with LPS (0.1 μg/ml) in the presence or absence of iNOS inhibitor, such as aminoguanidine (AG; 1 mM) or N-(3-(Aminomethyl)benzyl) acetamidine) (1400W; 50 μM), general NOS inhibitor L-Nω-nitroarginine methyl ester (L-NAME; 300 μM), or TLR4 inhibitor TAK-242 (1 μM) for 18 h and the iNOS expression, the production of NO (nitrite levels) and the effect on IL-6-induced (30 ng/ml 0.5 h) phosphorylation of STAT3 were analyzed in BV2 cells (B-i) or primary cultured rat microglia (B-ii). In addition, the levels of S-nitrosylated proteins (sno-Proteins) and S-nitrosylated and S-glutathionylated STAT3 levels (B-iii, iv) and cell proliferation (B-v) were analyzed in BV2 cells. (C) Following the transfection of CHO cells with empty pCMV vector (pCMV-mock) or pCMV vector expressing human iNOS (pCMV-iNOS), the production of NO (nitrite levels) (C-i) and the effect of IL-6 (30 ng/ml 0.5 h) on the phosphorylation of STAT3 (C-ii) and cell proliferation (C-iii) were analyzed. The vertical lines in A-iv, B-i, -ii, -iv, and -v, C-i and -iii indicate the standard error mean; *p<0.05; **p<0.01; ***p<0.001 compared with control group, ++p<0.01; +++p<0.001; ##p<0.01; not significant (N.S.) >0.05 compared with indicated group. IL-6, interleukin-6; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; NO, nitric oxide; STAT3, signal transducer and activator of transcription 3.
To further assess the role of iNOS expression and endogenous NO in the inhibition of STAT3 phosphorylation, BV2 cells (Fig. 1B-i) or primary cultured microglia (Fig. 1B-ii) were treated with iNOS-specific inhibitors, such as aminoguanidine (AG; 1 mM) or 1400W [N-(3-(Aminomethyl)benzyl)acetamidine; 50 μM], or general NOS inhibitor, L-Nω-nitroarginine methyl ester (L-NAME; 300 μM). Treatment of BV2 cells and primary microglia with AG, 1400W, and L-NAME completely inhibited LPS-induced NO production from these cells. As LPS increased NO production (Fig. 1B-i, ii), the cellular levels of S-nitrosylated proteins including STAT3 increased in BV2 cells (Fig. 1B-iii) and primary microglia (Fig. 4B-i), which were inhibited by L-NAME treatment (Fig. 1B-iii). In addition, treatment of BV2 cells or primary microglia with NOS inhibitors reversed the LPS-induced inhibition of IL-6 induced STAT3 phosphorylation (Fig. 1B-i, ii), therefore indicating a role for iNOS-generated endogenous NO in S-nitrosylation of STAT3 and inhibition of STAT3 phosphorylation. In this experiment, we also observed that treatment TAK-242 (1 μM), a TLR4 inhibitor, also abolished the inhibitory effect of LPS on IL-6-induced STAT3 phosphorylation (Fig. 1B-i), thereby indicating the participation of LPS/TLR4 signaling in iNOS expression and NO production and the regulation of STAT3 activity.
FIG. 4.

GSNO increases S-nitrosylation of STAT3. (A). The effects of GSNO (2 h treatment) on the S-nitrosylation of STAT3 or its upstream kinase JAK2 and receptor gp130 in BV2 cells were examined by biotin switch method. For analysis of total S-nitrosylated proteins, Western analysis for biotin-labeled proteins was performed (A-i). For analysis of S-nitrosylation of STAT3, JAK2, and receptor gp130, the biotinylated proteins were pulled down with avidin–agarose conjugate and the levels of STAT3, JAK2, and gp130 were determined by Western analysis (A-ii). (B) The effect of GSNO (0.5 mM for 2 h) and LPS (0.1 μg/ml for 12 h) treatment on S-nitrosylation of cellular proteins (B-i) and STAT3 (B-ii) was also examined in primary cultured rat microglia. (C) The effects of GSNO (500 μM for 2 h) on IL-6-induced (30 ng/mL for 0.5 h) phosphorylation of STAT3 (Tyr705) and JAK2 (Tyr1007/1008) were examined by the Western analysis (C-i). The phosphorylation of gp130 was examined by immunoprecipitation of phospho-Tyr containing proteins and following Western analysis of gp130 (C-ii). The levels of β-actin were used as internal loading control. (D) The effect of GSNO (2 h) on S-glutathionylation of STAT3 and other cellular proteins in BV2 cells were analyzed by immunoprecipitation and Western analysis. For positive control, the control cell lysates were incubated with oxidized and reduced glutathione mixture (GSSG/GSH; 5 mM each; pH 7.0) for 1 h at room temperature in the presence or absence of 20 mM dithiothreitol (DTT). (E) STAT3 S-nitrosylation (E-i) and S-glutathionylation (E-ii) in BV2 cells treated with GSNO (0.5 mM for 2 h) were further analyzed by sandwich ELISA using antibodies specific to S-nitrosocysteine or GSNO and STAT3. The vertical lines in E indicate the standard error mean; **p>0.01; not significant (N.S.)>0.05 compared with vehicle (VHC) treated group. ELISA, enzyme-linked immunosorbent assay; JAK2, Janus-activated kinase 2.
Previously, S-glutathionylation of STAT3 was also reported to regulate its activity under oxidative stress conditions (51). Since LPS is known to induce oxidative stress in microglia (14), we analyzed whether LPS treatment increases STAT3 S-nitrosylation and S-glutathionylation. Sandwich enzyme-linked immunosorbent assay (ELISA) for STAT3 S-nitrosylation and S-glutathionylation (Fig. 1B-iv) shows that LPS treatment increased S-nitrosylation as well as S-glutathionylation of STAT3. The maximum increases in S-nitrosylation of STAT3 were observed at 12 h after LPS treatment and which was well correlated with the data shown in biotin switch assay (Fig. 1A-iii, iv). On the other hand, the maximum increases in S-glutathionylation were observed at 24 h following LPS treatment. In addition, the LPS-induced increases in both S-nitrosylation and S-glutathionylation of STAT3 were significantly reduced by co-treatment of LPS with iNOS inhibitor AG, thereby indicating a role for LPS-induced iNOS expression in the regulation of STAT3 S-nitrosylation as well as S-glutathionylation.
Figure 1B-v shows that the increase in microglial proliferation by IL-6 treatment was decreased by LPS pretreatment, and these decreases were reversed by L-NAME pretreatment, indicating the role of endogenous NO signaling in the regulation of STAT3 activation and microglial proliferation. In addition, in support of the role of endogenous NO in the regulation of STAT3 activation, we also observed that overexpression of recombinant iNOS resulted in an increased production of NO (Fig. 1C-i) that in turn inhibited IL-6-induced phosphorylation of STAT3 (Fig. 1C-ii) and cell proliferation (Fig. 1C-iii). These data with increased LPS-induced cellular production of NO as well as increased production of NO from cells with overexpressed iNOS document inhibitory role of cellular endogenous NO in the regulation of STAT3 activity and thus cell proliferation.
