

Abstract
A fundamental question regarding the gating mechanism of voltage-activated K+ (Kv) channels is how five positively charged voltage-sensing residues in the fourth transmembrane (TM) segment1, 2 are energetically stabilized, as they operate in a low-dielectric cell membrane. The simplest solution would be to pair them with negative charges3. However, too few negatively charged channel residues are positioned for such a role4, 5. Recent studies suggest that some of the channel’s positively charged residues are exposed to cell membrane phospholipids and interact with their head groups5–9. A key question nonetheless remains: Is the phospho-head of membrane lipids necessary for proper function of the voltage sensor itself? Here, we find that a given type of Kv channel may interact with several species of phospholipid, and that enzymatic removal of their negatively charged phospho-head creates an insuperable energy barrier for the positively charged voltage sensor to move through the initial gating step(s), thus immobilizing it, and also raises the energy barrier for the downstream step(s).
Kv2.1 channels, expressed in Xenopus oocytes, interact with sphingomyelin8 present mainly in the outer leaf of plasma membranes. To investigate the importance of phospho-head groups of membrane lipids in Kv channel gating we employed bacterial sphingomyelinases C and D (SMases C and D)10, 11. Both enzymes specifically hydrolyze sphingomyelin but in different ways (Fig. 1a): SMase D removes only choline and leaves the lipid ceramide-1-phosphate behind in the membrane whereas SMase C removes phosphocholine, leaving ceramide behind12, 13. A comparison of the effects of these two enzymes on the channels will thus help reveal the functional significance of the phosphodiester group in voltage gating.
Figure 1.

Reaction schemes of lipid hydrolysis and the SMase D effect on Kv2.1 channels. a, b, Hydrolysis of sphingomyelin by SMases C or D (a) and of phosphatidylcholine by PC-PLC (b). R and R= represent acyl chains. c, Amplitude of Kv2.1 current, repeatedly elicited by stepping membrane voltage from -80 to -40 mV, where the arrow indicates addition of recombinant SMase D to the bath. d, G–V relations obtained before SMase D treatment (squares), and 30 min (circles) or 24 h (triangles) after a 10-min exposure of oocytes to SMase D. e, Oocytes were exposed to SMase D for 30 min and then washed with enzyme-free solution before being injected with Kv2.1 cRNA. Data were collected from each oocyte 18 hr after the injection (circles), and again after an additional 15-min SMase D treatment (triangles). The control G–V relation was obtained without SMase D treatment (squares). All data in d and e are presented as mean ± s.e.m. (n = 5), and the controls were pooled.
SMase D of Corynebacterium pseudotuberculosis11 shifts the conductance-voltage (G-V) relation of Kv2.1 by about -30 mV8 (Fig. 1d), allowing channels to be activated at a negative voltage where they otherwise remain largely deactivated (Fig. 1c). The effect is maximal within 2 minutes and persists for at least 24 hours (Fig. 1c, d; cells cannot regenerate sphingomyelin from ceramide-1-phosphate). It does not require direct exposure of channels to SMase D as it also occurred in Kv2.1-expressing oocytes that had been treated with SMase D and then thoroughly washed prior to injection with Kv2.1 cRNA (Fig. 1e). Additional exposure of such oocytes to SMase D, as expected, caused no further shift. Thus, SMase D acts through its lipase activity, not by direct binding to the channel. Our previous study8 supports an electrostatic mechanism whereby removal of the positively charged choline favors the activated state of the positively charged voltage sensors.
Unlike SMase D, SMase C removes the negatively charged phosphodiester group as well as choline. SMase C of Bacillus anthracis14 (BaSMase C) reduces Kv2.1 current ~90% (Fig. 2a–c) and the reduction is voltage independent for both partial and complete enzymatic treatment (Fig. 2d). SMase C of Staphylococcus aureus15 (SaSMase C) caused a comparable level of inhibition but with faster kinetics, and only the kinetics – not the extent – of inhibition are enzyme-concentration dependent (Fig. 2b). Thus, SMase C inhibits the channels via its enzymatic activity rather than by direct binding (more evidence below).
Figure 2.

