

Abstract
The extracellular matrix (ECM) is a complex entity containing a large portfolio of structural proteins, signaling molecules, and proteases. Changes in the overall integrity and activational state of these ECM constituents can contribute to tissue structure and function, which is certainly true of the myocardium. Changes in the expression patterns and activational states of a family of ECM proteolytic enzymes, the matrix metalloproteinases (MMPs), have been identified in all forms of LV remodeling and can be a contributory factor for the progression to heart failure. However, new clinical and basic research has identified some surprising and unpredicted changes in MMP profiles in LV remodeling processes, such as with pressure or volume overload, as well as with myocardial infarction. From these studies, it has become recognized that proteolytic processing of signaling molecules by certain MMP types, particularly the transmembrane MMPs, may actually facilitate ECM accumulation as well as modulate fibroblast transdifferentiation – both critical events in adverse LV remodeling. Based upon the ever increasing substrates and diversity of biological actions of MMPs, it is likely that continued research regarding the relationship of LV remodeling to this family of proteases will yield new insights into the ECM remodeling process itself as well as new therapeutic targets.
Keywords: matrix metalloproteinases, heart failure, myocardial remodeling, extracellular matrix, transforming growth factor, fibroblast
Overview and Introduction
The clinical presentation of heart failure (HF) is generally that of a common set of clinical signs and symptoms. However, the underlying mechanical and structural basis responsible for the development of the HF syndrome is quite diverse. Identifying the causal mechanisms responsible for the development and progression of the HF process is crucial for the purposes of developing preventive and therapeutic strategies for this disease. Essential milestones in the development and progression of HF include changes in the structure, composition, and geometry of the left ventricular (LV) myocardium which has been generically termed LV remodeling. There are fairly distinct patterns of LV remodeling that occur and are dependent upon the initial pathophysiological stimulus, but once instigated, LV remodeling is an important predictor for the development and progression of HF.1–5 LV remodeling entails changes in the structure and function of the cardiocyte, the vascular compartment, and extracellular matrix (ECM), whereby changes within all of these entities occur as a continuum during the initiation and progression to HF. There is no question that fundamental defects in cardiocyte function, structure, and viability play a major role in the development and progression to HF. However, there is growing recognition that the ECM mediates both mechanical and biological signals that contribute to this process.6–15 For example, the ECM provides the critical interface for force transmission and alignment of myocardial fascicles, and also provides the substrate for transmembrane adhesion of cardiocytes.11,13–16 Thus, a loss of normal ECM structure and function can directly alter transduction of contractile force and intracellular signaling of cardiocytes, which in turn will change LV systolic function. In contrast, excessive ECM accumulation can directly alter myocardial passive stiffness properties, which will directly affect LV diastolic function.11,17 In certain instances, significant heterogeneity in ECM remodeling occurs whereby a loss of normal ECM structure and function is accompanied by abnormal ECM accumulation, which can impair both LV systolic and diastolic function.18–20 Through transgenic, pharmacological, and clinical observational studies, it is apparent that changes in ECM structure and function directly contribute to the adverse LV remodeling process, which accompanies the development and progression of HF.9–11,15–17,21–55 Finally, the dynamic nature of the ECM in terms of trafficking, processing, and compartmentalizing critical biological molecules that modulate myocardial growth and function, from growth factors to microRNAs (miRs), is becoming appreciated.6–8,24,27,30,56–61 Accordingly, the overall goal of this review is to provide a current perspective on a specific aspect of ECM proteolysis as it relates to the LV remodeling process and the progression to HF.
LV Remodeling Patterns and Heart Failure
For the purposes of this review, some generalizations regarding the structural basis of the HF process will be made, and while an oversimplification, can provide a means of classifying some commonalities with respect to LV remodeling. Classification of the LV remodeling process would include: (1) the pressure overloaded myocardium, such as that with hypertension or aortic stenosis, which causes LV hypertrophy and can give rise to increased collagen accumulation (i.e. “fibrosis”);9,10,17,19,31,34,37–44 (2) the volume overloaded myocardium, such as that with valvular regurgitation, aortocaval fistulae, and dilated cardiomyopathies, which is characterized by significant LV dilation and potentially a loss of normal collagen matrix structure and function;29,46,50,51,54,55 (3) the injured myocardium, most notably that of myocardial infarction (MI), whereby a heterogeneous remodeling process can occur simultaneously within the LV myocardium, giving rise to myocardial hypertrophy and fibrosis within the remote viable region, as well as mural wall thinning, expansion, and loss of normal collagen matrix structure and function within the MI region.20–28,32,47–49,52,53 These generalized LV remodeling categories will be utilized for the purposes of putting into context how ECM proteolytic activity may cause a “feed-forward” process to accelerate the development and progression of LV remodeling. Specifically, this review will first provide a brief overview of a refined perspective on the structure and function of the ECM and of the proteases and pathways that affect LV remodeling. Then a more focused review will be provided on ECM proteolytic systems, the matrix metalloproteinases (MMPs), with particular attention to the transmembrane MMPs and the relation to the LV remodeling process. One of the main goals of this review will be to identify how past concepts regarding MMPs solely causing ECM degradation was an oversimplification, and to examine new functionalities for this highly diverse family of proteolytic enzymes relevant to LV remodeling and progression to HF.
Myocardial Extracellular Matrix Redefined and MMP Relevance
The emergence of studies regarding the biology and function of MMPs in the progression of HF has resulted in an overall reappraisal of the ECM in terms of normal myocardial structure and function and contributory roles in adverse remodeling processes. The myocardial ECM was historically considered to be a relatively static structure composed of fibrillar collagens with relatively slow turnover rates, but it is now apparent that the ECM is a complex and dynamic entity. All of these ECM constituents are vulnerable to proteolytic processing directly or indirectly by MMPs,65–69 which in turn will alter the interactions between these different structural and biologically active interstitial molecules, and thereby determine the overall structure and function of the ECM. Changes in the structure and function of the ECM hold physiological implications in terms of both LV systolic and diastolic performances. Regarding systolic function, biological modeling and structure-function studies have demonstrated that the ECM plays a critical role in mechanotransduction of sarcomere shortening into myocardial contractile force.11,12,15,16,63 For example, activation of endogenous MMPs acutely altered fibrillar collagen content in papillary muscles and directly affected active tension development, whereas the contractility of myocytes isolated from these preparations was unaffected.16 When examined from a biophysical-mechanical basis, it is clear that the ECM contributes to stiffness properties of muscle in general and holds particular relevance to the myocardium.9–12,17,19,39,42,44,63,71–73 For example, correlative studies, such as those reported by Borg et al, demonstrated that species-specific differences in the structure and composition of fibrillar collagen was associated with changes in myocardial stiffness.70 In clinical studies of LV hypertrophy secondary to a prolonged pressure overload, significant collagen accumulation was directly associated with indices reflective of increased LV myocardial stiffness.9,10,37–44,71,72 In basic mechanistic studies, inhibition of fibrillar collagen cross-linking, for example, directly reduced myocardial stiffness properties.73,74 The effects on the stiffness modulus are nonlinear,12,15,16,63,72 and therefore, small changes in one direction or the other in terms of collagen content and architecture will result in significant and physiologically relevant changes in LV myocardial stiffness, and in turn, overall LV diastolic performance. Thus, proteolytic alterations of the ECM, such as those through the induction and activation of the MMPs, not only would contribute to overall LV myocardial architecture, but also directly affect LV myocardial systolic and diastolic function.
The ECM is also an important entity in terms of providing for the deposition, processing, trafficking, and transmembrane transduction of biological signals.6–8,13,14,30,57,75–83 The classic ligand-transmembrane receptor-intracellular transduction pathway is dependent upon the rate and extent that a ligand can traverse the ECM and interact with the extracellular domain of the cognate receptor. Moreover, the release of exosomes containing mRNA or miRs into contact with the ECM, as well as the direct release of miRs into the interstitial space, can provide a cell-cell communication pathway.83,84 The MMPs and associated proteases process transmembrane receptors, including integrins, and thereby alter receptor transduction pathways as well as cell-ECM interactions.65,66,75,76 Thus, proteolytic induced changes in the composition and structure of the ECM would directly alter cell signaling/communication. In terms of processing bioactive molecules within the ECM, examples would include cytokine processing, such as tumor necrosis factor (TNF) and transforming growth factor (TGF).24,32,76–82,85,86 A specific illustration of the potential significance of the interaction between TGF and certain MMP types is discussed in a subsequent section.
MMPs: A Small Family on a Large Tree
While this review will focus upon the certain MMP types in terms of LV remodeling, it must be recognized that this is but one family of the large class of metzincin proteases; a highly diverse set of proteolytic enzymes exist within the ECM. In terms of relevance and interaction with the MMPs, a family of associated proteinases would include the disintegrin and metalloproteinases (ADAMs) and the ADAM with thrombospondin motifs (ADAMTS). 75,76 For example, ADAM-10 processes membrane bound TNF to a soluble form, and in turn can cause the induction of MMPs.75.76 Since a number of MMPs can process membrane bound TNF as well, this can form a potent, localized induction loop within the myocardium. Among a number of other substrates, ADAMTSs are engaged in proteolytic removal of the procollagen N-terminal domain – a critical step in collagen maturation. These particular ADAMTSs would therefore also reduce the susceptibility of newly formed collagen to proteolytic degradation, and in turn would increase collagen stability and cross linking. In light of the fact that certain transmembrane type MMPs can be co-localized to ADAM/ADAMTS and integrin complexes, 13,14,75,76 it is likely that a strong interplay exists between these families of ECM proteases.
The initial description of a matrix degrading enzyme was based upon the seminal observations whereby the excised tadpole tail degraded a collagen gel – hence the term collagenase.86 There are now approximately 23 MMP types expressed within humans, and the distribution, functionality, and substrates are diverse as discussed in several broader reviews.65–69,88–92 Moreover, the term “MMP function” yielded only 10 publications in 1990, whereas this same search term yielded over 2,200 publications in 2011 alone.(PubMed, National Library of Medicine, Search Performed July 14, 2012) Thus, in order to provide focus, only those MMP types that have been most intensely investigated with respect to LV remodeling will be discussed. Initially, the MMPs were classified based upon recognized substrates, and while this nomenclature no longer holds relevance, it is still commonly used to group the MMP types. This classification scheme would include the Collagenases, such as MMP-1, MMP-13, and MMP-8; the Gelatinases which would include MMP-2 and MMP-9; the Stromelysins/Matrilysins which would include MMP-3 and MMP-7; and the Membrane Type MMPs which would include MMP-14. This is hardly the exhaustive list of MMP types that are likely expressed within the mammalian myocardium. Moreover, this list does not include MMP types that are relevant within the vasculature, such as MMP-12. Nevertheless, this simplified list and classification scheme provides a suitable context by which to examine the current field in terms of LV remodeling. This review will focus upon proteolytic interactions of the MMPs, such as the membrane type MMPs (i.e. MMP-14), with particular attention to the differential biological effects and substrates relevant to the LV remodeling that occurs in pressure overload, volume overload, and myocardial infarction.
Differential Expression and Effects of MMPs in Pressure Overload
In general, whether due to systemic hypertension or a fixed outflow obstruction, such as aortic stenosis, LV pressure overload causes significant myocardial growth, paralleled by increased ECM accumulation – of particular note, fibrillar collagen.9,10,17,19,41,71,72 This would lead to the assumption that MMP expression, and in turn ECM proteolysis, would be reduced with LV hypertrophy and ECM accumulation. However, in contrast to canonical thought, certain MMP types are actually induced with LV hypertrophy and ECM accumulation, notably with the progression of diastolic dysfunction, whereas other MMP types are reduced or relatively unchanged.31,38,40–44,92,93 Some of the basic and clinical studies that provide support for a multidimensional role of MMPs in LV pressure overload and the progression to HF are summarized below.