GSNO, an endogenous NO carrier, inhibits phosphorylation of STAT3 (Tyr705) in microglia
GSNO is one of most characterized and abundant endogenous RSNOs that mediate protein S-nitrosylation (8). Therefore, we next investigated the role of exogenous GSNO in the regulation of STAT3 activation. Treatment of BV2 microglia with GSNO decreased the IL-6-induced STAT3 phosphorylation in a dose-dependent manner (Fig. 2A-i) at all the time points tested (0.5, 1, 3, and 6 h) (Fig. 2B). Under these experimental conditions, GSNO had no significant effects on total STAT3 protein levels (Fig. 2A-i, B) and cell viability up to 24 h (Fig. 2A-ii and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars). GSNO itself is not cell permeable and its cellular transport is mediated by its cleavage by the action of γ-glutamyl transpeptidase (59). In this study, we also tested the effect of S-nitroso-N-acetyl cysteine (NACSNO), a low mass cell-permeable SNO donor (55) (Fig. 2C), and S-nitrosocysteine (CysNO) and S-nitroso-N-acetylpenicillamine (SNAP) (Supplementary Fig. S2) as controls for S-nitroso donor. Similar to the activity of GSNO, NACSNO, CysNO, and SNAP also inhibited the phosphorylation of STAT3, whereas GSNO metabolic derivatives, such as decomposed GSNO (agGSNO; aged GSNO under light for 7 days), cell permeable GSH (GSH monoethyl ester; meGSH), sodium nitrite (NaNO2), and sodium nitrate (NaNO3), have no significant effects on IL-6-induced STAT3 phosphorylation in BV2 microglia (Fig. 2C-i) and primary cultured rat microglia (Fig. 2C-ii). These data document that GSNO, an endogenous NO carrier, inhibits STAT3 phosphorylation by S-nitrosothiol-based mechanisms (GSNO/NACSNO), but not by GSH-dependent mechanisms.
FIG. 2.
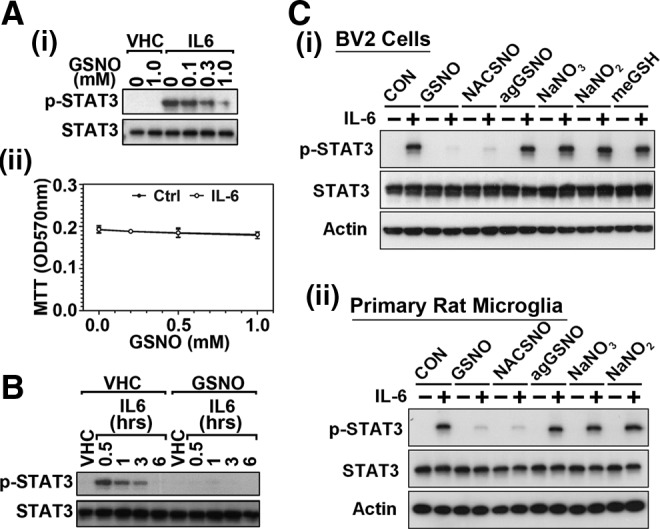
GSNO inhibits phosphorylation of STAT3 (Tyr705). (A) Cultured BV2 murine microglial cells were pretreated with the increasing concentrations of GSNO for 2 h in the dark and stimulated with IL-6 (30 ng/mL) for 30 min. Phosphorylated (Tyr705) and pan STAT3 protein levels in nuclear extract were determined by Western analysis as described under experimental procedures (A-i). Under the same experimental conditions, cell viability was also examined by MTT assay (A-ii). (B) Effect of GSNO pretreatment (500 μM for 2 h) on the time course changes in STAT3 phosphorylation was also examined following the IL-6 (30 ng/ml) treatment. (C) Effect of GSNO-related compounds, such as S-nitroso-N-acetyl cysteine (NACSNO; cell permeable S-nitroso donor; 500 μM) or GSNO-related metabolites, such as aged (decomposed) GSNO (agGSNO; 500 μM), cell permeable GSH (GSH monoethyl ester; meGSH; 500 μM), sodium nitrite (NaNO2; 500 μM), and sodium nitrate (NaNO3; 500 μM), on IL-6 (30 ng/ml 0.5 h)-induced STAT3 phosphorylation was examined in BV2 cells (C-i) or in primary cultured rat microglia (C-ii). GSH, glutathione; GSNO, S-nitrosoglutathione; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
GSNO inhibits IL-6-induced STAT3 transactivation and microglial proliferation
Because endogenous NO and exogenous GSNO inhibited STAT3 Tyr705 phosphorylation (Figs. 1 and 2), we next examined whether GSNO treatment inhibits the target DNA binding activity of STAT3 by gel-shift assay. Figure 3A-i shows that the treatment of BV2 microglia with GSNO (500 μM for 2 h) efficiently reduced the IL-6-induced (30 ng/ml for 30 min) STAT3 DNA binding activity. To further establish the role of GSNO in the regulation of STAT3 transactivity, BV2 microglia transfected with STAT3 responsive luciferase reporter construct (pSTAT3/R-Luc) were treated with IL-6 in the presence or absence of GSNO. As shown in Figure 3A-ii, GSNO treatment decreased IL-6-induced STAT3-mediated transcription activity. Accordingly, GSNO treatment inhibited the IL-6-induced expressions of cell cycle regulator Cyclin D1 and cell survival regulator Bcl-2 (Fig. 3B-i). Although Cyclin D1 and Bcl-2 are well-known downstream targets of STAT3 (4, 42), we further evaluate the role of STAT3 in GSNO-mediated regulation of expression of these proteins following transfection with constitutive active STAT3 construct. We also observed that GSNO-mediated inhibition of Cyclin D1 and Bcl-2 expressions were completely restored by transfection of cells with constitutive active STAT3 mutant (Supplementary Fig. S3), documenting the involvement of STAT3 in GSNO-induced inhibition of Cyclin D1 and Bcl-2 expression. Furthermore, GSNO treatment also decreased IL-6-induced microglial proliferation as demonstrated by 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (Fig. 3B-ii). Therefore, these data describe GSNO-mediated mechanism as a potential regulator of STAT3 signaling pathways that play critical role in microglia activation and proliferation.
FIG. 3.
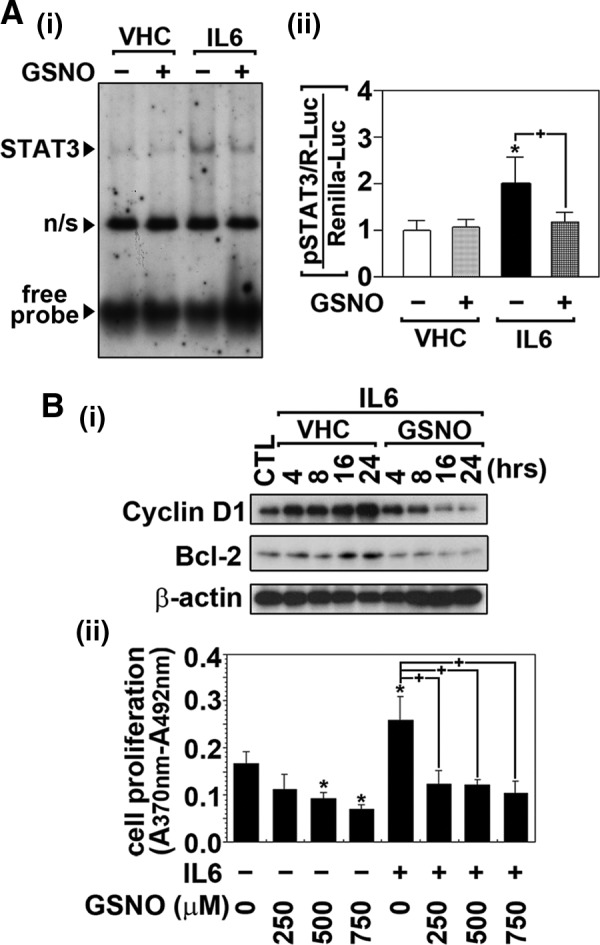
GSNO inhibits target DNA interaction of STAT3 and its transactivation by IL-6. (A) The effects of GSNO on IL-6-induced STAT3 target DNA binding activity and its transactivity were examined by gel-shift and reporter gene assays. For the gel-shift assay, BV2 microglia were pretreated with GSNO (500 μM) for 2 h and then treated with IL-6 (30 ng/mL) for 30 min. Nonspecific reaction is indicated by n/s (A-i). For the reporter gene assay, BV2 microglia transfected with STAT3-responsive luciferase construct (pSTAT3/R-Luc) and renilla luciferase contruct (phRL-CMV) as transfection control were pretreated with GSNO (500 μM) for 2 h and then treated with IL-6 (30 ng/mL) for 24 h. STAT3 transactivities were analyzed by luciferase activity assay as described under experimental procedure (A-ii). (B) The effects of GSNO on the expression of STAT3 downstream gene expression and cell proliferation were examined in BV2 microglia. The cells were treated with GSNO (500 μM) for 2 h and then treated with IL-6 (30 ng/mL) for indicated time points. The cellular levels of cyclin D1 and Bcl-2 were then analyzed by Western analysis (B-i). The effect of pretreatment of BV2 cells with increasing concentrations of GSNO for 2 h on IL-6-induced (30 ng/mL for 14 h) cell proliferation was analyzed by BrdU incorporation assay (B-ii). The vertical lines in A-ii and B-iii indicate the standard error of mean; *p<0.05 compared with vehicle-treated (VHC; dimethylsulfoxide or phosphate-buffered saline) control groups; +p<0.01 compared with IL-6-treated groups. BrdU, 5-bromo-2′-deoxyuridine.