Effects of SMase C on Kv2.1, Kir1.1 and a KcsA-Kir2.1 chimera. a, Kv2.1 currents elicited at 5 second intervals with the voltage protocol shown, gradually declining following addition of recombinant BaSMase C to the bath. Dotted line indicates zero current level. b. Time course of current, where the arrow indicates addition of 14 ng/μl (open circles, left) or 1.4 ng/μl (open triangles, middle) SaSMase C, or 40 ng/μl BaSMase C (closed squares, right). c, G–V curves of Kv2.1 before (squares) and after (dots) SMase C treatment. d, Fraction of current remaining at various times after addition of SaSMase C, plotted against voltage: top three data sets correspond to different levels of partial enzymatic treatment and the bottom set to complete treatment. The currents were normalized to those before addition of SMase C at the corresponding voltages. e, G–V curves without (open symbols) or with (closed symbols) 2 h incubation in a 5 mM MβCD-containing solution, and before (squares) or after (circles) SaSMase C treatment. f, Fraction of current remaining after addition of 3 μM HaTx (squares, top) and subsequent treatment with SaSMase C (circles, middle), or after SaSMase C treatment alone (triangles, bottom). g–j, Kv2.1 currents plotted against time where the arrows labeled 1 indicate addition of 10 mM phosphocholine (g), 0.25 mg/ml ceramide (C8) pre-dissolved in ethanol (h), 10 mM choline (i), or 0.25 mg/ml ceramide-1-phosphate (C-1-P; C12) pre-dissolved in 1: 49 dodecane/methanol (j). SMase C (g, h) or SMase D (i, j) were used as positive controls (arrow labeled 2). Similar results were observed in at least three experiments for each case. k, I–V curves of Kir1.1 before (squares) or after (circles) SMase C treatment, and following subsequent application of 0.5 μM pore-blocking tertiapin-Q (triangles). l, I–V curves of a KcsA-Kir2.1 chimera before (squares) and after (circles) SMase C treatment. All data in c–f, k and l are presented as mean ± s.e.m. (n = 5 - 8).
Sphingomyelin and cholesterol may form membrane domains called “lipid rafts”. We used the cholesterol-extracting agent methyl-β-cyclodextrin (MβCD)16 to test whether disruption of sphingomyelin-cholesterol interactions underlies the SMase C effect. A 2-hour exposure of oocytes to 5 mM MβCD (a common regimen) affected little the G-V curves obtained before or after SMase C treatment (Fig. 2e). Thus, the channel-inhibitory effect of SMase C does not appear to simply reflect disruption of sphingomyelin-cholesterol interactions.
Inhibition of Kv2.1 function by removal of the phospho-head of sphingomyelin is consistent with the observation that the voltage-gated (6TM) bacterial channel KvAP, unlike the non-voltage-gated (2TM) bacterial channel MthK, is non-functional when reconstituted in a non-phospholipid bilayer9, an observation suggesting that voltage sensors, not the ion conduction pore, require phospholipids for proper functioning. Pursuing that reasoning, we tested the effect of SMase C on two other non-voltage-gated (2TM) K+ channels: Kir1.1 and a KcsA pore with the Kir2.1 cytoplasmic termini attached17. Our finding that SMase C profoundly inhibits K+ conduction in both channels (Fig. 2k, l) indicates that sphingomyelin head groups are in fact critical for the proper function of these 2TM channels that lack voltage sensors. More direct approaches are thus needed to assess how SMase C inhibits voltage-gated Kv2.1 channels.
Looking for evidence that the observed SMase C inhibition of Kv2.1 results from removal of sphingomyelin phospho-heads around voltage sensors, we tested whether hanatoxin (HaTx), known to bind to voltage sensors18, 19, prevents SMase C from reaching the lipids and thus mitigates its effect (kinetics of HaTx inhibition and recovery18 are slow compared to the rate of SMase C-caused inhibition). Exposure to 3 μM HaTx inhibits Kv2.1 current ~70% at 30 mV and ~30% at 60 mV (Fig. 2f). (Extent of current reduction does not reflect extent of HaTx binding: this is because a channel bound with HaTx can still be activated by stronger-than-usual depolarization19, 20.) In the absence of HaTx, SMase C reduces current by 90% at all voltages but, in its presence, the observed current reduction is much smaller (Fig. 2f). Thus, HaTx binding protects a large fraction of channels against SMase C, in support of the notion that SMase C inhibits the channels by removing sphingomyelin phospho-heads around voltage sensors.