LV Pressure Overload and Transmembrane MMP Interactions
Cyclic strain has been shown to induce a number of MMPs, such as the gelatinase MMP-2.94 In myocardial biopsies taken from patients with LV pressure overload secondary to aortic stenosis, increased MMP-2 expression and activity was identified.92 In patients with LV hypertrophy and HF with a history of hypertension, plasma levels of MMP-2 were significantly increased compared to age matched control subjects.10,40–42,44 For example, in patients with established LV hypertrophy, the relative levels of plasma MMP-2 could be utilized in a diagnostic model with respect to progression to HF.44 Moreover, a meta-analysis demonstrated that elevated plasma MMP-2 levels were a consistent finding in patients with LV hypertrophy and developing HF.41 In a murine model of LV pressure overload, genetic deletion of MMP-2 reduced the degree of myocardial fibrillar collagen accumulation and improved indices of LV diastolic function.95 It is well-established that the primary mechanism for conversion of pro-MMP-2 to active MMP-2 is through complex formation with the membrane type MMP, MMP-14.89–91,96 MMP-14 expression is sensitive to changes in mechanical load, whereby increased wall tension proliferated MMP-14 promoter activity in-vitro.97 Increased myocardial MMP-14 expression has been identified in patients with LV pressure overload.92 In animal models of LV pressure overload, an early and sustained induction of MMP-14 has been identified.31,93,98 For example, increased MMP-14 promoter activity and subsequently MMP-14 proteolytic activity has been reported following the induction of LV pressure overload in mice.31 Moreover, these studies have identified an association between changes in myocardial ECM remodeling, notably increased collagen accumulation to that of MMP-14 induction.31,93,98 These observations would suggest that increased MMP-14 contributes to adverse ECM remodeling, which is a pivotal structural event in terms of LV pressure overload and progression to HF. The pathways by which MMP-14 contributes to adverse ECM remodeling likely include facilitating proteolysis of interstitial molecules directly (such as integrins), amplification of active MMP-2 causing ECM instability, and abnormal architecture, as well as through enhancing profibrotic signaling pathways. As discussed in a subsequent section, an important proteolytic relationship likely exists between MMP-14 and the subsequent activation of the profibrotic signaling molecule TGF, 31,88,93–98 which would hold particular relevance in the context of LV pressure overload.
LV Pressure Overload Causes Diversity in MMP Induction
While certain MMP types, such as MMP-2 and MMP-14, appear to be uniformly increased with LV pressure overload, other MMP types, such as the interstitial collagenase MMP-13, have been reported to be decreased.38,40 For example, detectable plasma levels of MMP-13 were reduced in patients with LV hypertrophy and HF.40 Other MMP types, such as MMP-1 and MMP-3, appear to be either unchanged or reduced with LV pressure overload.10,38,40–42,44,92,98 Interestingly, transgenic expression of human MMP-1 in mice (this MMP type is absent in rodents) and induction of LV pressure overload resulted in a relative reduction in myocardial fibrillar collagen content and improved indices of LV function.34 These findings suggest that the loss of normal constitutive levels of certain MMP types, or failure of an induction of certain MMP types with LV pressure overload, may facilitate abnormal ECM accumulation and adverse myocardial remodeling. This process does not occur in isolation, and key matricellular proteins and signaling molecules, some of which are identified in a preceding section, significantly change with LV pressure overload, as do the constitutive inhibitors of the MMPs that are described briefly in a subsequent section. It must also be recognized that the induction/repression of certain MMP types is likely to be a dynamic process and therefore would be a function of both time and magnitude of the LV load.
MMPs in Volume Overload/Cardiomyopathies
In terms of significant LV dilation and ECM remodeling that are hallmarks of the dilated cardiomyopathies and volume overload states, a robust expression and activation of MMPs have been clearly identified.46,50–51,54–55,99–107 For example, in patients with end-stage dilated cardiomyopathy, myocardial samples obtained at the time of transplant revealed significantly higher levels of MMPs from all of the representative MMP classes.46,107 The MMPs elevated in patients with dilated cardiomyopathy included MMPs associated with an inflammatory response such as MMP-7, MMP-8, and MMP-9.45,107 Of note, the most robust levels for any of the MMP types was that of MMP-14.46 On the other hand, relative levels for MMP-1 were reduced in dilated cardiomyopathy samples.107 In a more recent clinical study of stress induced cardiomyopathy (takotsubo), a similar reduction in plasma MMP-1 levels was observed.108 One of the limitations of these clinical observations is that the measurements were performed when severe HF had become manifest, and therefore, whether and to what degree these changes contributed to disease progression remained unclear.
Volume Overload and MMP Induced ECM Proteolysis – Evidence from Animal Models
However, using animal models of LV dilation and dysfunction, a mechanistic role for MMP induction in this process has emerged.50,54–55,99–103 Using the rapid pacing model of developing dilated cardiomyopathy, an early and time dependent induction of MMPs has been reported, which paralleled changes in LV geometry and function.55 More importantly, the early MMP induction and LV dilation preceded any significant change in isolated myocyte contractility,55 suggesting that MMP mediated ECM remodeling is an early structural event in this LV remodeling process. In the canine model of volume overload secondary to mitral regurgitation, LV dilation was accompanied by an early and robust increase in MMP-9, followed by a persistent increase in relative MMP-2 levels, as assessed by substrate zymography.99,100 In the rodent model of aortocaval fistula, the progression of LV dilation and dysfunction is concordant with myocardial MMP induction.54,102–103 In a similar murine model of volume overload, transgenic deletion of MMP-9 attenuated the degree of LV dilation and dysfunction.106 Using broad spectrum MMP pharmacological inhibition at the onset of the pathological stimulus (either rapid pacing or fistula), the progression of LV dilation and dysfunction attenuated significantly and reduced indices of ECM turnover and instability.50,54 Taken together, these findings provide a cause-effect relation between MMP proteolytic activity and LV dilation with a volume overload stimulus.
LV Dilation and Volume Overload; MMP and Signaling Interactions
In the aortocaval fistula model, the MMP induction-activation cascade is likely facilitated by a resident interstitial cell, the mast cell.101,105 There is a strong interrelationship between bioactive signaling molecules within the ECM, such as the cytokine TNF and MMP induction/activation.30,82,89–91 Mast cells can secrete significant amounts of MMPs as well as TNF, and both of these pathways have been implicated in volume overload induced remodeling.54,100,104–105 As such, it is likely that cross-talk occurs between interstitial cell types, such as the mast cell and fibroblast, in terms of cytokine processing and signaling, which in turn would perpetuate the LV remodeling process in terms of MMP induction and ECM proteolytic events. Indeed, interruption of mast cell function directly or inhibition of the TNF signaling pathways in turn reduced the magnitude of LV remodeling with volume overload.104,105 Interestingly, the magnitude of LV remodeling and MMP induction with volume overload appear to be estrogen dependent.101,102 Uniformly, the initiation of LV dilation with a volume overload stimulus is a robust increase in active MMP-2,54,99–103 and while not directly measured in these past investigations, would putatively suggest a concordant induction of MMP-14.96 Through in-silico mapping, the MMP-14 promoter contains estrogen response elements,109,110 and estrogen exposure directly modifies MMP-14 levels within the myocardium.93 In the estrogen receptor knockout mouse, MMP-14 expression was increased, as was MMP-2 activation.111 These observations would support the hypothesis that estrogen may modify MMP-14 expression, and as a consequence, the proteolytic events that would facilitate ECM remodeling and LV dilation with a volume overload stimulus.
MMPs in Myocardial Infarction
In regards to LV remodeling, post-MI remodeling may hold the greatest complexity in terms of the temporal and regional heterogeneity of events occurring within the ECM.4,5,18–20,115,116,119 Nevertheless, through both clinical observational studies and the use of transgenic/pharmacological studies, it is quite apparent that MMPs play pivotal roles in both the requisite healing process following myocardial injury, as well as the adverse remodeling process commonly termed “infarct expansion”.21–28,32,47–49,52,53,55,56,111–114 In the early time period following MI (first 72 hours), a more classical wound healing response occurs with the appropriate amplification of cytokines, influx of inflammatory cells, and proliferation/transdifferentiation of fibroblasts to a myofibroblast phenotype.64,115–118 A diverse number of MMP types released from both inflammatory cells and myofibroblasts facilitate proteolysis of ECM components and allow for nascent scar formation.64,115–118 However, unlike the prototypical wound healing response in more static tissue interfaces, continuous and persistent release of MMPs occur well beyond this initial wound healing period. There is a shift in MMPs synthesized primarily by the expanded population of transdifferentiated myofibroblasts,117,118 which results in ECM instability particularly at the infarct-viable myocardial border.4,5,20 However, the remote viable myocardium also undergoes significant ECM remodeling, which includes proliferation of fibroblasts, MMP induction, and fibrillar collagen accumulation.18,19,24,28,32,47–49,52,115,116 Thus, the early elaboration of certain MMP types that appear to facilitate a physiological wound healing response are followed by a more maladaptive induction of MMPs within the MI, border, and remote myocardium, which in turn contribute to adverse remodeling within all of these regions. The challenge that remains is to identify those MMP types that facilitate an appropriate wound healing response versus those that contribute to the development of post-MI remodeling and the progression to HF.
Post-MI Remodeling and MMP Induction
Some of the first studies to demonstrate a mechanistic relationship between MMP activation and LV remodeling following MI were performed in rodents.23,53 For example, Heymans and colleagues demonstrated in different transgenic constructs a relationship between MMP activation and post-MI remodeling.23 Rhode et al reported pharmacological MMP inhibition in mice attenuated the invariable LV dilation that occurred following surgically induced MI.53 Transgenic deletion of MMP-9, MMP-2, or MMP-7 in murine constructs of MI have all been shown to alter the post-MI remodeling process, but in some particularly unique ways.21,22,26,27 For example, targeted deletion of MMP-7 appeared to affect myocardial conduction pathways and proteins and underscored the diversity of substrates that are likely relevant in the context of post-MI remodeling.27,120 Using transgenic constructs that facilitated MMP activation along with pharmacological rescue provided further support for a mechanistic relationship between MMP induction and activation as well as adverse post-MI remodeling.23,25 Finally, direct molecular imaging approaches in murine models of MI have been performed, which provided a temporal map of MMP induction and activation within the LV during the remodeling process.28,47 For example, using transgenic MMP reporter constructs, increased MMP-2 and MMP-9 promoter activation was identified within the MI and border zones early post-MI, but interestingly, increased MMP-2 promoter activation was identified within the viable remote region as well as in the atrium.28,47 Taken together, these murine studies have provided novel insight into those MMP types that may be causative in LV remodeling, as well as the regional and temporal aspects of MMP induction post-MI.
Differential MMP Induction Post-MI – Refining MMP Targets
Using large animal post-MI models and MMP profiling, past studies have demonstrated that certain MMP types, such as MMP-1, were significantly reduced within the MI region, whereas other MMP types, such as MMP-14, were significantly increased within all regions post-MI.48,49,119 These past observations would suggest that certain MMP types, such as MMP-1, may not be contributory to the adverse LV remodeling process post-MI, and indeed pharmacological “MMP-1” sparing inhibitors were examined in pre-clinical HF models.48,50 Using appropriate dosing titration, the plasma concentrations of these MMP inhibitors would effectively be an order of magnitude lower than that necessary to achieve MMP-1 inhibition, but would effectively inhibit MMP types such as MMP-2, MMP-9, and MMP-14.50 The main finding from these studies was that it was not necessary to perform “global” MMP inhibition, and that targeting a potential subset of MMP types may be an effective strategy.121 Moreover, these past studies suggested that pharmacological MMP inhibition is not necessary immediately following MI, but can be successfully instituted after the completion of the initial wound healing period.48 The relative distribution of MMP-14 within the LV at 14 days post-MI (when presumably resolution of the acute wound healing phase has subsided) and the relation to cardiocytes and fibroblasts, respectively, is shown in Figure 1. In this figure, it can be appreciated that MMP types, such as MMP-14, are ubiquitously expressed in a number of cell types including myocytes, fibroblasts, and endothelial cells. Within the remote myocardium, MMP-14 is expressed within myocytes and fibroblasts and likely contributes to a profibrotic response within this region, as discussed in the next paragraph. On the other hand, MMP-14 induction appears to be robust within proliferating fibroblasts in both the border and MI regions, where significant and persistent ECM proteolytic activity and turnover occurs. This would suggest that MMP-14 and subsequent activation of soluble MMP types, such as MMP-2, would cause continued ECM instability within these regions post-MI and thereby contribute to LV remodeling, dilation, and eventually dysfunction. This is but one illustration that emphasizes how the development of future strategies to interrupt the adverse LV remodeling process post-MI will need to be region, temporally, and MMP type specific.
