

Abstract
Interpretation of high throughput screening (HTS) data in cell-based assays may be confounded by cytotoxic properties of screening compounds. Therefore, assessing cell toxicity in real time during the HTS process itself would be highly advantageous. Here, we investigate the potential of putatively impermeant, fluorescent, DNA-binding dyes to give cell toxicity readout during HTS. Amongst 19 DNA-binding dyes examined, three classes were identified that were (1) permeant, (2) cytotoxic, or (3) neither permeant nor cytotoxic during 3-day incubation with a macrophage cell line. In the last class, four dyes (SYTOX Green, CellTox Green, GelGreen, and EvaGreen) gave highly robust cytotoxicity data in 384-well screening plates. As proof of principle, successful combination with a luminescence-based assay in HTS format was demonstrated. Here, both intracellular growth of Legionella pneumophila (luminescence) and host cell viability (SYTOX Green exclusion) were assayed in the same screening well. Incorporation of membrane-impermeant, DNA-binding, fluorescent dyes in HTS assays should prove useful by allowing evaluation of cytotoxicity in real time, eliminating reagent addition steps and effort associated with endpoint cell viability analysis, and reducing the need for follow-up cytotoxicity screening.
Introduction
Traditionally, eukaryotic cell toxicity is measured in an “endpoint” assay. Specifically, a reagent is added after a defined time period to measure a metabolic or physical property. Classic examples include measurement of lactate dehydrogenase release through addition of substrate, cellular ATP through use of ATP-dependent luminescent chemistry, and permeability to membrane-impermeant dyes such as propidium iodide.1 These assays have proven extremely useful. However, their amenability to a high throughput format is suboptimal. Specifically, as endpoint assays, they require several additional steps to be performed at the end of the screening process. Many cytotoxicity assays also have a time-constrained readout; for example, measurement of an enzyme-dependent color reaction that varies with time. Therefore, multiplied over the large number of screening plates in high throughput screening (HTS), the additional effort required for these endpoint assays may prove considerable or impractical.2–4
Accordingly, the ability to add cytotoxicity test reagents during assay setup, akin to inclusion of fluorescent probes in real-time polymerase chain reaction (PCR) assays,5–7 would confer great advantage. Impermeant DNA-binding dyes from a theoretical standpoint would seem ideal for this purpose. These dyes enter and bind to nuclear DNA as dying eukaryotic cells lose membrane permeability. Several demonstrate little to no solution fluorescence and greatly increased quantum yield when bound to DNA.8,9 Therefore, fluorescent signal correlates with loss of eukaryotic cell viability and can be normalized to positive (i.e., permeabilized) and negative controls to give a fractional measure of cell death or cell viability.
The use of DNA-binding dyes in this way will depend on satisfying several conditions: first, that the dyes remain nonpermeable for an extended period of time; second, that the dyes themselves are nontoxic; and third, that dyes have spectral characteristics that allow efficient detection in available high throughput microplate readers. We therefore sought to examine a large number of commercially available, impermeant, DNA-binding dyes to assess suitability for use as real-time HTS cytotoxicity probes.
Materials and Methods
Reagents
SYTOX Blue™, SYTOX Green™, SYTOX Orange™, SYTOX Red™, TOTO-3™, BOBO-3™, POPO-1™, YO-PRO-1™, YO-PRO-3™, SYTO-9™, SYTOX AADvanced™, YOYO-1™, YOYO-3™, and propidium iodide were purchased from Life Technologies (Grand Island, NY). CellTox Green™ was purchased from Promega (Madison, WI); DRAQ7™ was purchased from Biostatus Limited (Leicestershire, United Kingdom). RedDot2™, EvaGreen™, GelGreen™, and GelRed™ were purchased from Biotium, Inc. (Hayward, CA). DRAQ7, EvaGreen, GelGreen, and GelRed were purchased as aqueous stock solutions. The remaining dyes were purchased as DMSO stock solutions. Dyes were diluted in experiments to final concentrations listed in Table 1. Saponin, levofloxacin, azithromycin, and DMSO (cell culture grade) were purchased from Sigma-Aldrich (St. Louis, MO).