We next examined whether SMase C affects gating current, a capacitive current arising from voltage sensor movement21. Unfortunately, measuring Kv2.1’s gating current is technically challenging due to the lack of high-expression mutants that produce large gating currents but little ionic current. To circumvent this limitation, we employed the well-studied Shaker(-IR22) Kv channel for ionic current, and its non-conducting V478W mutant23 for gating current measurements. SMase C treatment reduces both ionic and gating currents by about half (Fig. 3a–d). Such a proportional reduction could be a reflection of the fact that about half the channels interact with sphingomyelin in such a manner that removal of the negatively charged phosphate groups creates an insuperable energy barrier for the positive gating charges to move during early gating transitions, effectively precluding activation by experimentally accessible depolarizations. Alternatively, the proportional reduction might be a coincidence peculiar to this particular Kv subtype, where half of the gating charges in individual channels remain functional and these channels, hence, susceptible to partial activation. However, as shown below, we similarly observed an approximately proportional reduction of ionic and gating currents in Kv1.3 channels. Additionally, SMase C does not reduce the slope of the Q-V curve (Fig. 3f), a parameter related to the effective number of gating charges. It is thus more probable that in about half the Shaker channels (more than half in the case of Kv1.3 and Kv2.1), sphingomyelin phospho-heads play an essential role in allowing voltage sensors to undergo early transitions involving the bulk of gating-charge movement. Phospholipids other than sphingomyelin presumably fulfill that role in the remaining channels. The two channel populations may exist in membrane domains that differ in their sphingomyelin content.
Figure 3.

Effect of SMase C and PC-PLC on ionic and gating currents of Shaker channels. a, b, Ionic currents of Shaker(-IR) (a) and gating currents of the V478W Shaker mutant (b) elicited with the protocols shown, gradually decreasing on addition of recombinant BaSMase C. c, d, G–V (c) and Qon-V (d) relations before (squares) or after (filled circles) SMase C treatment. The data are presented as mean ± s.e.m. (n = 6–11); the error bars are generally smaller than the symbols. e, Ionic current trace after SMase C treatment normalized in amplitude to that before treatment in a. f, Normalized Q–V curves before (squares) and after (filled circles) SMase C treatment in d. g, h, Integrals of on (g) and off (h) gating charges (before or after SMase C) versus time; all traces are normalized in amplitude. i–k, On-gating currents of Shaker’s V478W mutant versus time (similar results were observed in at least three experiments for each case). Gating currents were collected by stepping voltage from -100 mV to 0 mV (i, k) or -60 mV (j). The arrows indicate the addition and washout of SMase C (i); addition and washout of SMase D and subsequent addition of SMase C (j); addition and washout of purified native PC-PLC (k). The current was on average reduced 11.2 ± 2.8% following addition of PC-PLC (mean ± s.e.m., n =3). l, Q–V curves of control (closed squares) or after SMase D (open triangles) and subsequent SMase C (open circles) treatment (mean ± s.e.m, n = 5).
The above inference that the head groups of other phospholipids besides sphingomyelin enable early gating transitions in about half of Shaker channels expressed in Xenopus oocytes does not exclude the possibility that voltage sensors in these channels still interact with sphingomyelin, but in a manner important for late (downstream) transitions only. Rather, the very existence of channels whose early transitions do not require sphingomyelin permits investigation of the impact of sphingomyelin phospho-head groups on late transitions. We found that the kinetics of ionic currents that remain after SMase C treatment are markedly slowed (Fig. 3e) whereas those of the remaining gating currents are barely affected (Fig. 3g, h). Removal of phospho-head groups of sphingomyelin therefore must also raise (but not insuperably) the energy barrier for one or more gating transitions downstream from the bulk of the gating charge movement. This modest increase in the transition energy for one or more late rate-limiting steps is consistent with only 5–10% of total gating charge movement occurring late in the gating sequence24, 25. The resulting small change in the shallow part of the normalized Q-V curve at more depolarized potentials (Fig. 3f) is reminiscent of a S4 mutant (L370V) whose late transition is altered24.