Figure 1.

Representative LV myocardial sections taken from remote, border, and MI regions at 14 days post-MI in adult pigs, using methods described previously.48,49 (TOP) Immunofluorescent images of these 3 regions following staining with an MMP-14 antisera (Cy3-red). As reported previously in both animal and human LV specimens,46,49,52 robust expression of MMP-14 can be observed in both the myocyte and non-myocyte (fibroblast) cell populations. (BOTTOM) Multiple fluorescent labeling was performed whereby nuclear staining was performed using DAPI (blue), phalloidin for actin (green), and a specific antisera for collagen type I (yellow) was performed. Co-localization of MMP-14 will appear as yellow regions. Within the remote region, significant MMP-14 localization can be observed within myocytes and the interstitial space consistent with the transmembrane nature of this MMP type. Within the border region, localization can be appreciated in both viable myocardial cells and proliferating fibroblasts (upper part of panel). Within the MI region, phalloidin positive cells, consistent with myofibroblasts, predominated and co-localized with MMP-14.
(Images obtained using a BioRad MRC1024 Confocal Scanning Laser Microscope by Dr. Robert Price, Instrumentation Resource Facility, USC School of Medicine). Scale bar = 30 microns; MMP-14 antisera: Abcam, ab3897, Collagen I antsera: Santa Cruz sc-87048, both used at 1:100 dilution.
MMP Proteolysis and Profibrosis: Two Sides of the Same Coin
It is apparent that MMP induction and activation is not an “all or none” process in terms of ECM proteolysis, and in fact, certain MMP types may actually facilitate ECM synthesis and accumulation; i.e. fibrosis. One of the MMP types with a diverse proteolytic portfolio that likely plays a role in ECM degradation and instability, as well as ECM profibrotic signaling, is MMP-14.24,32,77–80,85,86,88,89,91,117 Uniformly, increased myocardial levels of MMP-14 have been identified in LV remodeling with pressure overload, a dilated LV phenotype, or post-MI.31,46,47,49,50,52,98,119 While most certainly MMP-14 is not the only MMP type with a multiplicity of actions, this particular MMP will be discussed for the purposes of illustration and is schematically presented in Figure 2. First, MMP-14 is localized to the cell membrane surface and provides for focal ECM degradation of basement membrane components and thereby alters cell-cell interactions. Second, MMP-14 likely works in a cooperative manner with ADAMs to cause integrin proteolysis and shedding.75,76,91,123 Third, MMP-14 directly causes proteolysis of a number of ECM constituents, which in turn results in the formation of smaller fragments (i.e. matricryptins), and thereby alters a number of intracellular signaling cascades.68,124,125 Fourth, as stated previously, MMP-14 constitutes a predominant pathway by which other soluble MMP types, such as MMP-2, are activated within the ECM.96 Finally, MMP-14 can directly facilitate ECM synthesis by enhancing and amplifying profibrotic signaling molecules, such as TGF.24,32,78–80
Figure 2.
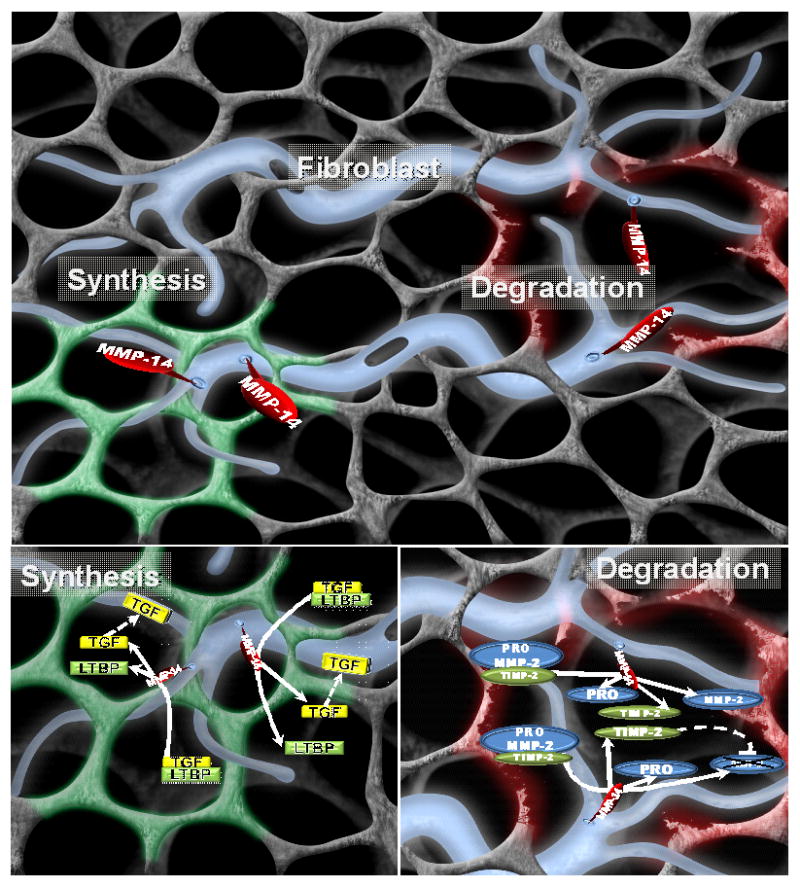
(TOP) Schematic of the fibroblast (blue) and ECM in terms of the functional diversity of MMPs; in this case, the representative membrane type MMP, MMP-14. MMP-14 is a transmembrane MMP with a short cytoplasmic tail that likely holds significance for intracellular signaling and potential regulation. The extracellular domain of MMP-14 can cause localized proteolysis of a wide portfolio of ECM proteins, cause a loss of normal ECM-integrin engagement, and activate other MMPs. Thus, MMP-14 can cause a robust and localized amplification of ECM degradation, and in turn, instability. On the other hand, MMP-14 can process profibrotic signaling molecules, such as the release of active TGF, and in turn promote increased fibrillar collagen synthesis and accumulation. In the context of cancer,86,89,90 activated cells demonstrate a high degree of MMP polarization and would suggest that these diverse proteolytic actions of MMP-14 can be occurring simultaneously within different ECM locations. With adverse ECM remodeling, such as that with pressure or volume overload or following MI, these ECM degradation and synthesis events can occur concomitantly and be polarized to different subcellular locations as well as to different regions of the LV. Thus, depending upon the context and substrate, MMP-14 can facilitate a loss of normal ECM and replacement fibrosis (such as with pressure overload), a loss of normal ECM and structural support (such as with volume overload), or a combination of both of these proteolytic events (such as with MI).
(LOWER LEFT) While MMPs, such as MMP-14, were considered to strictly cause ECM proteolysis, MMP-14 can directly induce a profibrotic cascade involving TGF. MMP-14 can cause proteolysis of the latency binding protein-1 (LTBP-1), which holds TGF in an inactive state, and thereby directly induce a TGF mediated profibrotic signaling cascade.77–80
(LOWER RIGHT) Another critical function of MMP-14 is the complex formation with pro-MMP-2 and TIMP-2, which will result in an active form of MMP-2. Moreover, TIMP-2 can then bind to the active site of MMP-2, which will extinguish proteolytic activity. This localized activation-inhibition cycle can provide for precise ECM proteolysis. Thus, the type and location of the MMP-14 substrate very likely dictates the effects upon ECM structure and function. (Initial image provided courtesy of Shaun Riffle, USC School of Medicine, and annotation by Craig P. Novack).
The diversity of biological events that can be induced in whole or in part by MMP-14 may also provide insight into how one MMP type may have significantly different effects upon ECM structure and function in the context of the different LV remodeling patterns. For example, in LV pressure overload, the signals and substrates for MMP-14 would include profibrotic signaling molecules and processing of growth factors. In LV volume overload, the mechanical and biological signals likely coalesce to provoke increased release of soluble MMP types such as MMP-2, which in turn would be processed by MMP-14. In post-MI remodeling, it is likely that these MMP-14 mediated events that are operative in both pressure and volume overload are occurring simultaneously, but in a region and time specific manner.
Normal ECM Degradation and Profibrotic Signaling by MMP-14
Through a number of in-vitro and proteolytic assays, it has been demonstrated that MMP-14 processes latent TGF binding protein-1 (LTBP-1), which will cause release of biologically active TGF and the binding to local TGF receptor complexes.32,78–80 Further studies have demonstrated that MMP-14 can proteolytically alter the TGF co-receptor, endoglin (CD150), which in turn will modify TGF mediated signaling.8,80 In the context of LV pressure overload, it is likely that MMP-14 contributes to the acceleration of myocardial fibrillar collagen accumulation and the development of diastolic dysfunction.31 While remaining associative, it is likely that MMP-14 induces profibrotic signaling through a TGF dependent pathway.81 In the volume overload-cardiomyopathies, robust MMP-2 activation is a hallmark in the progressive LV dilation and the loss of ECM mediated support. In this case, it is likely that significantly increased MMP-14 mediated activation of MMP-2 would be a critical proteolytic pathway whereby heightened ECM proteolysis occurs.19,46,50,51,54,55,99–101 In the context of post-MI LV remodeling, it is very likely that MMP-14 plays critical roles in both ECM proteolysis and instability of the MI region, and at the same time, increases ECM accumulation and fibrosis within the border region.24,32,52 In a transgenic mouse model of cardiac restricted MMP-14 overexpression, adverse post-MI remodeling, such as increased fibrosis, has been observed.24,32 In contrast, transgenic mediated reduction in MMP-14 expression reduced the degree of post-MI remodeling and ECM accumulation.32 In both of these past studies, concordant directional changes in MMP-14 mediated LTBP-1 hydrolysis were observed. The liberation of TGF from the latency binding proteins results in binding to a transmembrane receptor complex, and ultimately the phosphorylation of receptor transduction elements, the Smad proteins. In the murine model of MI, the heightened MMP-14 specific LTBP-1 hydrolysis was directly associated with increased Smad phosphorylation, indicative of increased TGF receptor stimulation.24,32 With either increased or decreased levels of MMP-14, Smad phosphorylation changed in a parallel fashion and in turn was associated with concordant changes in fibrillar collagen expression.24,32 In the murine model of pressure overload, MMP-14 promoter induction and ultimately MMP-14 expression was directly associated with the time dependent changes in LV myocardial fibrillar collagen expression and accumulation.31 While these studies are far from conclusive, and the TGF signaling pathway can be complex, these observations as well as those from other model systems78–80 suggest that MMPs such as MMP-14 can not only cause degradation of normal ECM structure and thereby function, but also induce a potent profibrotic signaling cascade, such as through the proteolytic release of TGF (Figure 2). These proteolytic events can be occurring simultaneously in a specific region or in different regions, which in turn would facilitate adverse ECM remodeling.
Fibroblast Proliferation, Transdifferentiation, and MMP-14
The diverse actions of MMP-14 in terms of simultaneously causing ECM proteolysis as well as processing biological signaling molecules have received considerable attention in regards to the tissue remodeling and mesenchymal cell transdifferentiation, which occurs in cancer.80,86,88–91,117 There are similarities in the signaling pathways that are evoked during this mesenchymal transdifferentiation process to that of fibroblast-myofibroblast transdifferentiation.115–118,126,127 In terms of LV remodeling such as that with pressure overload or MI, it is likely that this transdifferentiation process occurs in that an increased density of myofibroblasts commonly occurs. While an incomplete definition, myofibroblasts are those that express key markers such as alpha-smooth muscle actin.118,127 In this case, transformed fibroblasts have been identified with unabated expression of ECM proteolytic enzymes, such as MMP-14.128 The specific cell type of origin with respect to the myofibroblast remains unclear and may arise from a stem-cell type, pericyte, or a clonal expansion of endogenous fibroblasts. For example, in models of MI and angiotensin II induced hypertrophy, it has been identified that mesenchymal stem cells transmigrate, proliferate, and differentiate to a myofibroblast phenotype.129,130 The transmigration of mesenchymal cells likely requires an upregulation of MMPs as well as processing of chemokines, whereby MMP-14 may play a role in both of these processes.65,86,123,131,132 With aging, cardiac restricted overexpression of MMP-14 increased cell markers of myofibroblast transformation as well as myocardial fibrosis.24 TGF has been shown to play a role in mesenchymal transdifferentiation.117,133 Taken together, these observations would suggest that MMPs, such as that by MMP-14, can contribute to selective degradation of basement membrane components and activation of biological signaling molecules – all critical events that would contribute to fibroblast transdifferentiation and expansion in the context of LV remodeling. However, this remains an intriguing postulate, and additional research in this area is required.