GSNO increases S-nitrosylation of STAT3
To identify the molecular target of NO/GSNO in inhibition of STAT3 Tyr705 phosphorylation, we examined whether GSNO treatment would increase S-nitrosylation of STAT3 or its upstream regulators, such as JAK2 and gp130. As expected, Figure 4A-i and B-i shows that GSNO treatment of BV2 cells and primary cultured microglia increased S-nitrosylation of a variety of cellular proteins as observed by Western analysis for S-biotinylated proteins. The S-biotinylated proteins were further enriched by avidin–biotin pull-down assay, and the levels of S-nitrosylated (SNO) STAT3, JAK2, and gp130 were analyzed. Figure 4A-ii and B-ii show that GSNO treatment increased S-nitrosylation of STAT3 in BV2 cells and microglia, whereas under the similar conditions, no S-nitrosylation of JAK2 and gp130 was detected. In a parallel set of experiments, GSNO treatment inhibited IL-6-induced phosphorylation of STAT3 (Fig. 4C-i) but had no effect on the phosphorylation of JAK2 (Tyr1007/1008) (Fig. 4C-i) or gp130 (Fig. 4C-ii), indicating that GSNO inhibits STAT3 phosphorylation by direct S-nitrosylation of STAT3 rather than by altering the S-nitrosylation and activity of upstream regulators JAK2 and gp130.
To examine the effects of GSNO on STAT3 S-glutathionylation, BV2 cells were treated with GSNO (0.5 and 1 mM) for 2 h and the S-glutathionylation of STAT3 was analyzed by immunoprecipitation assay (Fig. 4D) as well as sandwich ELISA (Fig. 4E). For the positive control of STAT3 glutathionylation in immunoprecipitation assay, the control cell lysates were incubated with 5 mM of GSSG/GSH mix for 1 h at room temperature in the presence or absence of dithiothreitol (20 mM). Figure 4D shows that GSNO up to 1 mM did not increase any detectable STAT3 S-glutathionylation, whereas GSSG/GSH robustly increased S-glutathionylation of STAT3 and other cellular components. In addition, the estimation of STAT3 S-nitrosylation and S-glutathionylation by ELISA further indicated that GSNO increases STAT3 S-nitrosylation without affecting its S-glutathionylation (Fig. 4E-i, ii).
GSNO inhibits STAT3 phosphorylation by direct S-nitrosylation of STAT3
To further support the conclusions that NO/GSNO inhibits STAT3 phosphorylation by direct S-nitrosylation of STAT3, we performed in vitro kinase assay using purified recombinant JAK2 (recJAK2) and STAT3 (recSTAT3). Figures 5A and B show that GSNO or NACSNO treatment of kinase reaction mixture containing purified recJAK2 and recSTAT3 decreased the recSTAT3 phosphorylation in a dose-dependent manner. Consistent with data shown in Figure 2C, GSNO-related compounds, such as agGSNO, GSH, sodium nitrate, and sodium nitrite had no effect on recSTAT3 phosphorylation except GSSG/GSH (1 mM each) mixture (Fig. 5B) that mimics the cellular oxidative stress, documenting the involvement of S-nitrosylation as well as previously described redox-dependent mechanism (e.g., S-glutathionylation) in the regulation of STAT3 phosphorylation (51). Next, we treated the purified recJAK2 or recSTAT3 separately with GSNO and residual GSNO was removed by ultrafiltration as shown in experimental design in Figure 5C-i. Incubation of recJAK2 and recSTAT3 (lanes 1 and 2) without GSNO treatment as control resulted in the phosphorylation of STAT3. Incubation of GSNO-treated recJAK2 and untreated recSTAT3 (lanes 5 and 6) followed by mixing them in kinase assay also resulted in the phosphorylation of recSTAT3 indicating that GSNO treatment of recombinant JAK2 had no effect on its activity for STAT3 phosphorylation (Fig. 5C-ii). On the other hand, GSNO treatment of recSTAT3 before incubation with recJAK2 treated without (lanes 3 and 4) or with GSNO (lanes 7 and 8) was unable to phosphorylate recSTAT3 documenting that direct effect of GSNO treatment on STAT3 inhibits the ability of JAK2 to phosphorylate STAT3 (Fig. 5C-ii).
FIG. 5.

GSNO inhibits JAK2-mediated STAT3 phosphorylation in cell-free in vitro kinase assay system. (A) To examine whether GSNO is able to inhibit JAK2-mediated STAT3 phosphorylation without any involvement of other regulatory factors, in vitro kinase assay was performed using purified recombinant STAT3 (recSTAT3) and JAK2 (recJAK2). The purified recSTAT3 and recJAK2 mixture was incubated with increasing concentration of GSNO for 30 min, and kinase assay was initiated by the addition of reaction buffer without (w/o) or with (w/) ATP. The resulted phosphorylated recSTAT3 (Tyr705) levels were analyzed by Western blot. (B) To examine the effect of GSNO-related compounds on the phosphorylation of recSTAT3 in cell-free system, GSNO (100 μM), NACSNO (100 μM), aged GSNO (100 μM agGSNO; decomposed GSNO), NaNO3 (100 μM), NaNO2, (100 μM). glutathione (GSH/100 μM), or GSSG/GSH (1 mM) were added to reaction mixture before initiation of kinase reaction for 30 min. (C) To specify the target molecule of GSNO in inhibition of recSTAT3 phosphorylation, recSTAT3 or recJAK2 was preincubated with GSNO (100 μM for 30 min) individually and the residual GSNO was removed with Centricon (YM-10) before initiate the kinase reaction to minimize the unwanted effects of residual GSNO on untreated component as shown in the flow diagram (C-i). Following the in vitro kinase reaction, phosphorylated recSTAT3 was analyzed by Western blot (C-ii). For lane 1 and 2 (a), untreated recJAK2 and recSTAT3 were used. For lane 3 and 4 (b), untreated recJAK2 and GSNO-treated recSTAT3 were used. For lane 5 and 6 (c), GSNO-treated recJAK2 and untreated recSTAT3 were used. For lane 7 and 8 (d), GSNO-treated recJAK2 and recSTAT3 were used. The vertical lines in A, B, and C-ii indicate the standard error of mean; *p<0.05; **p<0.01; ***p<0.001; not significant (N.S.)>0.05 compared with vehicle-treated (VHC; dimethylsulfoxide) control groups.
GSNO S-nitrosylates Cys259 in STAT3 and inhibits JAK2-mediated phosphorylation
Next, we performed studies to identify the Cys residue(s) that is S-nitrosylated by GSNO treatment. STAT3 contains 14 Cys residues (Fig. 6A-i). To identify the target Cys residue(s) for S-nitrosylation, two mutants with deletions of different sizes, f1 (aa 1-580) and f2 (aa 1-314), were generated from wild-type (wt) mouse STAT3 cDNA (aa 1-769) (Fig. 6A-i) and transfected into Chinese hamster ovary (CHO) cells. The cells expressing wt and mutant STAT3 (f1 or f2) were then treated with GSNO (500 μM for 2 h) and the levels of S-nitrosylated STAT3 proteins were analyzed. Figure 6A-ii shows that GSNO increases S-nitrosylation of wt STAT3 and its deletion mutants (f1 and f2) indicating that two size variant of STAT3 mutants contains target Cys residue for S-nitrosylation similar to wt STAT3 and that at least one of three Cys residues in shorter mutant f2 is the target Cys residue for GSNO-induced STAT3 S-nitrosylation. Western analysis of myc-tagged STAT3 in Figure 6A-iii shows that the cells were expressing equal amounts of each recombinant STAT3 protein between vehicle and GSNO-treated cells.