Consistent with our early conclusion that SMase C acts by means of its lipase activity, gating current did not recover after SMase C washout (Fig. 3i). For further confirmation, we exploited the fact that SMase C acts only on sphingomyelin, not on ceramide-1-phosphate. As expected, conversion of sphingomyelin to ceramide-1-phosphate with SMase D shifts the activation curve to the left (Fig. 3l), i.e., allows mobilization of gating charges at more negative voltages (Fig 3j). Indeed, pre-treatment with SMase D prevents subsequently added SMase C from causing current inhibition (Fig 3j, l). The differing effects of SMases C and D indicate that the lipid product of at least one enzyme, perhaps both, continues to interact with the channel protein for the duration of the experiment. The persistence probably reflects strong channel-lipid interactions and/or confinement of relevant lipid molecules to microdomains. Consistent with this view, channel current was not significantly affected by acute addition of products of sphingomyelin hydrolysis catalyzed by either enzyme (Fig. 2g–j).
To rule out the possibility that the observed SMase C effect results from non-specific hydrolysis of the predominant outer-leaf phospholipid phosphatidylcholine (PC), we tested a PC-specific phospholipase (PC-PLC) (Fig. 1b) from Bacillus cereus directly against the channels, and found only modest (11.2 ± 2.8%) gating current depression, compared to ~50% for SMase C (Fig. 3k). SMase C, therefore, does not cause channel inhibition by hydrolyzing PC. The small inhibition caused by PC-PLC is, again, consistent with some channels interacting with phospholipids other than sphingomyelin.
SMase C was originally termed β-hemolysin after its in vitro hemolytic effect10, yet its role in pathogenesis remains largely unknown. The fact that inhibition of Kv1.3 in lymphocytes26, 27 is immuno-suppressive28 motivated us to test SaSMase C against human Kv1.3. We found that SaSMase C eliminates well over half of the ionic current of Kv1.3 and of the gating current of its non-conducting W384F mutant29 (Fig. 4), in an approximately proportional manner as in the case of Shaker channels. This finding raises the intriguing possibility that the SMase C action against Kv1.3 helps S. aureus to neutralize host defenses.
Figure 4.
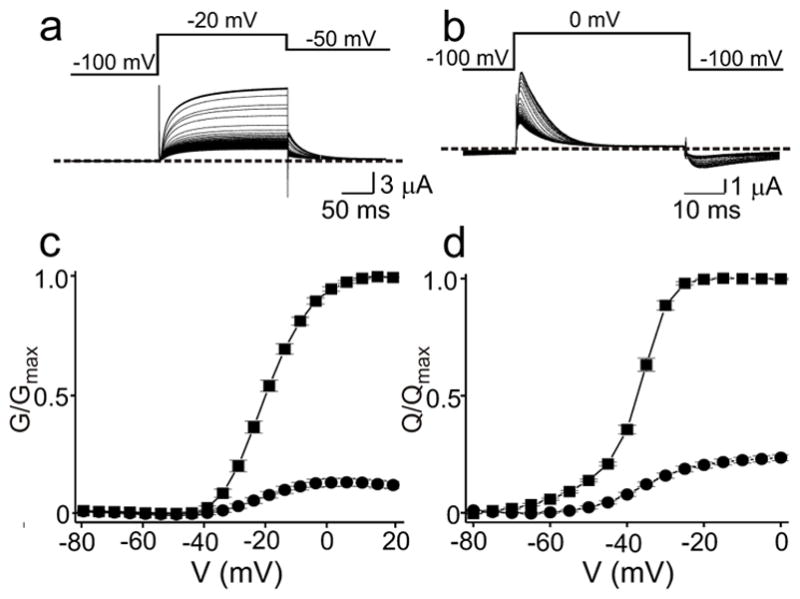
SMase C inhibition of Kv1.3. a, b, Ionic currents of wild-type Kv1.3 (a) and gating currents of the W384F mutant (b) elicited at 5 second intervals with the voltage protocol shown, gradually declining following addition of recombinant SaSMase C (the off-gating current is largely immobilized). c, d, G–V (c) and Qon-V (d) relations before (squares) and after (dots) treatment with SaSMase C. The data are presented as mean ± s.e.m. (n = 9–11).