Regulation of MMPs as Targets for Myocardial Remodeling
In light of the potent biological effects of MMP proteolytic activity, there are critical control points in terms of MMP transcription, synthesis, and post-translational modification that hold potential for therapeutic targeting.
Transcriptional Activity
MMP transcriptional activity is under the influence of upstream biological signals that are operative in the context of LV remodeling and HF, which would include bioactive molecules such as neurohormones and cytokines.30,82,89–91,134,135 Thus, pharmacological approaches that modify these signaling pathways would likely modify MMP transcription. MMP induction is also likely to be sensitive to mechanical signals, such as load, and it has been shown that an acute increase in LV afterload can induce MMPs, such as MMP-2 and MMP-14.31 Moreover, there are possible important interactions and integrations between mechanical and biological signals, which ultimately influence MMP induction. For example, an acute increase in LV load caused a robust increase in interstitial MMP activity, which could be modified, to some degree, by angiotensin II receptor inhibition.134 Following an acute MI in pigs, a similar pharmacological approach has been shown to modify myocardial MMP induction.135 The importance of MMP transcriptional activity in terms of LV remodeling and progression to HF can also be demonstrated from clinical studies of MMP polymorphisms.136–138 For example, a genetic variation within the MMP-9 promoter (C-1562T), which would result in higher MMP-9 transcriptional activity, was associated with elevated plasma MMP-9 levels and worsened post-MI survival.137 While these polymorphism studies can be problematic with respect to direct causality in specific MMP induction, it does emphasize the potential mechanistic importance of MMP promoter activity. Using promoter-reporter constructs,28,35 it may be possible to determine which signaling pathways play a predominant role in type-specific MMP transcriptional activity in the context of LV remodeling. Another likely and potentially unique mechanism for MMP transcriptional activation is through the extracellular matrix metalloproteinase inducer (EMMPRIN) protein.139,140 Significantly elevated myocardial levels of EMMPRIN have been identified in patients with significant LV remodeling and HF and is also increased in patients following MI.46,140 In both of these clinical observational studies, increased expression of EMMPRIN was coincident with MMPs likely contributory to the LV remodeling process, including MMP-2, MMP-9, and MMP-14. In a transgenic model of cardiac restricted over-expression of EMMPRIN, increased myocardial levels of MMP-2 and MMP-14 occurred and resulted in adverse LV remodeling as a function of age.33 However, it remains uncertain how EMMPRIN causes MMP transcription in the context of LV remodeling and to what degree this signaling pathway contributes to the progression of HF.
Post-Transcriptional Regulation
The microRNAs (miRs) can directly influence post-transcriptional events through a number of molecular interactions, which include binding to mRNA and interfering with initiation of translation and/or accelerating mRNA degradation. Thus, while the specificity of certain miRs in terms of mRNA targets remains to be fully developed, it is becoming apparent that certain miRs likely hold relevance to LV remodeling and to the ECM in particular.45,59–61,140–142 For example, in a mouse model of MI, changes in relative myocardial miR-29a levels had distinct and directional effects on ECM remodeling in terms of myocardial fibrosis.60 Other studies have provided direct evidence for a mechanistic relationship between relative miR-29a levels and MMP protein content.142 In other studies, overexpression of miR-133a in a transgenic model reduced the invariable changes within the ECM with LV pressure overload.141 While it is likely that specific miRs can directly influence ECM proteolytic pathways by post-transcriptional regulation of specific MMPs, it is also likely that changes in ECM composition and structure can, in turn, influence this post-transcriptional pathway. Specifically, miRs can be released into the ECM through several mechanisms, including a pathway involving exosomes.83,84 The trafficking of miRs within the ECM may constitute an important mechanism of cell-cell communication, and therefore, MMP mediated changes in ECM structure, function, and composition would directly affect this process. In addition, the egress of miRs into the ECM will also result in movement into the vascular compartment and allow for profiling specific miR levels through peripheral blood sampling. Using this approach, specific relationships between plasma profiles of specific miRs and MMPs in the context of LV remodeling, such as following an MI, may be possible.56,58
Post-Translational Control
A critical control point with respect to post-translational regulation of MMP activity is through the endogenous set of small molecular weight proteins, the tissue inhibitors of MMPs (TIMPs).89–91 There are four known TIMPs with a very high affinity for binding to active MMPs, which were initially thought to be of similar functional characteristics. However, there is now clear and specific functionality identified for each TIMP, and this has been the focus of several reviews.89–91,143 In general, however, there appears to be a discordant induction of TIMPs in the context of LV remodeling, which in turn would affect net MMP proteolytic capacity.38–44,51,56,98,108,119 For example, in LV pressure overload, relatively higher levels of TIMP-1 occur in relation to MMP levels, and this would favor a net reduction in ECM degradation.9,10,38–44 On the other hand, levels of TIMPs in the context of post-MI remodeling appear to be reduced, which would favor higher net MMP proteolytic activity.49,56,119 However, this is likely a significant oversimplification of the complex interaction between MMPs and TIMPs in terms of LV remodeling. For example, in pressure overload, transgenic mediated deletion of TIMP-2 or TIMP-3 actually accelerated the progression of LV remodeling and failure.29,30,144 Moreover, TIMPs may not only bind to the active MMP domain, but also bind to the pro-MMP domain, which can actually facilitate MMP activation.89–91,96,137 For instance, pro-MMP-2 and TIMP-2 form a complex, which ultimately interacts with MMP-14, yielding active MMP-2.96 This MMP-TIMP interaction would therefore suggest that increased levels of TIMP-2 may actually facilitate MMP activation and ECM proteolysis. Thus, similar to that of MMPs, there is significant functional diversity and complexity of the TIMPs. Understanding these functional interactions between specific MMP and TIMP types will likely yield novel and more specific strategies to regulate ECM proteolysis with LV remodeling.
One of the more exciting and perhaps disappointing areas regarding the regulation of ECM proteolysis has been in terms of post-translational control of MMP activity through pharmacological MMP inhibition. Pre-clinical models of broad spectrum, or “MMP-1 sparing” MMP inhibitors, uniformly demonstrated beneficial effects in terms affecting early adverse LV remodeling, particularly in the context of post-MI remodeling.21,23,25,48–50,53,54 In addition to providing further evidence for a cause-effect between MMP activation and adverse LV remodeling, these studies provided the impetus for moving to clinical feasibility. Early clinical trials of pharmacological MMP inhibitors in cancer patients were plagued by adverse systemic effects, notably of a musculoskeletal origin.121,145 Furthermore, it was also difficult to determine effective dosing for MMP inhibitors as there was not a biological response variable that could be easily measured.121,145 Nevertheless, a clinical trial of MMP inhibition in post-MI patients (Selective Matrix Metalloproteinase Inhibitor to Prevent Ventricular Remodeling After Myocardial Infarction (Prevention of Myocardial Infarction Early Remodeling-PREMIER)) was undertaken.122 The primary endpoint was that of changes in LV volumes at 6 months post-MI, whereby patients were randomized to a single dosing regimen of an “MMP-1 sparing” inhibitor or placebo. However, the dosing regimen was altered during the course of this study due to concerns regarding potential musculoskeletal effects. As such, a simulation of the pharmacokinetics of this specific MMP inhibitor and the dosing strategy used in the PREMIER study would suggest that a minimum concentration necessary to provide MMP inhibition for a majority of MMP types was not realized.121,146 Moreover, the overall net change in the primary response variable, LV end-diastolic volume, was surprisingly low compared to the majority of other post-MI studies (~10%), and thereby would attenuate the ability to detect a treatment effect. For these reasons, it was not surprising that the results of this initial MMP inhibition study were equivocal. However, due to the clear cause-effect relation between MMP mediated ECM proteolysis and the adverse tissue remodeling process in a number of pathological conditions, there is a resurgence of alternative and more targeted MMP inhibitors on the horizon.150–153 This is outlined in the following Future Directions section.
Future Directions in MMP Translational Research and Myocardial Remodeling
Diagnostics
One of the initially unsuspected outcomes from MMP translational research was the ability to measure MMPs from a peripheral blood sample.39–44,56,108,112–114 While the sources and origins of changes in peripheral levels of MMPs can be multifactorial, clinical studies have provided evidence that profiling MMPs as well as TIMPs can hold diagnostic and prognostic significance in the context of HF.44,112 As basic science studies on the biological roles of specific MMP types continues, it is likely that a refined set of MMP measurements from a peripheral blood sample would be useful biomarkers for identifying those patients at risk for more rapid and progressive LV remodeling and the development of HF. In addition, it may be possible to utilize these MMP/TIMP profiles in terms of a surrogate marker for assessing and developing dosing regimens. A more direct method for quantifying MMP activity and expression with respect to the LV remodeling process is through imaging.47,52,153 The increased spatial resolution of imaging systems in general, coupled with molecular probes such as an MMP specific imaging agent, is possible to directly identify areas of increased MMP activity with LV remodeling. For example, initial studies using a radiolabeled MMP tracer in a murine model of MI demonstrated specific and focal uptake within the MI and border zones.47 These initial studies were advanced to large animals whereby dual SPECT and CT imaging could be performed and thereby allow for spatial registration of LV anatomy and MMP activity with MI.52 Representative dual isotope SPECT/CT imaging in a large animal model of MI is shown in Figure 3. These imaging approaches will likely further our understanding of the regional and temporal aspects of MMP induction in the context of LV remodeling and allow for serial translational studies to be performed as it obviates the need for myocardial sampling.
Figure 3.

Dual isotope hybrid SPECT/CT imaging obtained using Thallium-201 (201Tl) and a technetium-99m labeled MMP targeted tracer (99mTc-RP805) in a canine model of MI induced by balloon coronary occlusion. The imaging approaches and validation have been reported previously.52 LV myocardial perfusion by 201TI is designated as green, and MMP radiotracer uptake is designated as red. Significant MMP radiotracer uptake could be observed within the LV myocardium, indicative of MMP proteolytic activity, at 3 and 14 days post-MI. Moreover, MMP activity appears to occur within normally perfused LV regions at later post-MI time points. (Images courtesy of Dr. Albert Sinusas, Professor of Medicine and Diagnostic Radiology Director, Cardiovascular Imaging Director, Yale Translational Research Imaging Center, Yale University School of Medicine).
Therapeutics
While broad-based pharmacological MMP inhibitors yielded problematic issues surrounding dosing efficacy as well as significant potential for systemic effects, important research directions in this area would involve more highly selective inhibitors. For example, the in-vivo use of a selective MMP-14 inhibitory antibody has been described.150 Another approach is to interfere with MMP substrate binding, rather than directly inhibiting the zinc-dependent catalytic domain.148 Finally, tetracycline derivatives remain a feasible possibility for MMP inhibition and have been advanced clinically in terms of preventing the MMP mediated tissue remodeling in gingivitis.145,147 Another potential approach is through the use of cell based, or more targeted therapies, which would obviate the need for systemic delivery. For example, implantation of TIMP transfected cells altered the time course of post-MI remodeling in rats.149 Other applications include MMP gene transfer through regional placement of biodegradable materials.151,152 While direct targeting of MMP induction and activation remains a potential area for relevant translational research, it may also be possible to exploit this family of proteases in other relevant therapeutic areas. For example, MMP mediated proteolysis of the ECM may be relevant in terms of the microenvironment and matrix scaffolding for stem cell engraftment.154,155
This review was based upon the premise that MMPs act upon structural and/or biological signaling molecules within the ECM. However, there is evidence for intracellular actions of MMPs that hold relevance to LV remodeling and the progression to HF.35,146,147,156 For example, MMP-2 has been localized to cardiocyte mitochondria and was inducible by oxidative stress. Moreover, this past study identified different MMP-2 isoforms existing within the intracellular matrix as opposed to the ECM.156 Thus, not only is there an ever growing number of MMP types that are expressed within the myocardial with LV remodeling, but there may also be isoforms for each of these MMP types. These discrete MMP expression-localization patterns would hold relevance in terms of substrate processing, whereby a number of intracellular substrates, both of the cytoskeletal and contractile protein domains, have been identified by degradomic profiling.65–69,120 Based upon the ever increasing substrates and diversity of biological actions of MMPs, it is likely that continued research regarding the relationship of LV remodeling to this family of proteases will yield new insights into the ECM remodeling process itself, as well as new therapeutic targets.