FIG. 6.

GSNO selectively S-nitrosylates STAT3 at Cys259. (A) Wild-type (wt) STAT3 contains 14 Cys residues, which are distributed throughout the 6 domains, namely NH2-terminal (NT) domain, coiled-coil (CC) domain, DNA binding domain (DBD), in linker domain (LD), Src homology 2 (SH2) domain, and COOH terminal (CT) domain. To identify the target Cys residue(s) for S-nitrosylation, wt STAT3 conjugated with Myc and 6xHis tags was sequentially deleted; the deletion mutants were designated as f1 and f2 (A-i). Chinese hamster ovary (CHO) cells were transfected and overexpressed with wt STAT3 or its deletion mutant (f1 or f2) and treated with GSNO (300 μM) for 2 h. The S-nitrosylation of wt or mutant STAT3 was analyzed by Western analysis of Myc-tagged STAT3 followed by the biotin switch procedure (A-ii). The protein levels of myc-tagged STAT3 in whole cell lysates were used to check for equal transfection and equal expression of recombinant STAT3s between GSNO-treated group and untreated group (A-iii). (B) Because GSNO was able to increase S-nitrosylation of f2 fragment, Cys residues localized in this fragment were individually mutated to Ala (C108A, C251A, and C259) and transfected into CHO cells as described under the Materials and Methods section. Empty vector (MOCK) was used for control of transfection. Following treatment of cells with GSNO (300 μM) for 2 h, total STAT3 protein levels as well as S-nitrosylated STAT3 levels were analyzed by biotin switch procedure and Western analysis using antibody specific to STAT3 (B-i) or myc-tag (B-ii). In each panel, upper bands correspond to recombinant STAT3 (rec), which is conjugated with myc and 6xHis tags; lower bands correspond to their endogenous counterparts (endo). (C). The effect of GSNO or GSSG/GSH mix (0–500 μM) treatment for 0.5 h on S-nitrosylation (C-i) and S-glutathionylation (C-ii) of recombinant wt STAT3 or recombinant C259A mutant STAT3 was analyzed by sandwich ELISA using respective antibodies specific to S-nitrosocysteine or glutathione. The purified recombinant wt STAT3 and its C259A mutant proteins in cell-free system were used for in vitro S-nitrosylation and S-glutathionylation. For negative control for S-nitrosylation and S-glutathionylation, 10 mM of dithiothreitol or HgCl2 with GSNO was used. rec, recombinant.
Above studies conclude that at least one of these Cys residues (C108, C251, or C259) present in shorter mutant f2 is the target Cys residue for GSNO-induced STAT3 S-nitrosylation. To identify the target Cys residue(s) for S-nitrosylation, each Cys residue present in f2 fragment was mutated to Ala (C108A, C251A, or C259A) and the resulted mutant constructs were individually overexpressed in CHO cells. Following GSNO treatment, Western analysis for total or S-nitrosylated STAT3 was performed using STAT3-specific antibody for the detection of endogenous (endo) and recombinant (rec) STAT3 or with myc-tag-specific antibody for the detection of only recSTAT3. Figure 6B-i shows that the GSNO treatment increased S-nitrosylation not only of the endogenous and recombinant wt STAT3 but also of the recombinant STAT3 bearing C108A and C251A mutations. However, GSNO treatment did not S-nitrosylate recombinant STAT3 bearing C259A mutant (Fig. 6B-i). Second, purified avidin–biotin protein complex (S-nitrosylated proteins) showed no signal for myc-tagged protein (recombinant STAT3) in the cells transfected with C259A mutant (Fig. 6B-ii). To further confirm the role of C259 in GSNO-mediated S-nitrosylation, we also performed point mutation of all other Cys residues but found no involvement of other Cys residue in GSNO-mediated STAT3 S-nitrosylation (Supplementary Fig. S4). In addition, we also perform ELISA for detection of STAT3 S-nitrosylation (Fig. 6C-i) and S-glutathionylation (Fig. 6C-ii) using purified recombinant wt STAT3 and its C259A mutant. GSNO treatment increased the S-nitrosylation of wt STAT3 in a dose-dependent manner but not that of C259A mutant. Moreover, GSNO treatment did not increase S-glutathionylation of either wt or C259A mutant STAT3 proteins. On the other hand, GSSG/GSH mix increased the S-glutathionylation of both wt and mutant STAT3 proteins. The decreased S-nitrosylation of wt STAT3 by treatment with HgCl2 or dithiothreitol (DTT) (Fig. 6C-i) as well as decreased S-glutathionylation by DTT treatment (Fig. 6C-ii) indicates the specificity of the assay for S-nitrosylation or S-glutathionylation. Overall, these observations support the conclusions that GSNO S-nitrosylates STAT3 specifically on Cys259 and that GSNO does not induce STAT3 S-glutathionylation. Moreover, the observed STAT3 S-glutathionylation under the oxidative stress conditions involves Cys residue(s) other than Cys259 residue.
To further assess the role of Cys259 in S-nitrosylation-mediated inhibition of STAT3 phosphorylation, we next examined the effect of GSNO treatment on the phosphorylation of wt and mutant STAT3s. Figure 7A-i shows that GSNO treatment inhibited IL-6-induced phosphorylation of endogenous STAT3, wt recSTAT3, and recSTAT3 bearing C108A or C251A mutation. However, GSNO did not affect IL-6 induced phosphorylation of recSTAT3 bearing C259A mutation (Fig. 7A-i). Figure 7A-ii shows the lack of effects of GSNO on phosphorylation of recSTAT3 with C259A mutation. These observations indicate that while S-nitrosylation at Cys259 abolishes the phosphorylation of wt STAT3, the lack of S-nitrosylation in C259A mutant abolished the effect of GSNO-mediated inhibition of recSTAT3 phosphorylation. Furthermore, we also investigated the effect of C259A mutation on STAT3 transactivity in the cells transfected with STAT3-responsive luciferase reporter construct (pSTAT3/R-Luc). Again, while GSNO treatment inhibits the transactivation of wt STAT3 but GSNO treatment failed to inhibit IL-6-induced STAT3 transactivity in C259A mutant transfected cells as demonstrated by luciferase-reporter gene analysis (Fig. 7B). These data provide evidence that GSNO inhibits IL-6 induced STAT3 phosphorylation via S-nitrosylation of Cys259 and, in turn, inhibits its transactivation for various genes required for inflammation and cell proliferation (Fig. 8).
FIG. 7.