In summary, the voltage sensor of Kv channels may interact strongly with multiple molecules of several kinds of phospholipid. In the present experiments on Xenopus oocytes, sphingomyelin appears to be preferred over PC (these lipids differ in hydrogen-bonding characteristics). The apparent preference probably reflects both inherent channel-lipid affinity and/or relative abundance in the channels’ lipid micro-environments. Some interactions between voltage-sensors and phospholipids are important for the early gating transitions, and others for late transitions. These findings strongly support the hypothesis that phospho-head groups of membrane lipids, together with certain acidic channel residues4, 5, provide the necessary countercharges for the positively charged voltage-sensing residues during individual steps of the voltage-sensor movement. This charge-neutralizing action lowers the free energy of the overall gating process as well as the energy barrier for individual transitions, so that a modest voltage can drive the charged sensors through a series of conformational steps in a low-dielectric cell membrane to open the channel gate.
Methods
The cDNAs of Kv2.1, Shaker-IR, Kir1.1 and the KcsA-Kir2.1 chimera were cloned in the pGEMHess vector, and Kv1.3 in the pSP64 vector. The mutant channel cDNAs were produced through PCR-based mutagenesis and conformed with DNA sequencing. The cRNAs were synthesized with T7 or SP6 polymerases using the corresponding linearized cDNAs as templates. Channel currents were recorded from whole oocytes (previously injected with cRNA encoding relevant channels) using a two-electrode voltage clamp amplifier (Warner OC-725C). The resistance of electrodes filled with 3 M KCl was 0.2 - 0.3 MΩ. Unless specified otherwise, the bath solution contained (in mM): 95/5 NaCl/KCl (or 80/20 for tail current measurements), 0.3 CaCl2, 1 MgCl2 and 10 HEPES; pH was adjusted to 7.6 with NaOH. Chemical reagent and lipase stock solutions (2 μl) were manually added to the 100 μl recording chamber. Ceramide-C8 (Cayman Chemicals) was pre-dissolved in ethanol, and ceramide-1-phosphate-C12 (Avanti Polar lipids) in 1: 49 dodecane/methanol. Unless specified otherwise, the final concentrations of BaSMase C, SaSMase C, SMase D, and PC-PLC were 40, 14, 4, and 50 ng/μl, respectively. PC-PLC from B. cereus was purchased from Sigma-Aldrich. To produce recombinant SMases C and D30, E. coli BL21 (DE3) cells were transformed with the respective cDNAs cloned into pET30 vector, grown in LB medium to ~0.6 OD at 600 nm, and induced with 1 mM IPTG for 2 hours. The bacteria were harvested, resuspended, and sonicated. The resulting samples were loaded onto a cobalt affinity column and eluted by stepping the imidazole concentration from 50 mM to 500 mM (all SMase proteins contain N- and C-terminal His tags). The imidazole was later removed by dialysis. HaTx was purified as previously described31. Unless specified otherwise, all chemicals were purchased from Sigma-Aldrich/Fluka.
Acknowledgments
We thank S. Billington for sharing SMase D cDNA, C. Deutsch for Kv1.3 cDNA, K. Ho and S. Hebert for Kir1.1, R. Joho for Kv2.1 cDNA, K. Swartz for Shaker-V478W cDNA, C. Armstrong for comments on our manuscript, and P. De Weer for review and discussion of our manuscript. This study was supported by a grant from NIGMS to Zhe Lu.