Table 1.
Summary of Fluorescent Dyes Tested by Microscopy and/or Plate Reading
| Dye | Final conc.a | Permeability/cytotoxicity characteristicsb | Excitation optimum (range) | Emission optimum (range) | Dichroic mirror cutoff or beam splitterc | Excitation filter, nm/bandwidthc | Emission filter, nm/bandwidthc |
|---|---|---|---|---|---|---|---|
| POPO-1 | 0.1 μM | Nonpermeable, nontoxic | 434 (395–451) | 456 (440–513) | Beam splitter (BS550, barcode 401) | 420/8 (Photometric 420, barcode 308) | 475/8 (Photometric 475, barcode 309) |
| SYTOX Blue | 1 μM | Nonpermeable, nontoxic | 444 (405–462) | 480 (452–521) | Beam splitter (BS550, barcode 401) | 420/8 (Photometric 420, barcode 308) | 475/8 (Photometric 475, barcode 309) |
| YOYO-1 | 0.1 μM | Cytoplasmic permeable, non-toxic | 491 (441–507) | 509 (491–573) | ND | ND | ND |
| YOPRO-1 | 0.1 μM | Cytoplasmic permeable, nontoxic | 491 (442–507) | 509 (491–570) | ND | ND | ND |
| GelGreen | 1× | Nonpermeable, nontoxic | 270 (200–300) 510 (460–530)d | 530 (510–565) | 505 (FITC, barcode 403) | 485/14 (FITC 485, barcode 102) | 535/25 (FITC 535, barcode 206) |
| EvaGreen | 0.5× | Nonpermeable, nontoxic | 500 (455–520) | 530 (505–565) | 505 (FITC, barcode 403) | 485/14 (FITC 485, barcode 102) | 535/25 (FITC 535, barcode 206) |
| SYTOX Green | 125 nM | Nonpermeable, nontoxic | 504 (460–522) | 523 (505–588) | 505 (FITC, barcode 403) | 485/14 (FITC 485, barcode 102) | 535/25 (FITC 535, barcode 206) |
| CellTox Green | 0.5× | Nonpermeable, nontoxic | 512 (460–522) | 532 (524–590) | 505 (FITC, barcode 403) | 485/14 (FITC 485, barcode 102) | 535/25 (FITC 535, barcode 206) |
| Propidium iodide | 30 μM | Toxic and permeable after 1 day of incubation | 536 (470–581) | 617 (579–652) | 555 (BODIPY TMR, barcode 405) | 530/10 (YFP 530, barcode 221) | 650/40 (Custom 650, barcode 227) |
| SYTOX AADvanced | 1 μM | ND | 546 (456–603) | 647 (596–732) | 555 (BODIPY TMR, barcode 405) | 530/10 (YFP 530, barcode 221) | 650/40 (Custom 650, barcode 227) |
| SYTOX Orange | 0.5 μM* 0.25 μM† | Nonpermeable, nontoxic | 547 (505–603) | 570 (545–614) | 555 (BODIPY TMR, barcode 405) | 535/25 (FITC 535, barcode 206) | 570/8 (Photometric 570, barcode 318) or 590/20 (Rhodamine 590, barcode 217) |
| BOBO-3 | 0.1 μM | Cytoplasmic permeable, nontoxic | 570 (516–602) | 602 (579–652) | ND | ND | ND |
| YOPRO-3 | 0.1 μM | Cytoplasmic permeable, nontoxic | 612 (547–637) | 631 (609–695) | Beam splitter (BS550, barcode 401) | 600/8 (Photometric 600, barcode 319) | 650/50 (no label, barcode 510) |
| YOYO-3 | 0.1 μM | Cytoplasmic permeable, nontoxic | 612 (556–633) | 631 (606–687) | ND | ND | ND |
| DRAQ7 | 3 μM | Nonpermeable, nontoxic | 633 (488–680) | 678 (650–755) | 658 (Cy5, barcode 420) | 600/8 (Photometric 600, barcode 319) | 665/7.5 (APC, barcode 205) |
| TOTO-3 | 0.1 μM | ND | 642 (577–667) | 660 (637–724) | 658 (Cy5, barcode 420) | 600/8 (Photometric 600, barcode 319) | 665/7.5 (APC, barcode 205) |