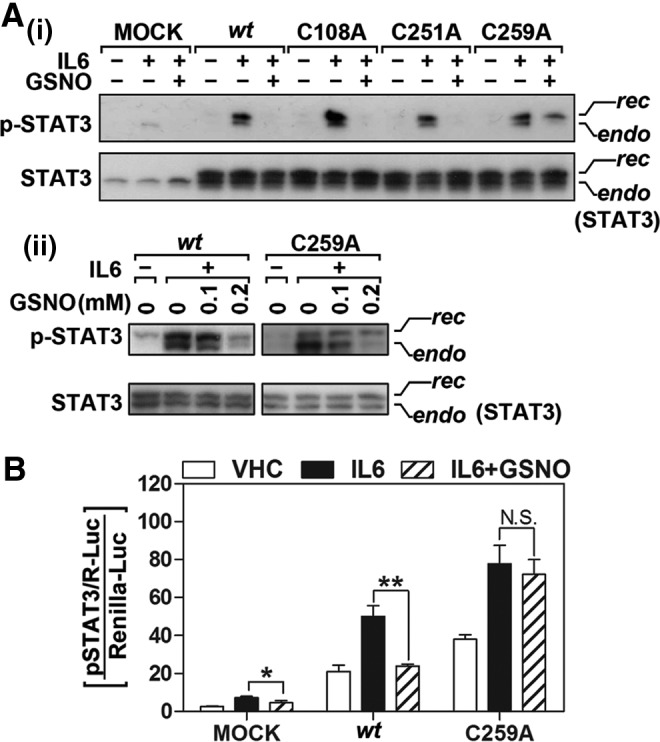
Point mutation of Cys259 to Ala abolishes the inhibitory action of GSNO on IL-6-induced STAT3 Tyr705 phosphorylation and its transactivity. (A) CHO cells were transfected and overexpressed with empty vector (MOCK), wt STAT3 or its point mutant (C108A, C251A, or C259). The cells were then pretreated with GSNO (300 μM for 2 h) followed by IL-6 (30 ng/ml) treatment for 30 min. The total and phospho-Tyr705 STAT3 levels were analyzed by Western analysis. The upper bands corresponds to recombinant STAT3 (rec), which is conjugated with myc and 6xHis tags; lower bands correspond to their endogenous counterparts (endo) (A-i). STAT3 phosphorylation in the cells expressing wtSTAT3 and mutant STAT3 (C259A) was also analyzed following the treatment of cells with lower concentration of GSNO (A-ii). (B) Effect of point mutation of Cys259 to Ala on the inhibitory role of GSNO in IL-6-induced STAT3 transactivation was examined by STAT3 responsive element luciferase assay. CHO cells transfected with STAT3-responsive luciferase construct (pSTAT3/R-Luc) and wt or C259A mutant STAT3 constructs were pretreated with GSNO (500 μM) for 2 h and then treated with IL-6 (30 ng/mL) for 24 h. STAT3 transactivities were analyzed by luciferase activity assay as described under experimental procedure. The renilla luciferase contruct (phRL-CMV) was used as a transfection control. The vertical lines in B indicate the standard error of mean; *p<0.05; **p<0.01; N.S.>0.05 compared with IL-6-treated groups.
FIG. 8.
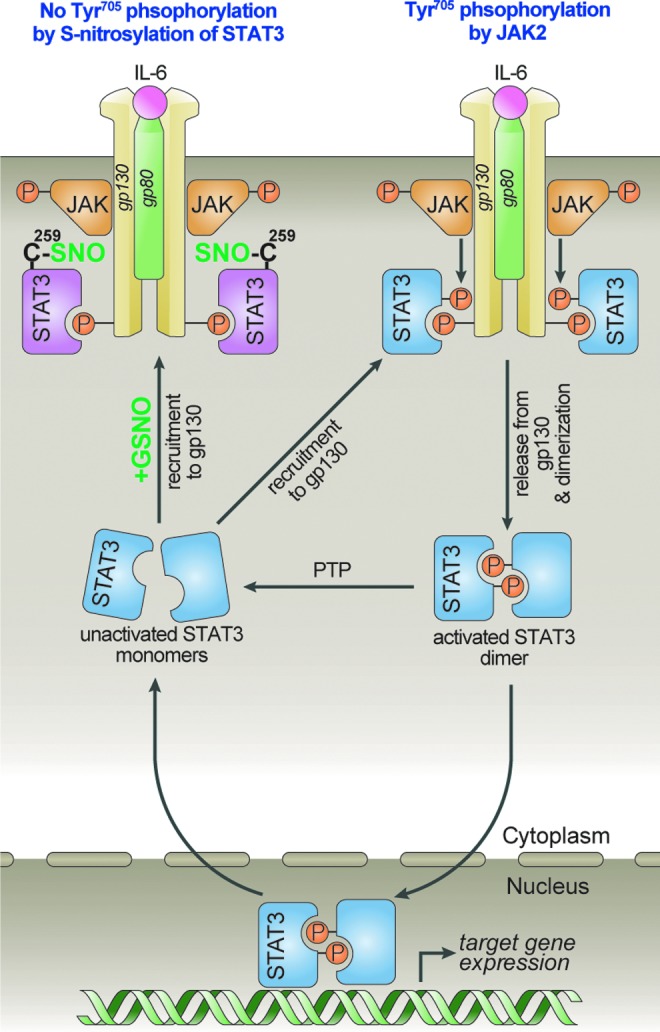
Regulation of STAT3 activation (Tyr705 phosphorylation) by S-nitrosylation-dependent mechanism. IL-6 induces STAT3 Tyr705 phosphorylation by following sequential steps: (i) homodimerization of gp130 and auto-phosphorylation of JAK2 (Tyr1007/1008), (ii) JAK2-mediated phosphorylation of Tyr residues in cytoplasmic part of gp130, (iii) recruitment of STAT3 to phospho-Tyr of gp130, (iv) STAT3 Tyr705 phosphorylation by JAK2, and (v) release of phospho-Tyr705-STAT3 from gp130 followed by STAT3 homodimerization through reciprocal interaction of the phospho-Tyr705 of one monomer and the SH2 domain of the corresponding monomer. Small molecular mass S-nitroso compounds including GSNO, which are endogenously formed by NO and thiol compounds, transfer their S-nitrosyl group to Cys259 residue of STAT3. The S-nitrosylation of Cys259 residue may inhibit JAK2-mediated phosphorylation of STAT3, which is recruited by gp130 receptor. Consequently, S-nitrosylation of STAT3 inhibits formation of STAT3 dimer and suppresses STAT3-dependent gene expression for cell cycle and cell survival.
Discussion
In this study, we report that endogenous NO produced by iNOS inhibited microglial proliferation via inhibiting the phosphorylation of STAT3 at Tyr705. We also report, for the first time, that phosphorylation is inhibited by S-nitrosylation of Cys259 in STAT3. These conclusions are supported by the following data: (i) endogenous NO produced by iNOS or exogenous GSNO treatment inhibits IL-6-induced phosphorylation of STAT3 at Tyr705 (Figs. 1 and 2) and repressed STAT3-dependent gene expression for cell proliferation (Cyclin D1) and survival (Bcl-2) (Fig. 3); (ii) GSNO treatment increases S-nitrosylation of STAT3 but has no effect on its upstream kinase (JAK2) or receptor (gp130) (Fig. 4); (iii) selective preincubation of purified recombinant STAT3 with GSNO inhibits STAT3 Tyr705 phosphorylation mediated by purified recombinant JAK2; however, selective preincubation of purified JAK2 has no effect on STAT3 Tyr705 phosphorylation (Fig. 5); (iv) GSNO selectively S-nitrosylates STAT3 at Cys259 (Fig. 6) and the mutation of Cys259 to Ala abolishes the inhibitory action of GSNO on IL-6-induced STAT3 Tyr705 phosphorylation and its transactivity (Fig. 7). Although these findings provide evidence that S-nitrosylation of STAT3 on Cys259 inhibits microglial proliferation, however, the possible structural changes of STAT3 produced by replacement of Cys with Ala are not ruled out.