References
- 1.Aggarwal SK, MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 2.Seoh SA, Sigg D, Papazian DM, Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong CM. Sodium channels and gating currents. Physiol Revs. 1981;61:644–683. doi: 10.1152/physrev.1981.61.3.644. [DOI] [PubMed] [Google Scholar]
- 4.Papazian DM, et al. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 5.Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 6.Cuello LG, Cortes DM, Perozo E. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 2004;306:491–495. doi: 10.1126/science.1101373. [DOI] [PubMed] [Google Scholar]
- 7.Freites JA, Tobias DJ, von Heijne G, White SH. Interface connections of a transmembrane voltage sensor. Proc Natl Acad Sci U S A. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramu Y, Xu Y, Lu Z. Enzymatic activation of voltage-gated potassium channels. Nature. 2006;442:696–699. doi: 10.1038/nature04880. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt D, Jiang QX, MacKinnon R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 2006;444:775–779. doi: 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- 10.Glenny AT, Stevens NF. Staphylococcal toxins and antitoxins. J Pathol Bacteriol. 1935;40:201–210. [Google Scholar]
- 11.McNamara PJ, Cuevas WA, Songer JG. Toxic phospholipases D of Corynebacterium pseudotuberculosis, C. ulcerans and Arcanobacterium haemolyticum: cloning and sequence homology. Gene. 1995;156:113–118. doi: 10.1016/0378-1119(95)00002-n. [DOI] [PubMed] [Google Scholar]
- 12.Kurpiewski G, Forrester LJ, Barrett JT, Campbell BJ. Platelet aggregation and sphingomyelinase D activity of a purified toxin from the venom of Loxosceles reclusa. Biochim Biophys Acta. 1981;678:467–476. doi: 10.1016/0304-4165(81)90128-8. [DOI] [PubMed] [Google Scholar]
- 13.Doery HM, Magnusson BJ, Cheyne IM, Sulasekharam J. A phospholipase in staphylococcal toxin which hydrolyses sphingomyelin. Nature. 1963;198:1091–1092. doi: 10.1038/1981091a0. [DOI] [PubMed] [Google Scholar]
- 14.Read TD, et al. The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature. 2003;423:81–86. doi: 10.1038/nature01586. [DOI] [PubMed] [Google Scholar]
- 15.Projan SJ, et al. Nucleotide sequence: the beta-hemolysin gene of Staphylococcus aureus. Nucleic Acids Res. 1989;17:3305. doi: 10.1093/nar/17.8.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kilsdonk EP, et al. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–17256. doi: 10.1074/jbc.270.29.17250. [DOI] [PubMed] [Google Scholar]
- 17.Lu Z, Klem AM, Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413:809–813. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- 18.Swartz KJ, MacKinnon R. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 1997;18:675–682. doi: 10.1016/s0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- 19.Phillips LR, et al. Voltage-sensor activation with a tarantula toxin as cargo. Nature. 2005;436:857–860. doi: 10.1038/nature03873. [DOI] [PubMed] [Google Scholar]
- 20.Swartz KJ, MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 1997;18:665–673. doi: 10.1016/s0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- 22.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 23.Kitaguchi T, Sukhareva M, Swartz KJ. Stabilizing the closed S6 gate in the Shaker Kv channel through modification of a hydrophobic seal. J Gen Physiol. 2004;124:319–332. doi: 10.1085/jgp.200409098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoppa NE, McCormack K, Tanouye MA, Sigworth FJ. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255:1712–1715. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- 25.Loboda A, Armstrong CM. Resolving the gating charge movement associated with late transitions in K channel activation. Biophys J. 2001;81:905–916. doi: 10.1016/S0006-3495(01)75750-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 27.Matteson DR, Deutsch C. K+ channels in T lymphocytes: a patch clamp study using monoclonal antibody adhesion. Nature. 1984;307:468–471. doi: 10.1038/307468a0. [DOI] [PubMed] [Google Scholar]
- 28.Chandy KG, et al. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perozo E, MacKinnon R, Bezanilla F, Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- 30.Ramu Y, Xu Y, Lu Z. Inhibition of CFTR Cl-channel function caused by enzymatic hydrolysis of sphingomyelin. Proc Natl Acad Sci U S A. 2007;104:6448–6453. doi: 10.1073/pnas.0701354104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swartz KJ, MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]