| SYTOX Red | 5 nM | ND | 640 (584–664) | 658 (635–721) | 658 (Cy5, barcode 420) | 600/8 (Photometric 600, barcode 319) | 665/7.5 (APC, barcode 205) |
| GelRed | 1×* 1× or 0.1ׇ | Nonpermeable, nontoxic | 270 (200–300) 515 (470–560)e | 595 (555–650) | 555 (BODIPY TMR, barcode 405) | 520/25 (Custom 520, barcode 506) | 590/20 (Rhodamine 590, barcode 217) |
| RedDot2 | 1×* 0.4׆ | Nonpermeable, nontoxic | ∼610 (488–647)f | 695 (647–760) | 658 (Cy5, barcode 420) | 600/8 (Photometric 600, barcode 319) | 720/8 (Photometric 720, barcode 322) |
aConcentration used was for both microscopy and plate reading unless otherwise indicated (*microscopy; †plate reading; ‡plate reading, tested only in white microplate dishes). Where fluorophore concentration was not available (i.e., proprietary), concentration used is indicated relative to the manufacturer's recommended 1× concentration.
bCharacteristics detected by microscopy.
cPerkin-Elmer designations for dichroic mirrors, filters, and beam splitters indicated in parentheses.
dSecond peak optimal.
eFirst peak has optimal absorbance, but only the second peak could be tested with available filters.
fA greater absorption peak exists at ∼660 nm, but is not optimal for fluorescent excitation.
ND, not determined.
Cell Culture
J774A.1 cells (ATCC, Manassas, VA) were passaged in suspension in RPMI 1640 medium (Corning Cellgro, Manassas, VA) containing 9% iron-supplemented calf serum (Atlanta Biologicals, Flowery Branch, GA). For microscopy experiments, J774 cells were plated in the same medium on dishes containing embedded #1.5 thickness, German glass cover slips (catalog # P35G-1.5-20-C; MatTek Corp., Ashland, MA) at approximately 5×107 cells/cm2. For microplate experiments, J774 cells were resuspended in the same medium, lacking phenol red and supplemented with 100 μg/mL thymidine, and plated at approximately 5×107 cells/cm2 onto black (Corning 3709) or white (Corning 3570) opaque, sterile, tissue culture-treated, 384-well plates. This plating density equates to 5×104 J774 cells/well or 1.92×106 cells per 384-well dish.
Microscopy
For microscopic assessment of dye permeability and cytotoxicity, coverslips were examined using a Nikon Eclipse Ti™ inverted epifluorescence microscope with standard DAPI, FITC, TRITC, and Cy5 filter cubes. Alterations in J774 cell morphology (cell loss, change in shape) were assessed by phase contrast microscopy. Dye permeability was scored based on fluorescence of the cell nucleus and cytoplasm (within 4 h after dye addition, then on days 1 and 2, and then on day 3 both before and after permeabilization of cells with saponin). Saponin was added to culture wells in one-third volume of additional medium without washing to achieve a final concentration of 0.15%. Microscopic images were captured using a CCD camera (QImaging, Surrey, BC). Paired fluorescent images for each dye (with or without saponin treatment) were processed in parallel in Photoshop CS6™ (Adobe, San Jose, CA) so that contrast adjustments would be applied equally.