Cys259 is localized within the coiled-coil (CC) domain, while Tyr705 is localized near the SH2 domain in C-terminal region (60) Studies have reported that the CC domain is involved in interdomain interactions by which the CC domain participates in interaction between SH2 domain of STAT3 and phospho-Tyr of gp130 and thus regulates JAK2-mediated STAT3 Tyr705 phosphorylation (57, 58). At present, it is not known whether S-nitrosylation of Cys259 inhibits STAT3 Tyr705 phosphorylation through inhibiting STAT3/gp130 interaction or by altering the function and structure of CC domain. The data from in vitro kinase assay using purified JAK2 and STAT3 demonstrate that GSNO inhibited JAK2-mediated STAT3 phosphorylation in the absence of gp130 receptors (Fig. 5), thereby suggesting that STAT3 S-nitrosylation on Cys259 may inhibit STAT3 Tyr705 phosphorylation by inhibiting its accessibility to JAK2. In support, we observed that GSNO treatment increased the STAT3 recruitment to gp130 receptor upon the stimulation with IL-6 (See Supplementary Fig. S5). IL-6 induces STAT3 Tyr705 phosphorylation is followed by: (i) homodimerization of gp130 and autophosphorylation of gp130, (ii) JAK2-mediated Tyr phosphorylation of gp130, (iii) recruitment of STAT3 to phospho-Tyr of gp130, (iv) STAT3 Tyr705 phosphorylation by JAK2, and (v) release of phospho-STAT3 from gp130 via reciprocal interaction of the phospho-Tyr705 of one STAT3 monomer and the SH2 domain of the corresponding monomer. In this process, GSNO-mediated S-nitrosylation of Cys259 in STAT3 appears to inhibit JAK2 accessibility to STAT3 in gp130 receptor complex and thus inhibits Tyr705 phosphorylation. Consequently, GSNO appears to inhibit STAT3 release from gp130 complex. Although Tyr705 phosphorylation has been suggested to be a crucial post-translational modification that regulates the dimerization and transactivity of STAT3, STAT3 is also modulated by multiple other types of post-translational modification. In addition to phosphorylation at Tyr705, STAT3 is also regulated by phosphorylation at Ser727 by MAPK (13). Unlike the effect on Tyr705 phosphorylation, we observed that GSNO treatment had no effect on STAT3 phosphorylation at Ser727 (data not shown). STAT3 activity is also regulated by methylation (Arg31 and Lys140) (52), acetylation (Lys49, Lys87, and Lys685) (41, 53), and S-glutathionylation (51). At present, it is not known whether S-nitrosylation of Cys259 regulates or overrides post-translational modifications other than Tyr705 phosphorylation. However, S-nitrosylation-mediated inhibition of STAT3 function (expression of Bcl-2 and Cyclin D1 in Fig. 3B) and thus cell proliferation indicates that S-nitrosylation of STAT3 of Cys259 negates all other STAT3 modifications associated with its transactivity.
Beside the role in inflammatory signaling pathways (5, 22, 38), STAT3 functions as a critical mediator of oncogenic signaling through transcriptional activation of genes encoding apoptosis inhibitors (e.g., Bcl-xL, Mcl-1, and survivin), cell cycle regulators (e.g., cyclin D1, Pim-1, and c-Myc), and inducers of angiogenesis (e.g., vascular endothelial growth factor) (4, 5, 42). Therefore, the activated STAT3 is now considered to play a master regulatory role in the progression and survival of human cancer, thereby being regarded as a promising relevant target for multiple cancer types (5). Efforts are currently under way to develop the therapeutics targeting of STAT3 for treating various types of cancer, but so far, no clinically relevant therapeutic agent has been identified (29, 33). Potential utility for S-nitrosylating agents in the regulation of cancer cell proliferation and survival has important implications for targeting STAT3 for treating cancers. Indeed, we have observed that GSNO and S-nitrosylated N-acetyl cysteine, a closely related S-nitrosylating compound, inhibits cell proliferation of various types of cancer cells via inhibiting STAT3 activation (See Supplementary Fig. S6). Growing body of evidence suggests that S-nitrosylation is one of the major signal transduction mechanisms regulating various cellular functions, similar to other post-translational modification. S-nitrosylation-mediated biological regulations under physiological conditions are mediated via low molecular mass RSNOs, such as GSNO (8) and relevant enzyme system and protein carrier, such as GSNO reductase, thioredoxin, thioredoxin reductase, and glyceraldehyde-3-phosphate dehydrogenase (1, 16, 40, 43).
S-glutathionylation is another type of redox-sensitive post-translational modification and involved in functional regulation of variety proteins (36) and a recent report described the regulation of STAT3 Tyr705 phosphorylation via S-glutathionylation under oxidative stress conditions (51). Studies also have shown that GSNO can induce S-nitrosylation or S-glutathionylation according to the nucleophilicity of the cysteine residues in some proteins (31). In this study, we observed that LPS treatment induced both S-nitrosylation and S-glutathionylation of STAT3 (Fig. 1B-iv). STAT3 S-nitrosylation is associated with iNOS-mediated production of NO and cellular GSNO, whereas S-glutathionylation is associated with cellular oxidative stress (i.e., GSSG/GSH ratio) (Figs. 4D and 6C). The significance of differential regulation of STAT3 by S-nitrosylation versus S-glutathionylation under two cellular oxidative stress conditions is not understood at present. Although biotin switch assay has been widely used in the detection of S-nitrosylation of protein thiols, the specificity of biotin switch has been questioned. The reduction of S-nitrosothiol by ascorbate seems very substrate dependent (47). Some S-nitrosyl moieties cannot be reduced by ascorbate efficiently, whereas under some circumstances, ascorbate has been shown to not only reduce SNO groups but also reduce disulfides (47). Therefore, we developed immunological detection of S-nitrosylation and S-glutathionylation of STAT3 with sandwich ELISA using specific respective antibodies (Figs. 1B-iv, 4D, and 6C). This antibody-based detection of S-nitrosylation and S-glutathionylation will be a useful tool in addition to biotin switch assay for study of protein S-nitrosylationhelpful for detection of other proteins in addition to biotin switch assay.
S-nitrosylation-mediated regulation of cellular mechanisms is an evolving scientific field as during the recent past more than 3000 proteins are reported to be nitrosylated (1, 16, 43, 48). Various protein kinases (10, 30, 56) and phosphatases (7, 20, 37) are now known to be regulated by S-nitrosylation mechanism (1, 16, 43, 48) and in case of kinase the activity may be regulated by direct inhibition or activation of its activity or by modulating the accessibility of the substrate to kinase. Our studies described here indicate that GSNO does not alter the JAK2 activity but rather accessibility of substrate (STAT3) to JAK2 (Supplementary Fig. S5). Similarly, S-nitrosylation of the MAPK-kinase ASK1 inhibited the binding and thus accessibility of its downstream target MKK3 and MKK6 (35). Moreover, the observed inactivation (dephosphorylation) of STAT3 by S-nitrosylation and its functional activity indicates that this S-nitrosylation base modification overrides or inhibits STAT3 activation.
In summary, studies described here document that S-nitrosylation of STAT3 by endogenous NO and exogenous RSNO (GSNO)-induced S-nitrosylation inhibits cell signaling pathways leading to microglial proliferation by the inhibition of STAT3 activity. Second, Cys259 in STAT3 is identified as the specific target for GSNO-mediated STAT3 S-nitrosylation as its mutation to Ala was able to abolish the inhibitory action of GSNO on IL-6-induced STAT3 Tyr705 phosphorylation. These findings are highly relevant to disease mechanisms in inflammation disease of cancer as well as neurodegenerative disorders.
Materials and Methods
Cell culture and treatment
Primary rat microglia were prepared as described in our previous report (18). BV2 a murine microglial cells and CHO cells were purchased from American Type Culture Collection (Rockville, MD) and maintained in 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) containing 10% heat-inactivated fetal bovine serum (Invitrogen), 100 units/ml of penicillin, and 100 μg/ml of streptomycin. GSNO (World Precision Instruments, Inc., Sarasota, FL) was dissolved in dimethylsulfoxide (DMSO) and kept in −80°C. Before the experiment, the concentration of GSNO was determined photometrically using a molar extinction coefficient of 900 M−1cm−1 at 336 nm as described previously (46).
Western blot analysis and antibodies
Western blot analysis were performed as described previously (50) by using antibodies against pan- and phospho-Tyr705 STAT3 (Cell Signaling, Beverly, MA), pan- and phospho-Tyr1007/1008 JAK2 (Cell Signaling), cyclin D1 (Abcam, Cambridge, MA), biotin (Abcam), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA).