Acknowledgments
This work was supported by the National Institute of Health grants HL057952, HL089944, HL095608. Drs. Spinale and Zile are supported by the Research Service of the Department of Veterans Affairs (5I01BX000168-05, SURG-001-07F, 5101CX000415-02, and 5101BX000487-04, respectively). The authors wish to acknowledge Dr. Robert Price, USC School of Medicine, for providing the immunofluorescent images shown in Figure 1; Shaun Riffle and Craig P. Novack, USC School of Medicine, for the illustration used as part of Figure 2; and Dr. Albert Sinusas for supplying the MMP imaging study used in Figure 3. The authors wish to express appreciation to Ashley A. Sapp, USC School of Medicine, for editorial assistance.
Table of Abbreviations
- HF
heart failure
- LV
left ventricular
- EMMPRIN
extracellular matrix metalloproteinase inducer protein
- ECM
extracellular matrix
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- miR
microRNA
- TNF
tumor necrosis factor
- TGF
transforming growth factor
- ADAMs
a disintegrin and metalloproteinases
- ADAMTS
ADAMs with thrombospondin motifs
- SPECT
single photon emission computed tomography
- CT
cine x-ray computed tomography
Bibliography and References Cited
- 1.Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. 2011 Jan;4(1):98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Quiñones MA, Greenberg BH, Kopelen HA, Koilpillai C, Limacher MC, Shindler DM, Shelton BJ, Weiner DH. Echocardiographic predictors of clinical outcome in patients with left ventricular dysfunction enrolled in the SOLVD registry and trials: significance of left ventricular hypertrophy. Studies of Left Ventricular Dysfunction. J Am Coll Cardiol. 2000 Apr;35(5):1237–44. doi: 10.1016/s0735-1097(00)00511-8. [DOI] [PubMed] [Google Scholar]
- 3.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987 Jul;76(1):44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 4.Weir RA, McMurray JJ, Velazquez EJ. Epidemiology of heart failure and left ventricular systolic dysfunction after acute myocardial infarction: prevalence, clinical characteristics, and prognostic importance. Am J Cardiol. 2006 May 22;97(10A):13F–25F. doi: 10.1016/j.amjcard.2006.03.005. Epub 2006 Apr 21. [DOI] [PubMed] [Google Scholar]
- 5.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000 Jun 27;101(25):2981–8. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Vita J, Ruiz-Ortega M, Rupérez M, Esteban V, Sanchez-López E, Plaza JJ, Egido J. Endothelin-1, via ETA receptor and independently of transforming growth factor-beta, increases the connective tissue growth factor in vascular smooth muscle cells. Circ Res. 2005 Jul 22;97(2):125–34. doi: 10.1161/01.RES.0000174614.74469.83. Epub 2005 Jun 23. [DOI] [PubMed] [Google Scholar]
- 7.Filion RJ, Popel AS. Intracoronary administration of FGF-2: a computational model of myocardial deposition and retention. Am J Physiol Heart Circ Physiol. 2005 Jan;288(1):H263–79. doi: 10.1152/ajpheart.00205.2004. Epub 2004 Aug 26. [DOI] [PubMed] [Google Scholar]
- 8.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004 Dec 10;95(12):1167–73. doi: 10.1161/01.RES.0000150369.68826.2f. Epub 2004 Nov 11. [DOI] [PubMed] [Google Scholar]
- 9.López B, González A, Querejeta R, Larman M, Díez J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol. 2006 Jul 4;48(1):89–96. doi: 10.1016/j.jacc.2006.01.077. Epub 2006 Jun 12. [DOI] [PubMed] [Google Scholar]
- 10.Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007 Feb 20;115(7):888–95. doi: 10.1161/CIRCULATIONAHA.106.638569. Epub 2007 Feb 5. [DOI] [PubMed] [Google Scholar]
- 11.Fomovsky GM, Thomopoulos S, Holmes JW. Contribution of extracellular matrix to the mechanical properties of the heart. J Mol Cell Cardiol. 2010 Mar;48(3):490–6. doi: 10.1016/j.yjmcc.2009.08.003. Epub 2009 Aug 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacKenna DA, Vaplon SM, McCulloch AD. Microstructural model of perimysial collagen fibers for resting myocardial mechanics during ventricular filling. Am J Physiol. 1997 Sep;273(3 Pt 2):H1576–86. doi: 10.1152/ajpheart.1997.273.3.H1576. [DOI] [PubMed] [Google Scholar]
- 13.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002 Mar 8;90(4):458–64. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 14.Keller RS, Shai SY, Babbitt CJ, Pham CG, Solaro RJ, Valencik ML, Loftus JC, Ross RS. Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. Am J Pathol. 2001 Mar;158(3):1079–90. doi: 10.1016/S0002-9440(10)64055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kresh JY, Chopra A. Intercellular and extracellular mechanotransduction in cardiac myocytes. Pflugers Arch. 2011 Jul;462(1):75–87. doi: 10.1007/s00424-011-0954-1. Epub 2011 Mar 25. [DOI] [PubMed] [Google Scholar]
- 16.Stroud JD, Baicu CF, Barnes MA, Spinale FG, Zile MR. Viscoelastic properties of pressure overload hypertrophied myocardium: effect of serine protease treatment. Am J Physiol Heart Circ Physiol. 2002 Jun;282(6):H2324–35. doi: 10.1152/ajpheart.00711.2001. [DOI] [PubMed] [Google Scholar]
- 17.Abrahams C, Janicki JS, Weber KT. Myocardial hypertrophy in Macaca fascicularis. Structural remodeling of the collagen matrix. Lab Invest. 1987 Jun;56(6):676–83. [PubMed] [Google Scholar]
- 18.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995 Aug;147(2):325–38. [PMC free article] [PubMed] [Google Scholar]
- 19.Weber KT, Anversa P, Armstrong PW, Brilla CG, Burnett JC, Jr, Cruickshank JM, Devereux RB, Giles TD, Korsgaard N, Leier CV, et al. Remodeling and reparation of the cardiovascular system. J Am Coll Cardiol. 1992 Jul;20(1):3–16. doi: 10.1016/0735-1097(92)90130-f. [DOI] [PubMed] [Google Scholar]
- 20.Gajarsa JJ, Kloner RA. Left ventricular remodeling in the post-infarction heart: a review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev. 2011 Jan;16(1):13–21. doi: 10.1007/s10741-010-9181-7. [DOI] [PubMed] [Google Scholar]
- 21.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005 Mar;115(3):599–609. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000 Jul;106(1):55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999 Oct;5(10):1135–42. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 24.Spinale FG, Escobar GP, Mukherjee R, Zavadzkas JA, Saunders SM, Jeffords LB, Leone AM, Beck C, Bouges S, Stroud RE. Cardiac-restricted overexpression of membrane type-1 matrix metalloproteinase in mice: effects on myocardial remodeling with aging. Circ Heart Fail. 2009 Jul;2(4):351–60. doi: 10.1161/CIRCHEARTFAILURE.108.844845. Epub 2009 May 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikonomidis JS, Hendrick JW, Parkhurst AM, Herron AR, Escobar PG, Dowdy KB, Stroud RE, Hapke E, Zile MR, Spinale FG. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol. 2005 Jan;288(1):H149–58. doi: 10.1152/ajpheart.00370.2004. [DOI] [PubMed] [Google Scholar]
- 26.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Jr, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006 Jan;290(1):H232–9. doi: 10.1152/ajpheart.00457.2005. Epub 2005 Aug 26. [DOI] [PubMed] [Google Scholar]
- 27.Lindsey ML, Escobar GP, Mukherjee R, Goshorn DK, Sheats NJ, Bruce JA, Mains IM, Hendrick JK, Hewett KW, Gourdie RG, Matrisian LM, Spinale FG. Matrix metalloproteinase-7 affects connexin-43 levels, electrical conduction, and survival after myocardial infarction. Circulation. 2006 Jun 27;113(25):2919–28. doi: 10.1161/CIRCULATIONAHA.106.612960. Epub 2006 Jun 12. [DOI] [PubMed] [Google Scholar]
- 28.Mukherjee R, Mingoia JT, Bruce JA, Austin JS, Stroud RE, Escobar GP, McClister DM, Jr, Allen CM, Alfonso-Jaume MA, Fini ME, Lovett DH, Spinale FG. Selective spatiotemporal induction of matrix metalloproteinase-2 and matrix metalloproteinase-9 transcription after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006 Nov;291(5):H2216–28. doi: 10.1152/ajpheart.01343.2005. Epub 2006 Jun 9. [DOI] [PubMed] [Google Scholar]
- 29.Fedak PW, Smookler DS, Kassiri Z, Ohno N, Leco KJ, Verma S, Mickle DA, Watson KL, Hojilla CV, Cruz W, Weisel RD, Li RK, Khokha R. TIMP-3 deficiency leads to dilated cardiomyopathy. Circulation. 2004 Oct 19;110(16):2401–9. doi: 10.1161/01.CIR.0000134959.83967.2D. Epub 2004 Jul 19. [DOI] [PubMed] [Google Scholar]
- 30.Kassiri Z, Defamie V, Hariri M, Oudit GY, Anthwal S, Dawood F, Liu P, Khokha R. Simultaneous transforming growth factor beta-tumor necrosis factor activation and cross-talk cause aberrant remodeling response and myocardial fibrosis in Timp3-deficient heart. J Biol Chem. 2009 Oct 23;284(43):29893–904. doi: 10.1074/jbc.M109.028449. Epub 2009 Jul 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zile MR, Baicu CF, Stroud RE, Van Laer A, Arroyo J, Mukherjee R, Jones JA, Spinale FG. Pressure overload-dependent membrane type 1-matrix metalloproteinase induction: relationship to LV remodeling and fibrosis. Am J Physiol Heart Circ Physiol. 2012 Apr 1;302(7):H1429–37. doi: 10.1152/ajpheart.00580.2011. Epub 2012 Jan 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spinale FG, Mukherjee R, Zavadzkas JA, Koval CN, Bouges S, Stroud RE, Dobrucki LW, Sinusas AJ. Cardiac restricted overexpression of membrane type-1 matrix metalloproteinase causes adverse myocardial remodeling following myocardial infarction. J Biol Chem. 2010 Sep 24;285(39):30316–27. doi: 10.1074/jbc.M110.158196. Epub 2010 Jul 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zavadzkas JA, Plyler RA, Bouges S, Koval CN, Rivers WT, Beck CU, Chang EI, Stroud RE, Mukherjee R, Spinale FG. Cardiac-restricted overexpression of extracellular matrix metalloproteinase inducer causes myocardial remodeling and dysfunction in aging mice. Am J Physiol Heart Circ Physiol. 2008 Oct;295(4):H1394–402. doi: 10.1152/ajpheart.00346.2008. Epub 2008 Aug 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foronjy RF, Sun J, Lemaitre V, D’Armiento JM. Transgenic expression of matrix metalloproteinase-1 inhibits myocardial fibrosis and prevents the transition to heart failure in a pressure overload mouse model. Hypertens Res. 2008 Apr;31(4):725–35. doi: 10.1291/hypres.31.725. [DOI] [PubMed] [Google Scholar]
- 35.Wang GY, Bergman MR, Nguyen AP, Turcato S, Swigart PM, Rodrigo MC, Simpson PC, Karliner JS, Lovett DH, Baker AJ. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc Res. 2006 Feb 15;69(3):688–96. doi: 10.1016/j.cardiores.2005.08.023. Epub 2005 Sep 23. [DOI] [PubMed] [Google Scholar]