Microplate Dye Evaluation
For Z′ factor comparison, dyes were added at the final concentrations indicated in Table 1 to J774A.1 cells grown in 384-well microplates in a 50 μL well volume. Positive control wells received 0.15% saponin to permeabilize cell membranes, and negative control wells received the same concentration of DMSO solvent (0.2% final concentration). To allow screening of a large number of dyes contemporaneously, 8 positive control wells and 34 negative DMSO control wells were tested for each dye comparison. A larger number of negative control wells was used in order to detect intrinsic dye permeability with greater confidence (i.e., permeability in the absence of saponin). To detect intrinsic solution fluorescence, 8–12 wells lacking J774A.1 cells were also tested for each dye. Plates were incubated for the indicated amount of time at 37°C with 5% CO2, and read on an EnVision™ Multi-Label Reader (Perkin Elmer, Waltham, MA). Filter combinations used for each dye are listed in Table 1. Z′ were calculated as follows:
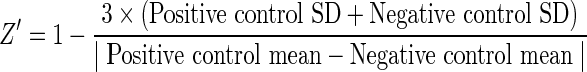 |
Proof of Principle HTS Infection Experiments
J774 cells were seeded in white, Corning #3570, 384-well microplates to achieve approximately 90% confluence on the day of the experiment. Stock solutions (500×) of saponin, levofloxacin, azithromycin, and DMSO control were introduced respectively at 0.1 μL/well by pin transfer using a customized, Epson robot, each into 96 wells in duplicate microplates (thus 192 wells in total for each test compound). Luminescent Legionella pneumophila, strain Lp02:flaA:lux, a thymidine auxotroph containing the Photorhabdus luminescens luxCDABE operon, was originally constructed in the laboratory of Bill Dietrich10 and provided by Andrew Olive (Harvard Medical School, Boston, MA). Prior to use in infection experiments, L. pneumophila was grown as confluent patches on buffered charcoal yeast extract agar plates supplemented with 0.1% α-ketoglutarate and 100 μg/mL thymidine for 1–2 days. Bacteria were then suspended in tissue culture medium supplemented with 100 μg/mL thymidine and SYTOX Green™ and then added, immediately following pin transfer described above, to J774A.1 cells to achieve a multiplicity of infection of approximately one bacterium per two macrophages. Final concentrations of compounds in microplates after pin transfer and addition of bacteria were 0.15% saponin, 10 μg/mL levofloxacin, or 10 μg/mL azithromycin, and 0.2% DMSO and 125 nM SYTOX Green in all wells. Infected 384-well plates were incubated at 37°C in a 5% CO2 atmosphere and read on an EnVision™ Multi-Label Reader (Perkin Elmer) at indicated time points. See Table 2 for summary of HTS assay setup.
Table 2.
High Throughput Assay Protocol Table
| Step | Procedure | Value | Description |
|---|---|---|---|
| 1 | Plate J774A.1 cells | 30 μL per well | 5×104 J774A.1 cells/well |
| 2 | Passage bacteria | Legionella pneumophila strain Lp02-Lux-flaA | Bacterial strain expresses luminescence operon that permits relative quantification of bacterial number over time |
| 3 | Incubate J774A.1 cells and bacteria separately overnight | Approximately 20 h | Incubate J774A.1 cells at 37°C with 5% CO2 to achieve approximately 90% confluence; incubate bacteria at 37°C in ambient air |
| 4 | Pin transfer of test compounds | 500× concentrates in DMSO; 0.1 μL/well | Saponin, mammalian cell lytic positive control; levofloxacin and azithromycin bacterial inhibition controls |
| 5 | Infection and SYTOX Green addition | 20 μL per well | Final concentration of 5×104 bacteria/well and 125 nM SYTOX Green |
| 6 | Pre-incubation readout of bacterial number | Luminescence | Quantify bacteria by luminescence |
| 7 | Pre-incubation readout of mammalian cell lysis | SYTOX Green Fluorescence | Cells that have lost membrane integrity exhibit fluorescent DNA staining |
| 8 | Repeat steps 6 and 7 after 1, 2, and 3 day incubation | As in steps 6 and 7 | Incubate at 37°C with 5% CO2 between readings |
Step Notes
1. Cells suspended in RPMI 1640 medium containing 9% iron-supplemented calf serum without phenol red, supplemented with 100 mg/mL thymidine; plated in Corning #3570 (white) or #3709 (black) dishes (384 wells each), growth area 0.072 cm2/well.