ELISA for STAT3 S-nitrosylation and S-glutathionylation
96-well microtiter plates were coated with mouse or rabbit antibody specific to STAT3 (1 μg/100 μl/well; Santa Cruz #sc-482 or Cell Signaling #9139, respectively) for 1 h and blocked with 1% BSA in PBS. Purified wt or mutant STAT3 (0.1 μg/100 μl/well) reacted with GSNO or GSSG/GSH or cell lysates extracted from GSNO- or LPS-treated cells were added and incubated at dark place at room temperature for 1 h. The plates were then reacted with anti-S-nitrosocysteine rabbit IgG (1:500 dilution; Sigma-Aldrich) or anti-glutathione mouse IgG (0.1 μg/100 μl/well; Virogen, Watertown, MA) (1 μg/100 μl/well) for 0.5 h, and further reacted with horseradish peroxidase-conjugated secondary antibody for 0.5 h and followed by colorimetric development with TMB component (Pierce, Rockfort, IL). The specificities of STAT3 antibodies were confirmed by Western immunoblot (Supplementary Fig. S7); one major band at 88 kDa representing more than 93% of total band intensities and minor bands with lower molecular weight representing its degradation products.
Immunoprecipitation
For detection of gp130 Tyr phosphorylation and STAT3 S-glutathionylation, the cells were lysed in 50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 1 mM Na3VO4, clarified by normal serum, and protein-A/G (Santa Cruz Biotechnology) and further incubated with anti-phospho-Tyr antibody (Cell Signaling) or anti-glutathione antibody (Virogen) with protein-A/G. The resulting complexes were precipitated and proteins bound to the beads were separated by SDS-PAGE, followed by immunoblotting with anti-glycoprotein130 antibody (gp130; Santa Cruz Biotechnology.) or STAT3 (Cell Signaling).
Cell viability and proliferation assay
Cell viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays as described previously (50). BV2 microglia proliferation was measured by BrdU incorporation assay. Briefly, BV2 microglia treated with GSNO for 2 h and IL-6 for 14 h were further incubated with 0.5 mM BrdU for 2 h and washed with the growth medium. The DNA incorporated BrdU was quantified by colorimetric BrdU ELISA kit (Roche Applied Science, Mannheim, Germany).
STAT3 gel-shift assay and reporter gene assay
The EMSA reaction was performed as described previously (50) with of DNA probe sequences (5′-AGA TCC TTC TGG GAA TTC CTA GAT C-3′). For STAT3 reporter gene assay, BV2 cells were transfected with STAT3-responsive luciferase construct (1.5 μg/well; Panomics, Inc., Redwood City, CA), which encodes firefly luciferase reporter gene, and phRL-CMV (0.1 μg/well; Promega, Madision, WI) construct, which encodes renilla luciferase under the control of a CMV immediate early enhancer/promoter for an internal control for transfection efficiencies. Transfection was mediated by using lipofectamine-Plus (Invitrogen), according to the manufacturer's instructions. IL-6 (30 ng/ml) was treated a day after transfection. Next day, the activities of luciferases were assayed by using dual-luciferase reporter system (Promega) according to the manufacturer's instructions.
Biotin switch assay for detection of S-nitrosylated proteins
Protein S-nitrosylation was detected using the biotin switch method with slight modification as described previously (39). Cells were lysed in 250 mM HEPES, pH 7.7, 1 mM EDTA, 0.1 mM neocuproine, 1% Nonidet P-40, 150 mM NaCl, 1 mM PMSF, 20 μM methyl methanethiosulfonate (MMTS), 80 μM carmustine, protease inhibitor mixture (Sigma), and mixed with an equal volume of 25 mM HEPES, pH 7.7, 0.1 mM EDTA, 10 μM neocuproine, 5% SDS, 20 μM MMTS and incubated at 50°C for 20 min. After acetone precipitation, the precipitates were resuspended in 25 mM HEPES, pH 7.7, 0.1 mM EDTA, 10 μM neocuproine, 1% SDS and mixed with two volumes of 20 mM HEPES, pH 7.7, 1 mM EDTA, 100 mM NaCl, and 0.5% Triton X-100. The S-nitrosylated proteins were then modified with biotin in 25 mM HEPES, pH 7.7, 0.1 mM EDTA, 1% SDS, 10 μM neocuproine, 10 mM ascorbate sodium salt, and 0.2 mM N-[6-(biotinamido)hexyl]-30-(20-pyridyldithio) propionamide (biotin-HPDP; Pierce). After acetone precipitation, biotinylated proteins were pull down with neutravidin-agarose.
Cell-free in vitro STAT3 kinase assay
Purified recombinant JAK2 (recJAK2; Active Motif, Carlsbad, CA) and STAT3 (recSTAT3; Active Motif) were washed with reaction buffer (60 mM HEPES pH 7.5, 5 mM MgCI2, 5 mM MnCl2, 3 μM Na3VO4) by using Centricon YM-10 (molecular weight cutoff, 10,000) (Amicon, Danvers, MA) to remove DTT. The resulted reaction mixtures were incubated with GSNO or other related agents for 30 min in the dark at room temperature, and then, the kinase reaction was initiated by addition of 1 mM ATP. Following the incubation at 37°C for 10 min, the reaction was terminated by addition of 2×SDS sample loading buffer for Western analysis. To specify the target molecules of GSNO in inhibition of STAT3 phosphorylation, recSTAT3 or recJAK2 were preincubated separately with GSNO and washed with Centricon YM-10 before their mixing for minimizing the undesired effects of residual GSNO on other components.
Construction of STAT3 mammalian expression plasmids and site-directed mutagenesis
Vectors expressing myc-His tagged wt STAT3 and its deletion mutants, f1 (aa 1-580) and f2 (aa 1-314), were constructed from a full-length mouse STAT3 cDNA (pBS-mSTAT3, Themo Fisher Scientific Open Biosystem) (Fig. 6A-i). For identification of target Cys for S-nitroyslation of STAT3, each Cys residue was replaced with Ala by site-directed mutagenesis of Cys via overlap extension. Please see Supplementary Materials and Methods for detail.
Statistical analysis
All values shown in the figures are expressed as the means±SEM of n determinations, obtained from at least three independent experiments. The results were examined by one- and two-way ANOVA; then individual group means were compared with the Bonferroni test. A p value of<0.05 was considered significant.