- 36.Janssens S, Lijnen HR. What has been learned about the cardiovascular effects of matrix metalloproteinases from mouse models? Cardiovasc Res. 2006 Feb 15;69(3):585–94. doi: 10.1016/j.cardiores.2005.12.010. Epub 2006 Jan 19. [DOI] [PubMed] [Google Scholar]
- 37.Díez J, Laviades C, Mayor G, Gil MJ, Monreal I. Increased serum concentrations of procollagen peptides in essential hypertension. Relation to cardiac alterations. Circulation. 1995 Mar 1;91(5):1450–6. doi: 10.1161/01.cir.91.5.1450. [DOI] [PubMed] [Google Scholar]
- 38.Heymans S, Schroen B, Vermeersch P, Milting H, Gao F, Kassner A, Gillijns H, Herijgers P, Flameng W, Carmeliet P, Van de Werf F, Pinto YM, Janssens S. Increased cardiac expression of tissue inhibitor of metalloproteinase-1 and tissue inhibitor of metalloproteinase-2 is related to cardiac fibrosis and dysfunction in the chronic pressure-overloaded human heart. Circulation. 2005 Aug 23;112(8):1136–44. doi: 10.1161/CIRCULATIONAHA.104.516963. Epub 2005 Aug 15. [DOI] [PubMed] [Google Scholar]
- 39.Lindsay MM, Maxwell P, Dunn FG. TIMP-1: a marker of left ventricular diastolic dysfunction and fibrosis in hypertension. Hypertension. 2002 Aug;40(2):136–41. doi: 10.1161/01.hyp.0000024573.17293.23. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006 May 2;113(17):2089–96. doi: 10.1161/CIRCULATIONAHA.105.573865. Epub 2006 Apr 24. [DOI] [PubMed] [Google Scholar]
- 41.Marchesi C, Dentali F, Nicolini E, Maresca AM, Tayebjee MH, Franz M, Guasti L, Venco A, Schiffrin EL, Lip GY, Grandi AM. Plasma levels of matrix metalloproteinases and their inhibitors in hypertension: a systematic review and meta-analysis. J Hypertens. 2012 Jan;30(1):3–16. doi: 10.1097/HJH.0b013e32834d249a. [DOI] [PubMed] [Google Scholar]
- 42.Mak GJ, Ledwidge MT, Watson CJ, Phelan DM, Dawkins IR, Murphy NF, Patle AK, Baugh JA, McDonald KM. Natural history of markers of collagen turnover in patients with early diastolic dysfunction and impact of eplerenone. J Am Coll Cardiol. 2009 Oct 27;54(18):1674–82. doi: 10.1016/j.jacc.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 43.Tayebjee MH, Nadar SK, MacFadyen RJ, Lip GY. Tissue inhibitor of metalloproteinase-1 and matrix metalloproteinase-9 levels in patients with hypertension Relationship to tissue Doppler indices of diastolic relaxation. Am J Hypertens. 2004 Sep;17(9):770–4. doi: 10.1016/j.amjhyper.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 44.Zile MR, Desantis SM, Baicu CF, Stroud RE, Thompson SB, McClure CD, Mehurg SM, Spinale FG. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ Heart Fail. 2011 May;4(3):246–56. doi: 10.1161/CIRCHEARTFAILURE.110.958199. Epub 2011 Feb 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gladka MM, da Costa Martins PA, De Windt LJ. Small changes can make a big difference - microRNA regulation of cardiac hypertrophy. J Mol Cell Cardiol. 2012 Jan;52(1):74–82. doi: 10.1016/j.yjmcc.2011.09.015. Epub 2011 Sep 24. [DOI] [PubMed] [Google Scholar]
- 46.Spinale FG, Coker ML, Heung LJ, Bond BR, Gunasinghe HR, Etoh T, Goldberg AT, Zellner JL, Crumbley AJ. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation. 2000 Oct 17;102(16):1944–9. doi: 10.1161/01.cir.102.16.1944. [DOI] [PubMed] [Google Scholar]
- 47.Su H, Spinale FG, Dobrucki LW, Song J, Hua J, Sweterlitsch S, Dione DP, Cavaliere P, Chow C, Bourke BN, Hu XY, Azure M, Yalamanchili P, Liu R, Cheesman EH, Robinson S, Edwards DS, Sinusas AJ. Noninvasive targeted imaging of matrix metalloproteinase activation in a murine model of postinfarction remodeling. Circulation. 2005 Nov 15;112(20):3157–67. doi: 10.1161/CIRCULATIONAHA.105.583021. Epub 2005 Nov 7. [DOI] [PubMed] [Google Scholar]
- 48.Yarbrough WM, Mukherjee R, Escobar GP, Mingoia JT, Sample JA, Hendrick JW, Dowdy KB, McLean JE, Lowry AS, O’Neill TP, Spinale FG. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation. 2003 Oct 7;108(14):1753–9. doi: 10.1161/01.CIR.0000091087.78630.79. Epub 2003 Sep 15. [DOI] [PubMed] [Google Scholar]
- 49.Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation. 2003 Feb 4;107(4):618–25. doi: 10.1161/01.cir.0000046449.36178.00. [DOI] [PubMed] [Google Scholar]
- 50.King MK, Coker ML, Goldberg A, McElmurray JH, 3rd, Gunasinghe HR, Mukherjee R, Zile MR, O’Neill TP, Spinale FG. Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res. 2003 Feb 7;92(2):177–85. doi: 10.1161/01.res.0000052312.41419.55. [DOI] [PubMed] [Google Scholar]
- 51.Li J, Schwimmbeck PL, Tschope C, Leschka S, Husmann L, Rutschow S, Reichenbach F, Noutsias M, Kobalz U, Poller W, Spillmann F, Zeichhardt H, Schultheiss HP, Pauschinger M. Collagen degradation in a murine myocarditis model: relevance of matrix metalloproteinase in association with inflammatory induction. Cardiovasc Res. 2002 Nov;56(2):235–47. doi: 10.1016/s0008-6363(02)00546-1. [DOI] [PubMed] [Google Scholar]
- 52.Sahul ZH, Mukherjee R, Song J, McAteer J, Stroud RE, Dione DP, Staib L, Papademetris X, Dobrucki LW, Duncan JS, Spinale FG, Sinusas AJ. Targeted imaging of the spatial and temporal variation of matrix metalloproteinase activity in a porcine model of postinfarct remodeling: relationship to myocardial dysfunction. Circ Cardiovasc Imaging. 2011 Jul;4(4):381–91. doi: 10.1161/CIRCIMAGING.110.961854. Epub 2011 Apr 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, McClure KF, Mitchell PG, Libby P, Lee RT. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999 Jun 15;99(23):3063–70. doi: 10.1161/01.cir.99.23.3063. [DOI] [PubMed] [Google Scholar]
- 54.Chancey AL, Brower GL, Peterson JT, Janicki JS. Effects of matrix metalloproteinase inhibition on ventricular remodeling due to volume overload. Circulation. 2002 Apr 23;105(16):1983–8. doi: 10.1161/01.cir.0000014686.73212.da. [DOI] [PubMed] [Google Scholar]
- 55.Spinale FG, Coker ML, Thomas CV, Walker JD, Mukherjee R, Hebbar L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res. 1998 Mar 9;82(4):482–95. doi: 10.1161/01.res.82.4.482. [DOI] [PubMed] [Google Scholar]
- 56.Webb CS, Bonnema DD, Ahmed SH, Leonardi AH, McClure CD, Clark LL, Stroud RE, Corn WC, Finklea L, Zile MR, Spinale FG. Specific temporal profile of matrix metalloproteinase release occurs in patients after myocardial infarction: relation to left ventricular remodeling. Circulation. 2006 Sep 5;114(10):1020–7. doi: 10.1161/CIRCULATIONAHA.105.600353. Epub 2006 Aug 21. [DOI] [PubMed] [Google Scholar]
- 57.Multani MM, Ikonomidis JS, Kim PY, Miller EA, Payne KJ, Mukherjee R, Dorman BH, Spinale FG. Dynamic and differential changes in myocardial and plasma endothelin in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2005 Mar;129(3):584–90. doi: 10.1016/j.jtcvs.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 58.Zile MR, Mehurg SM, Arroyo JE, Stroud RE, DeSantis SM, Spinale FG. Relationship between the temporal profile of plasma microRNA and left ventricular remodeling in patients after myocardial infarction. Circ Cardiovasc Genet. 2011 Dec;4(6):614–9. doi: 10.1161/CIRCGENETICS.111.959841. Epub 2011 Sep 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Rooij E, Olson EN. Searching for miR-acles in cardiac fibrosis. Circ Res. 2009 Jan 30;104(2):138–40. doi: 10.1161/CIRCRESAHA.108.192492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008 Sep 2;105(35):13027–32. doi: 10.1073/pnas.0805038105. Epub 2008 Aug 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009 Jan 30;104(2):170–8. doi: 10.1161/CIRCRESAHA.108.182535. 6p following 178. Epub 2008 Dec 18. [DOI] [PubMed] [Google Scholar]
- 62.Jenniskens GJ, Veerkamp JH, van Kuppevelt TH. Heparan sulfates in skeletal muscle development and physiology. J Cell Physiol. 2006 Feb;206(2):283–94. doi: 10.1002/jcp.20450. [DOI] [PubMed] [Google Scholar]
- 63.Gillies AR, Lieber RL. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve. 2011 Sep;44(3):318–31. doi: 10.1002/mus.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Widgerow AD. Cellular/extracellular matrix cross-talk in scar evolution and control. Wound Repair Regen. 2011 Mar-Apr;19(2):117–33. doi: 10.1111/j.1524-475X.2010.00662.x. [DOI] [PubMed] [Google Scholar]
- 65.Butler GS, Overall CM. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry. 2009 Nov 24;48(46):10830–45. doi: 10.1021/bi901656f. [DOI] [PubMed] [Google Scholar]
- 66.Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010 Jan;1803(1):39–54. doi: 10.1016/j.bbamcr.2009.09.015. Epub 2009 Oct 1. [DOI] [PubMed] [Google Scholar]
- 67.Gao YA, Agnihotri R, Vary CP, Liaw L. Expression and characterization of recombinant osteopontin peptides representing matrix metalloproteinase proteolytic fragments. Matrix Biol. 2004 Nov;23(7):457–66. doi: 10.1016/j.matbio.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Maurer P, Göhring W, Sasaki T, Mann K, Timpl R, Nischt R. Recombinant and tissue-derived mouse BM-40 bind to several collagen types and have increased affinities after proteolytic activation. Cell Mol Life Sci. 1997 May;53(5):478–84. doi: 10.1007/s000180050059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Symoens S, Renard M, Bonod-Bidaud C, Syx D, Vaganay E, Malfait F, Ricard-Blum S, Kessler E, Van Laer L, Coucke P, Ruggiero F, De Paepe A. Identification of binding partners interacting with the α1-N-propeptide of type V collagen. Biochem J. 2011 Jan 15;433(2):371–81. doi: 10.1042/BJ20101061. [DOI] [PubMed] [Google Scholar]
- 70.Borg TK, Ranson WF, Moslehy FA, Caulfield JB. Structural basis of ventricular stiffness. Lab Invest. 1981 Jan;44(1):49–54. [PubMed] [Google Scholar]
- 71.Weidemann F, Herrmann S, Störk S, Niemann M, Frantz S, Lange V, Beer M, Gattenlöhner S, Voelker W, Ertl G, Strotmann JM. Impact of myocardial fibrosis in patients with symptomatic severe aortic stenosis. Circulation. 2009 Aug 18;120(7):577–84. doi: 10.1161/CIRCULATIONAHA.108.847772. Epub 2009 Aug 3. [DOI] [PubMed] [Google Scholar]
- 72.Krayenbuehl HP, Hess OM, Monrad ES, Schneider J, Mall G, Turina M. Left ventricular myocardial structure in aortic valve disease before, intermediate, and late after aortic valve replacement. Circulation. 1989 Apr;79(4):744–55. doi: 10.1161/01.cir.79.4.744. [DOI] [PubMed] [Google Scholar]
- 73.Bing OH, Fanburg BL, Brooks WW, Matsushita S. The effect of lathyrogen beta-amino proprionitrile (BAPN) on the mechanical properties of experimentally hypertrophied rat cardiac muscle. Circ Res. 1978 Oct;43(4):632–7. doi: 10.1161/01.res.43.4.632. [DOI] [PubMed] [Google Scholar]
- 74.Kato S, Spinale FG, Tanaka R, Johnson W, Cooper G, 4th, Zile MR. Inhibition of collagen cross-linking: effects on fibrillar collagen and ventricular diastolic function. Am J Physiol. 1995 Sep;269(3 Pt 2):H863–8. doi: 10.1152/ajpheart.1995.269.3.H863. [DOI] [PubMed] [Google Scholar]