2. Transfer a visible amount of bacterial mass from a first passage plate to a second passage plate to create a growth patch (buffered charcoal yeast extract agar supplemented with 100 mg/mL thymidine).
3. Bacteria should develop into lawn prior to harvesting.
4. Control compound 500× stock solutions dissolved in DMSO (saponin, 75 mg/mL; levofloxacin, 5.0 mg/mL; azithromycin, 5.0 mg/mL). Pin transfer tip array washed in 20% methanol solution by sonication between uses.
5. Bacteria and SYTOX Green suspended in tissue culture medium and transferred to microplates using a Matrix Technologies/Thermo Fisher Scientific (Cambridge, MA) WellMate programmable multichannel peristaltic pump.
6. Perkin-Elmer EnVision “USLum” protocol used for high-sensitivity luminescence detection.
7. Perkin-Elmer EnVision fluorescence reading with Excitation 485/14, Emission 535/25, Dichroic Mirror 505 nm, minimum gain, minimum transmittance, and “High Concentration Mode” selected to prevent detector saturation.
Dose–Response
An HP D300 high performance dispensing system (Hewlett-Packard, Palo Alto, CA) was used to inoculate test wells with azithromycin in a twofold dilution series ranging from 16 μg/mL to 4 ng/mL final concentration. Experiments were otherwise performed as described in the preceding section. IC50 values were calculated in Prism 6 (GraphPad Software, Inc., San Diego, CA) using nonlinear, four parameter regression analysis.
HTS Pilot
Commercial bioactive libraries were from Biomol (now Enzo Life Sciences, Farmingdale, NY), Microsource (Gaylordsville, CT), Sigma-Aldrich, Biofocus (Saffron Walden, United Kingdom); Enzo Life Sciences, Prestwick (Illkirch, France), and Chembridge (San Diego, CA). Compounds were dissolved in DMSO. Experiments were performed in the same manner as described in Table 2 with the following modification. Pin transfer for screening compounds was performed into 384 well screening plates in duplicate. Final concentrations of compounds were usually at either 4 μg/mL or 20 μM. Library arrays were from the ICCB-Longwood collections of known bioactive compounds.11 Screening compounds were added to columns 1–22 in plates by pin transfer as described above. Column 23 was used for infected negative controls that were not treated with compound, 16 wells per plate. Column 24 contained eight interleaved positive controls each for luminescence (levofloxacin) and fluorescence (saponin). In contrast to proof of principle experiments, control compounds were added manually with a multichannel pipette. Modified z scores were calculated according to the formulae z=(x−μ)/σ (fluorescence) or z=(μ−x)/σ (luminescence), where x is the luminescent or fluorescence readout for each test well, and μ and σ are the mean and standard deviation values for the negative control wells within each respective screening plate. We specifically opted not to use the μ and σ values from the test wells for comparison, as the expected large number of positive hits in the known bioactive libraries would exaggerate experimental variability and lead to erroneous reduction in z score values. This strategy has been suggested previously to compensate for a high hit frequency in screening plates.12,13 The μ and x were reversed in the luminescence z score formula so that reduction in luminescence would yield a positive value. Scatter plots were created and analyzed using the Vortex software package (Dotmatics, San Diego, CA).
Results
Permeability and Toxicity
Fluorescent nucleic acid-binding dyes, described by their manufacturers as cell impermeant, were pre-screened by microscopy for permeability and toxicity during a 3-day incubation with J774 cells, a murine monocyte/macrophage cell line. In these and subsequent experiments, the positive permeability control was treatment with saponin, a cholesterol-binding detergent, that efficiently permeabilizes eukaryotic cells.14 Dyes were subsequently categorized into three classes: cytoplasmic-permeable, noncytotoxic (class I); nucleus-permeable, cytotoxic (class II); and nonpermeable, nontoxic (class III) as summarized in Table 1. Representative images for each class of dye are shown in Figure 1.