Supplementary Material
Abbreviations Used
- AG
aminoguanidine
- BrdU
5-bromo-2′-deoxyuridine
- CC
coiled-coil
- CHO
Chinese hamster ovary
- CysNO
S-nitrosocysteine
- ELISA
enzyme-linked immunosorbent assay
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- IL-6
interleukin-6
- iNOS
inducible nitric oxide synthase
- JAK
Janus-activated kinase
- L-NAME
L-Nω-nitroarginine methyl ester
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MMTS
methyl methanethiosulfonate
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NACSNO
S-nitroso-N-acetyl cysteine
- NO
nitric oxide
- rec
recombinant
- RSNO
S-nitrosothiol compound
- sGC
soluble guanylyl cyclase
- SH2
Src homology 2
- SHP
SH2 domain-containing protein tyrosine phosphatase
- SNAP
S-nitroso-N-acetylpenicillamine
- SNP
sodium nitroprusside
- STAT
signal transducer and activator of transcription
- TC-PTP
T-cell protein tyrosine phosphatases
- wt
wild type
Acknowledgments
This work was supported by grants from NIH and VA (NS072511, BX001062, NS037766 and BX001072). We also acknowledge Ms. Joyce Bryan and Ms. Chara Williams for their help in procurement of animals and supplies.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Anand P. and Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med (Berl) 90: 233–244, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker TL, Booden MA, and Buss JE. S-Nitrosocysteine increases palmitate turnover on Ha-Ras in NIH 3T3 cells. J Biol Chem 275: 22037–22047, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Block ML. and Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76: 77–98, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, and Darnell JE., Jr.Stat3 as an oncogene. Cell 98: 295–303, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Campbell IL. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res Brain Res Rev 48: 166–177, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Ceron PI, Cremonez DC, Bendhack LM, and Tedesco AC. The relaxation induced by S-nitroso-glutathione and S-nitroso-N-acetylcysteine in rat aorta is not related to nitric oxide production. J Pharmacol Exp Ther 298: 686–694, 2001 [PubMed] [Google Scholar]
- 7.Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, Khoo KH, and Meng TC. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem 283: 35265–35272, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiueh CC. and Rauhala P. The redox pathway of S-nitrosoglutathione, glutathione and nitric oxide in cell to neuron communications. Free Radic Res 31: 641–650, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Chung J, Uchida E, Grammer TC, and Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 17: 6508–6516, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curcio MF, Batista WL, Linares E, Nascimento FD, Moraes MS, Borges RE, Sap J, Stern A, and Monteiro HP. Regulatory effects of nitric oxide on Src kinase, FAK, p130Cas, and receptor protein tyrosine phosphatase alpha (PTP-alpha): a role for the cellular redox environment. Antioxid Redox Signal 13: 109–125, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Davis KL, Martin E, Turko IV, and Murad F. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol 41: 203–236, 2001 [DOI] [PubMed] [Google Scholar]
- 12.de Belder AJ, MacAllister R, Radomski MW, Moncada S, and Vallance PJ. Effects of S-nitroso-glutathione in the human forearm circulation: evidence for selective inhibition of platelet activation. Cardiovasc Res 28: 691–694, 1994 [DOI] [PubMed] [Google Scholar]
- 13.Decker T. and Kovarik P. Serine phosphorylation of STATs. Oncogene 19: 2628–2637, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal 7: 1223–1233, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Foster MW, Hess DT, and Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med 15: 391–404, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster MW, McMahon TJ, and Stamler JS. S-nitrosylation in health and disease. Trends Mol Med 9: 160–168, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Fukushima A, Loh K, Galic S, Fam B, Shields B, Wiede F, Tremblay ML, Watt MJ, Andrikopoulos S, and Tiganis T. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes 59: 1906–1914, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giri S, Nath N, Smith B, Viollet B, Singh AK, and Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 24: 479–487, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gow AJ, Buerk DG, and Ischiropoulos H. A novel reaction mechanism for the formation of S-nitrosothiol in vivo. J Biol Chem 272: 2841–2845, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Guan W, Sha J, Chen X, Xing Y, Yan J, and Wang Z. S-Nitrosylation of mitogen activated protein kinase phosphatase-1 suppresses radiation-induced apoptosis. Cancer Lett 314: 137–146, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, and Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334 (Pt 2): 297–314, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, and Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28: 7231–7243, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hess DT, Patterson SI, Smith DS, and Skene JH. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature 366: 562–565, 1993 [DOI] [PubMed] [Google Scholar]
- 24.Hess DT. and Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem 287: 4411–4418, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kacimi R, Giffard RG, and Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 8: 7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawahara K, Gotoh T, Oyadomari S, Kuniyasu A, Kohsaka S, Mori M, and Nakayama H. Nitric oxide inhibits the proliferation of murine microglial MG5 cells by a mechanism involving p21 but independent of p53 and cyclic guanosine monophosphate. Neurosci Lett 310: 89–92, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Khan M, Sekhon B, Giri S, Jatana M, Gilg AG, Ayasolla K, Elango C, Singh AK, and Singh I. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J Cereb Blood Flow Metab 25: 177–192, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Kim OS, Park EJ, Joe EH, and Jou I. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J Biol Chem 277: 40594–40601, 2002 [DOI] [PubMed] [Google Scholar]
- 29.Lai SY. and Johnson FM. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist Updat 13: 67–78, 2010 [DOI] [PubMed] [Google Scholar]
- 30.Marshall HE. and Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry 40: 1688–1693, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Mohr S, Hallak H, de Boitte A, Lapetina EG, and Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem 274: 9427–9430, 1999 [DOI] [PubMed] [Google Scholar]
- 32.Nath N, Morinaga O, and Singh I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J Neuroimmune Pharmacol 5: 240–251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Page BD, Ball DP, and Gunning PT. Signal transducer and activator of transcription 3 inhibitors: a patent review. Expert Opin Ther Pat 21: 65–83, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Papapetropoulos A, Rudic RD, and Sessa WC. Molecular control of nitric oxide synthases in the cardiovascular system. Cardiovasc Res 43: 509–520, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Park HS, Yu JW, Cho JH, Kim MS, Huh SH, Ryoo K, and Choi EJ. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J Biol Chem 279: 7584–7590, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Pastore A. and Piemonte F. S-Glutathionylation signaling in cell biology: progress and prospects. Eur J Pharm Sci 46: 279–292, 2012 [DOI] [PubMed] [Google Scholar]
- 37.Pei DS, Sun YF, and Song YJ. S-nitrosylation of PTEN Invovled in ischemic brain injury in rat hippocampal CA1 region. Neurochem Res 34: 1507–1512, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Pfitzner E, Kliem S, Baus D, and Litterst CM. The role of STATs in inflammation and inflammatory diseases. Curr Pharm Des 10: 2839–2850, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Prasad R, Giri S, Nath N, Singh I, and Singh AK. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia 55: 65–77, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Que LG, Yang Z, Stamler JS, Lugogo NL, and Kraft M. S-nitrosoglutathione reductase: an important regulator in human asthma. Am J Respir Crit Care Med 180: 226–231, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ray S, Boldogh I, and Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 129: 1616–1632, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, and Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21: 7611–7618, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Sengupta R. and Holmgren A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid Redox Signal 18: 259–269, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Seth D. and Stamler JS. The SNO-proteome: causation and classifications. Curr Opin Chem Biol 15: 129–136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stamler JS, Lamas S, and Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell 106: 675–683, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, and Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89: 444–448, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H. and Xian M. Chemical methods to detect S-nitrosation. Curr Opin Chem Biol 15: 32–37, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Z. Protein S-nitrosylation and cancer. Cancer Lett 320: 123–129, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, and Lucius R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J 17: 500–502, 2003 [DOI] [PubMed] [Google Scholar]
- 50.Won JS, Im YB, Key L, Singh I, and Singh AK. The involvement of glucose metabolism in the regulation of inducible nitric oxide synthase gene expression in glial cells: possible role of glucose-6-phosphate dehydrogenase and CCAAT/enhancing binding protein. J Neurosci 23: 7470–7478, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie Y, Kole S, Precht P, Pazin MJ, and Bernier M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 150: 1122–1131, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, Chance MR, Chen X, Du Y, Wang Y, An L, Wang Q, Lu T, Zhang X, Wang Z, and Stark GR. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A 107: 21499–21504, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan ZL, Guan YJ, Chatterjee D, and Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307: 269–273, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Zaman K, Carraro S, Doherty J, Henderson EM, Lendermon E, Liu L, Verghese G, Zigler M, Ross M, Park E, Palmer LA, Doctor A, Stamler JS, and Gaston B. S-nitrosylating agents: a novel class of compounds that increase cystic fibrosis transmembrane conductance regulator expression and maturation in epithelial cells. Mol Pharmacol 70: 1435–1442, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Zaman K, Palmer LA, and CGaston B. S-nitrosothiol signaling and gene regulation in pulmonary pathophysiology. In: Nitric oxide, Cell Signaling, and Gene Expression, edited by Lamas S. and Cadenas E. Boca Ranton, FL: CRC Press; 2006, pp. 321–330 [Google Scholar]
- 56.Zhang P, Yu PC, Tsang AH, Chen Y, Fu AK, Fu WY, Chung KK, and Ip NY. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J Neurosci 30: 14366–14370, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang T, Kee WH, Seow KT, Fung W, and Cao X. The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol Cell Biol 20: 7132–7139, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang T, Seow KT, Ong CT, and Cao X. Interdomain interaction of Stat3 regulates its Src homology 2 domain-mediated receptor binding activity. J Biol Chem 277: 17556–17563, 2002 [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y. and Hogg N. The mechanism of transmembrane S-nitrosothiol transport. Proc Natl Acad Sci U S A 101: 7891–7896, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong Z, Wen Z, and Darnell JE, Jr., Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264: 95–98, 1994 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.