- 75.Manso AM, Elsherif L, Kang SM, Ross RS. Integrins, membrane-type matrix metalloproteinases and ADAMs: potential implications for cardiac remodeling. Cardiovasc Res. 2006 Feb 15;69(3):574–84. doi: 10.1016/j.cardiores.2005.09.004. Epub 2005 Oct 25. [DOI] [PubMed] [Google Scholar]
- 76.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008 Dec;8(12):929–41. doi: 10.1038/nrc2459. Epub 2008 Nov 13. [DOI] [PubMed] [Google Scholar]
- 77.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010 Jun 11;106(11):1675–80. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 78.Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem. 2002 Jun 14;277(24):21352–60. doi: 10.1074/jbc.M111663200. Epub 2002 Apr 2. [DOI] [PubMed] [Google Scholar]
- 79.Tatti O, Vehviläinen P, Lehti K, Keski-Oja J. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp Cell Res. 2008 Aug 1;314(13):2501–14. doi: 10.1016/j.yexcr.2008.05.018. Epub 2008 Jun 6. [DOI] [PubMed] [Google Scholar]
- 80.Hawinkels LJ, Kuiper P, Wiercinska E, Verspaget HW, Liu Z, Pardali E, Sier CF, ten Dijke P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010 May 15;70(10):4141–50. doi: 10.1158/0008-5472.CAN-09-4466. Epub 2010 Apr 27. [DOI] [PubMed] [Google Scholar]
- 81.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011 Jun;121(6):2301–12. doi: 10.1172/JCI44824. Epub 2011 May 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diwan A, Dibbs Z, Nemoto S, DeFreitas G, Carabello BA, Sivasubramanian N, Wilson EM, Spinale FG, Mann DL. Targeted overexpression of noncleavable and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes. Circulation. 2004 Jan 20;109(2):262–8. doi: 10.1161/01.CIR.0000109642.27985.FA. Epub 2003 Dec 29. [DOI] [PubMed] [Google Scholar]
- 83.Chen X, Liang H, Zhang J, Zen K, Zhang CY. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012 Mar;22(3):125–32. doi: 10.1016/j.tcb.2011.12.001. Epub 2012 Jan 17. [DOI] [PubMed] [Google Scholar]
- 84.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007 Jun;9(6):654–9. doi: 10.1038/ncb1596. Epub 2007 May 7. [DOI] [PubMed] [Google Scholar]
- 85.Alfranca A, López-Oliva JM, Genís L, López-Maderuelo D, Mirones I, Salvado D, Quesada AJ, Arroyo AG, Redondo JM. PGE2 induces angiogenesis via MT1-MMP-mediated activation of the TGFbeta/Alk5 signaling pathway. Blood. 2008 Aug 15;112(4):1120–8. doi: 10.1182/blood-2007-09-112268. Epub 2008 Jun 9. [DOI] [PubMed] [Google Scholar]
- 86.Strongin AY. Mislocalization and unconventional functions of cellular MMPs in cancer. Cancer Metastasis Rev. 2006 Mar;25(1):87–98. doi: 10.1007/s10555-006-7892-y. [DOI] [PubMed] [Google Scholar]
- 87.Gross J, Lapiere CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc Natl Acad Sci U S A. 1962 Jun 15;48:1014–22. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010 Apr 2;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2011 Jan;278(1):28–45. doi: 10.1111/j.1742-4658.2010.07920.x. Epub 2010 Nov 19. [DOI] [PubMed] [Google Scholar]
- 90.Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol. 2008;40(6–7):1362–78. doi: 10.1016/j.biocel.2007.12.006. Epub 2007 Dec 24. [DOI] [PubMed] [Google Scholar]
- 91.Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. 2010 Jun;20(3):161–8. doi: 10.1016/j.semcancer.2010.05.002. Epub 2010 May 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Polyakova V, Hein S, Kostin S, Ziegelhoeffer T, Schaper J. Matrix metalloproteinases and their tissue inhibitors in pressure-overloaded human myocardium during heart failure progression. J Am Coll Cardiol. 2004 Oct 19;44(8):1609–18. doi: 10.1016/j.jacc.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 93.Dai Q, Lin J, Craig T, Chou YM, Hinojosa-Laborde C, Lindsey ML. Estrogen effects on MMP-13 and MMP-14 regulation of left ventricular mass in Dahl salt-induced hypertension. Gend Med. 2008 Mar;5(1):74–85. doi: 10.1016/s1550-8579(08)80010-1. [DOI] [PubMed] [Google Scholar]
- 94.Gupta V, Grande-Allen KJ. Effects of static and cyclic loading in regulating extracellular matrix synthesis by cardiovascular cells. Cardiovasc Res. 2006 Dec 1;72(3):375–83. doi: 10.1016/j.cardiores.2006.08.017. Epub 2006 Sep 1. [DOI] [PubMed] [Google Scholar]
- 95.Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension. 2006 Apr;47(4):711–7. doi: 10.1161/01.HYP.0000208840.30778.00. Epub 2006 Feb 27. [DOI] [PubMed] [Google Scholar]
- 96.Woessner JF, Jr, Nagase H. Activation of the zymogen forms of MMPs. Matrix Metalloproteinases and TIMPs. 2000:72–86. [Google Scholar]
- 97.Ruddy JM, Jones JA, Stroud RE, Mukherjee R, Spinale FG, Ikonomidis JS. Differential effects of mechanical and biological stimuli on matrix metalloproteinase promoter activation in the thoracic aorta. Circulation. 2009 Sep 15;120(11 Suppl):S262–8. doi: 10.1161/CIRCULATIONAHA.108.843581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yarbrough WM, Mukherjee R, Stroud RE, Rivers WT, Oelsen JM, Dixon JA, Eckhouse SR, Ikonomidis JS, Zile MR, Spinale FG. Progressive induction of left ventricular pressure overload in a large animal model elicits myocardial remodeling and a unique matrix signature. J Thorac Cardiovasc Surg. 2012 Jan;143(1):215–23. doi: 10.1016/j.jtcvs.2011.09.032. Epub 2011 Nov 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nagatomo Y, Carabello BA, Coker ML, McDermott PJ, Nemoto S, Hamawaki M, Spinale FG. Differential effects of pressure or volume overload on myocardial MMP levels and inhibitory control. Am J Physiol Heart Circ Physiol. 2000 Jan;278(1):H151–61. doi: 10.1152/ajpheart.2000.278.1.H151. [DOI] [PubMed] [Google Scholar]
- 100.Stewart JA, Jr, Wei CC, Brower GL, Rynders PE, Hankes GH, Dillon AR, Lucchesi PA, Janicki JS, Dell’Italia LJ. Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003 Mar;35(3):311–9. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 101.Chancey AL, Gardner JD, Murray DB, Brower GL, Janicki JS. Modulation of cardiac mast cell-mediated extracellular matrix degradation by estrogen. Am J Physiol Heart Circ Physiol. 2005 Jul;289(1):H316–21. doi: 10.1152/ajpheart.00765.2004. Epub 2005 Feb 18. [DOI] [PubMed] [Google Scholar]
- 102.Gardner JD, Murray DB, Voloshenyuk TG, Brower GL, Bradley JM, Janicki JS. Estrogen attenuates chronic volume overload induced structural and functional remodeling in male rat hearts. Am J Physiol Heart Circ Physiol. 2010 Feb;298(2):H497–504. doi: 10.1152/ajpheart.00336.2009. Epub 2009 Nov 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hutchinson KR, Stewart JA, Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol. 2010 Mar;48(3):564–9. doi: 10.1016/j.yjmcc.2009.06.001. Epub 2009 Jun 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jobe LJ, Meléndez GC, Levick SP, Du Y, Brower GL, Janicki JS. TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol. 2009 Oct;297(4):H1462–8. doi: 10.1152/ajpheart.00442.2009. Epub 2009 Aug 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Levick SP, Gardner JD, Holland M, Hauer-Jensen M, Janicki JS, Brower GL. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J Mol Cell Cardiol. 2008 Jul;45(1):56–61. doi: 10.1016/j.yjmcc.2008.04.010. Epub 2008 May 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moshal KS, Rodriguez WE, Sen U, Tyagi SC. Targeted deletion of MMP-9 attenuates myocardial contractile dysfunction in heart failure. Physiol Res. 2008;57(3):379–84. doi: 10.33549/physiolres.931221. Epub 2007 May 30. [DOI] [PubMed] [Google Scholar]
- 107.Thomas CV, Coker ML, Zellner JL, Handy JR, Crumbley AJ, 3rd, Spinale FG. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation. 1998 May 5;97(17):1708–15. doi: 10.1161/01.cir.97.17.1708. [DOI] [PubMed] [Google Scholar]
- 108.Essa EM, Zile MR, Stroud RE, Rice A, Gumina RJ, Leier CV, Spinale FG. Changes in Plasma Profiles of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of MMPs in Stress-Induced Cardiomyopathy. J Card Fail. 2012 Jun;18(6):487–92. doi: 10.1016/j.cardfail.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Genomatix GmbH. Munich, Germany: DiAlign Professional Release 3.1.5. Estrogen Response Elements: Genomatix used on June 26, 2012. [Google Scholar]
- 110.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. SMAD and Elk-1 Transcription Factors: Sequence Alignment done with Clustal on July 9-10 2012. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 111.Elliot S, Catanuto P, Fernandez P, Espinosa-Heidmann D, Karl M, Korach K, Cousins SW. Subtype specific estrogen receptor action protects against changes in MMP-2 activation in mouse retinal pigmented epithelial cells. Exp Eye Res. 2008 Apr;86(4):653–60. doi: 10.1016/j.exer.2008.01.010. Epub 2008 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nilsson L, Hallén J, Atar D, Jonasson L, Swahn E. Early measurements of plasma matrix metalloproteinase-2 predict infarct size and ventricular dysfunction in ST-elevation myocardial infarction. Heart. 2012 Jan;98(1):31–6. doi: 10.1136/heartjnl-2011-300079. Epub 2011 Jul 4. [DOI] [PubMed] [Google Scholar]
- 113.Kelly D, Cockerill G, Ng LL, Thompson M, Khan S, Samani NJ, Squire IB. Plasma matrix metalloproteinase-9 and left ventricular remodelling after acute myocardial infarction in man: a prospective cohort study. Eur Heart J. 2007 Mar;28(6):711–8. doi: 10.1093/eurheartj/ehm003. Epub 2007 Mar 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kai H, Ikeda H, Yasukawa H, Kai M, Seki Y, Kuwahara F, Ueno T, Sugi K, Imaizumi T. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Cardiol. 1998 Aug;32(2):368–72. doi: 10.1016/s0735-1097(98)00250-2. [DOI] [PubMed] [Google Scholar]
- 115.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010 Mar;48(3):504–11. doi: 10.1016/j.yjmcc.2009.07.015. Epub 2009 Jul 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012 Jan 6;110(1):159–73. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nisticò P, Bissell MJ, Radisky DC. Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol. 2012 Feb 1;4(2) doi: 10.1101/cshperspect.a011908.. pii: a011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal. 2012 Feb;18(1):35–49. doi: 10.1017/S143192761101227X. Epub 2012 Jan 4. [DOI] [PubMed] [Google Scholar]
- 119.Wilson EM, Moainie SL, Baskin JM, Lowry AS, Deschamps AM, Mukherjee R, Guy TS, St John-Sutton MG, Gorman JH, 3rd, Edmunds LH, Jr, Gorman RC, Spinale FG. Region- and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling. Circulation. 2003 Jun 10;107(22):2857–63. doi: 10.1161/01.CIR.0000068375.40887.FA. Epub 2003 May 27. [DOI] [PubMed] [Google Scholar]
- 120.Chiao YA, Zamilpa R, Lopez EF, Dai Q, Escobar GP, Hakala K, Weintraub ST, Lindsey ML. In vivo matrix metalloproteinase-7 substrates identified in the left ventricle post-myocardial infarction using proteomics. J Proteome Res. 2010 May 7;9(5):2649–57. doi: 10.1021/pr100147r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Peterson JT. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc Res. 2006 Feb 15;69(3):677–87. doi: 10.1016/j.cardiores.2005.11.032. Epub 2006 Jan 17. [DOI] [PubMed] [Google Scholar]
- 122.Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, Lyon R, Quinones M, Theroux P, Sydlowski D, Kim HE, Garcia MJ, Jaber WA, Weaver WD. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol. 2006 Jul 4;48(1):15–20. doi: 10.1016/j.jacc.2006.02.055. Epub 2006 Jun 21. [DOI] [PubMed] [Google Scholar]
- 123.Hofmann UB, Westphal JR, Van Kraats AA, Ruiter DJ, Van Muijen GN. Expression of integrin alpha(v)beta(3) correlates with activation of membrane-type matrix metalloproteinase-1 (MT1-MMP) and matrix metalloproteinase-2 (MMP-2) in human melanoma cells in vitro and in vivo. Int J Cancer. 2000 Jul 1;87(1):12–9. doi: 10.1002/1097-0215(20000701)87:1<12::aid-ijc3>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 124.Davis GE. Matricryptic sites control tissue injury responses in the cardiovascular system: relationships to pattern recognition receptor regulated events. J Mol Cell Cardiol. 2010 Mar;48(3):454–60. doi: 10.1016/j.yjmcc.2009.09.002. Epub 2009 Sep 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ricard-Blum S, Ballut L. Matricryptins derived from collagens and proteoglycans. Front Biosci. 2011 Jan 1;16:674–97. doi: 10.2741/3712. [DOI] [PubMed] [Google Scholar]
- 126.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007 Aug;13(8):952–61. doi: 10.1038/nm1613. Epub 2007 Jul 29. [DOI] [PubMed] [Google Scholar]
- 127.Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol. 2011 Apr;57(4):376–9. doi: 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Spruill LS, Lowry AS, Stroud RE, Squires CE, Mains IM, Flack EC, Beck C, Ikonomidis JS, Crumbley AJ, McDermott PJ, Spinale FG. Membrane-type-1 matrix metalloproteinase transcription and translation in myocardial fibroblasts from patients with normal left ventricular function and from patients with cardiomyopathy. Am J Physiol Cell Physiol. 2007 Oct;293(4):C1362–73. doi: 10.1152/ajpcell.00545.2006. Epub 2007 Aug 1. [DOI] [PubMed] [Google Scholar]
- 129.Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, Wang Y. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol. 2011 Aug;301(2):H538–47. doi: 10.1152/ajpheart.01114.2010. Epub 2011 May 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carlson S, Trial J, Soeller C, Entman ML. Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc Res. 2011 Jul 1;91(1):99–107. doi: 10.1093/cvr/cvr061. Epub 2011 Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Benhamron S, Reiner I, Zcharia E, Atallah M, Grau A, Vlodavsky I, Mevorach D. Dissociation between mature phenotype and impaired transmigration in dendritic cells from heparanase-deficient mice. PLoS One. 2012;7(5):e35602. doi: 10.1371/journal.pone.0035602. Epub 2012 May 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zarrabi K, Dufour A, Li J, Kuscu C, Pulkoski-Gross A, Zhi J, Hu Y, Sampson NS, Zucker S, Cao J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem. 2011 Sep 23;286(38):33167–77. doi: 10.1074/jbc.M111.256644. Epub 2011 Jul 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012 Jan;347(1):177–86. doi: 10.1007/s00441-011-1222-6. Epub 2011 Aug 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deschamps AM, Apple KA, Leonardi AH, McLean JE, Yarbrough WM, Stroud RE, Clark LL, Sample JA, Spinale FG. Myocardial interstitial matrix metalloproteinase activity is altered by mechanical changes in LV load: interaction with the angiotensin type 1 receptor. Circ Res. 2005 May 27;96(10):1110–8. doi: 10.1161/01.RES.0000167830.12010.6b. Epub 2005 Apr 28. [DOI] [PubMed] [Google Scholar]
- 135.Sawicki G, Menon V, Jugdutt BI. Improved balance between TIMP-3 and MMP-9 after regional myocardial ischemia-reperfusion during AT1 receptor blockade. J Card Fail. 2004 Oct;10(5):442–9. doi: 10.1016/j.cardfail.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 136.Tanner RM, Lynch AI, Brophy VH, Eckfeldt JH, Davis BR, Ford CE, Boerwinkle E, Arnett DK. Pharmacogenetic associations of MMP9 and MMP12 variants with cardiovascular disease in patients with hypertension. PLoS One. 2011;6(8):e23609. doi: 10.1371/journal.pone.0023609. Epub 2011 Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Blankenberg S, Rupprecht HJ, Poirier O, Bickel C, Smieja M, Hafner G, Meyer J, Cambien F, Tiret L AtheroGene Investigators. Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation. 2003 Apr 1;107(12):1579–85. doi: 10.1161/01.CIR.0000058700.41738.12. [DOI] [PubMed] [Google Scholar]
- 138.Mizon-Gérard F, de Groote P, Lamblin N, Hermant X, Dallongeville J, Amouyel P, Bauters C, Helbecque N. Prognostic impact of matrix metalloproteinase gene polymorphisms in patients with heart failure according to the aetiology of left ventricular systolic dysfunction. Eur Heart J. 2004 Apr;25(8):688–93. doi: 10.1016/j.ehj.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 139.Toole BP. Emmprin (CD147), a cell surface regulator of matrix metalloproteinase production and function. Curr Top Dev Biol. 2003;54:371–89. doi: 10.1016/s0070-2153(03)54015-7. [DOI] [PubMed] [Google Scholar]
- 140.Schmidt R, Bültmann A, Ungerer M, Joghetaei N, Bülbül O, Thieme S, Chavakis T, Toole BP, Gawaz M, Schömig A, May AE. Extracellular matrix metalloproteinase inducer regulates matrix metalloproteinase activity in cardiovascular cells: implications in acute myocardial infarction. Circulation. 2006 Feb 14;113(6):834–41. doi: 10.1161/CIRCULATIONAHA.105.568162. Epub 2006 Feb 6. [DOI] [PubMed] [Google Scholar]
- 141.Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW., 2nd MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010 Jan 8;106(1):166–75. doi: 10.1161/CIRCRESAHA.109.202176. Epub 2009 Nov 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jones JA, Stroud RE, O’Quinn EC, Black LE, Barth JL, Elefteriades JA, Bavaria JE, Gorman JH, 3rd, Gorman RC, Spinale FG, Ikonomidis JS. Selective microRNA suppression in human thoracic aneurysms: relationship of miR-29a to aortic size and proteolytic induction. Circ Cardiovasc Genet. 2011 Dec;4(6):605–13. doi: 10.1161/CIRCGENETICS.111.960419. Epub 2011 Oct 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008 Jul 8;1(27):re6. doi: 10.1126/scisignal.127re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kandalam V, Basu R, Moore L, Fan D, Wang X, Jaworski DM, Oudit GY, Kassiri Z. Lack of tissue inhibitor of metalloproteinases 2 leads to exacerbated left ventricular dysfunction and adverse extracellular matrix remodeling in response to biomechanical stress. Circulation. 2011 Nov 8;124(19):2094–105. doi: 10.1161/CIRCULATIONAHA.111.030338. Epub 2011 Oct 10. [DOI] [PubMed] [Google Scholar]
- 145.Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002 Sep;2(9):657–72. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 146.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007 Oct;87(4):1285–342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 147.Castro MM, Kandasamy AD, Youssef N, Schulz R. Matrix metalloproteinase inhibitor properties of tetracyclines: therapeutic potential in cardiovascular diseases. Pharmacol Res. 2011 Dec;64(6):551–60. doi: 10.1016/j.phrs.2011.05.005. Epub 2011 May 31. [DOI] [PubMed] [Google Scholar]
- 148.Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta. 2010 Jan;1803(1):72–94. doi: 10.1016/j.bbamcr.2009.08.006. Epub 2009 Aug 25. [DOI] [PubMed] [Google Scholar]
- 149.Tian H, Huang ML, Liu KY, Jia ZB, Sun L, Jiang SL, Liu W, Kinkaid HY, Wu J, Li RK. Inhibiting matrix metalloproteinase by cell-based TIMP-3 gene transfer effectively treats acute and chronic ischemic cardiomyopathy. Cell Transplant. 2011 Sep 23; doi: 10.3727/096368911X601000. [DOI] [PubMed] [Google Scholar]
- 150.Devy L, Huang L, Naa L, Yanamandra N, Pieters H, Frans N, Chang E, Tao Q, Vanhove M, Lejeune A, van Gool R, Sexton DJ, Kuang G, Rank D, Hogan S, Pazmany C, Ma YL, Schoonbroodt S, Nixon AE, Ladner RC, Hoet R, Henderikx P, Tenhoor C, Rabbani SA, Valentino ML, Wood CR, Dransfield DT. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009 Feb 15;69(4):1517–26. doi: 10.1158/0008-5472.CAN-08-3255. Epub 2009 Feb 10. [DOI] [PubMed] [Google Scholar]
- 151.Gojgini S, Tokatlian T, Segura T. Utilizing cell-matrix interactions to modulate gene transfer to stem cells inside hyaluronic acid hydrogels. Mol Pharm. 2011 Oct 3;8(5):1582–91. doi: 10.1021/mp200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yamawaki-Ogata A, Hashizume R, Satake M, Kaneko H, Mizutani S, Moritan T, Ueda Y, Narita Y. A doxycycline loaded, controlled-release, biodegradable fiber for the treatment of aortic aneurysms. Biomaterials. 2010 Dec;31(36):9554–64. doi: 10.1016/j.biomaterials.2010.08.069. [DOI] [PubMed] [Google Scholar]
- 153.Wagner S, Breyholz HJ, Faust A, Höltke C, Levkau B, Schober O, Schäfers M, Kopka K. Molecular imaging of matrix metalloproteinases in vivo using small molecule inhibitors for SPECT and PET. Curr Med Chem. 2006;13(23):2819–38. doi: 10.2174/092986706778522002. [DOI] [PubMed] [Google Scholar]
- 154.Wall ST, Yeh CC, Tu RY, Mann MJ, Healy KE. Biomimetic matrices for myocardial stabilization and stem cell transplantation. J Biomed Mater Res A. 2010 Dec 15;95(4):1055–66. doi: 10.1002/jbm.a.32904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Godier-Furnémont AF, Martens TP, Koeckert MS, Wan L, Parks J, Arai K, Zhang G, Hudson B, Homma S, Vunjak-Novakovic G. Composite scaffold provides a cell delivery platform for cardiovascular repair. Proc Natl Acad Sci U S A. 2011 May 10;108(19):7974–9. doi: 10.1073/pnas.1104619108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lovett DH, Mahimkar R, Raffai RL, Cape L, Maklashina E, Cecchini G, Karliner JS. A novel intracellular isoform of matrix metalloproteinase-2 induced by oxidative stress activates innate immunity. PLoS One. 2012;7(4):e34177. doi: 10.1371/journal.pone.0034177. Epub 2012 Apr 3. [DOI] [PMC free article] [PubMed] [Google Scholar]