Fig. 1.

Three classes of DNA-bindings dyes. J774 cells were incubated with indicated dyes for 3 days, and scored by phase contrast and epifluorescence microscopy without or with treatment with saponin (following 3-day incubation) to permeabilize cells. Representative images are shown. Note that in the nonpermeabilized photomicrographs, an attempt was made to capture one spontaneously permeable, apoptotic cell nucleus in the lower left-hand corner as an internal reference point for fluorescence comparisons. Dyes were scored as cytoplasmic permeable, nontoxic (class I); permeable, toxic (class II); or nonpermeable, nontoxic (class III).
In the first class, we noted several that stained the cytoplasm of J774 cells during prolonged incubation. This staining occurred without overt morphological alterations, suggesting lack of significant cytotoxicity. Although these dyes appeared to be largely excluded from the cell nucleus, the intensity of cytoplasmic staining appeared greater than or equal to nuclear staining following saponin permeabilization (e.g., Fig. 1, YOYO-3), predicting lack of utility in microplate assays. In contrast, propidium iodide (class II, Fig. 1), a dye used to assess cell death in both eukaryotic and prokaryotic endpoint assays,15,16 was initially impermeant to J774 cells at up to 1 day of incubation, but then became permeant to J774 cells by 2 days of incubation—that is, staining equal to saponin-treated controls, staining nuclei intensely (Fig. 1 and data not shown). It also caused J774 cells to round up and detach from dishes (Fig. 1), indicating significant cytotoxicity. Importantly, most of the dyes tested proved to be neither overtly permeable nor cytotoxic to J774 cells, and were accordingly candidates for use in real-time cytotoxicity assays.
Suitability for High Throughput Readout
Therefore, nontoxic, long-term impermeant dyes were tested for their ability to yield robust cytotoxicity data in 384-well black and white opaque tissue culture plates (Table 3). Several dyes with green or orange emission spectra yielded high Z′>0.5 (SYTOX Green and Orange, CellTox Green, EvaGreen, and GelGreen), generally with somewhat better performance in white dishes. However, dyes with red or far-red emission spectra uniformly performed poorly on a multimode plate reader, despite giving distinct permeability readout by microscopy. Specifically, SYTOX AADvanced, SYTOX Red, and RedDot2 (Table 3) yielded very low signal and therefore little separation between positive and negative controls. YOPRO-3 and TOTO-3 demonstrated modest but still insufficient separation (Z′<0). Signal-to-noise ratio aside, anomalous readings for DRAQ7 and, to a lesser extent, RedDot2 (higher signal in the absence of J774 cells than in their presence) also raised further concern about reliability. Finally, GelRed saturated the detector under all conditions tested. For blue fluorescent dyes, SYTOX Blue and POPO-1, band-pass filters alone did not enable sufficient discrimination between fluorescence emission and reflected excitation light, resulting in detector saturation under all conditions tested. Had appropriate dichroic mirrors been available to enhance discrimination further, it is possible that these dyes would have exhibited a tractable signal-to-noise ratio.
Table 3.
Z′ for Nonpermeable Dyes Over Time
| Day 0 | Day 1 | Day 2 | Day3 | |
|---|---|---|---|---|
| SYTOX Green | ||||
| Black dish | ND | 0.02 | 0.56 | 0.48 |
| White dish | 0.87 | 0.78 | 0.61 | 0.56 |
| SYTOX Orange | ||||
| Black dish | 0.25 | 0.52 | 0.48 | 0.21 |
| White dish | 0.64 | 0.51 | 0.41 | 0.21 |
| CellTox Green | ||||
| Black dish | ND | 0.36 | 0.49 | 0.48 |
| White dish | 0.79 | 0.58 | 0.79 | 0.84 |
| EvaGreen | ||||
| Black dish | 0.28 | 0.64 | 0.59 | 0.43 |
| White dish | 0.82 | 0.64 | 0.88 | 0.69 |
| GelGreen | ||||
| Black dish | 0.64 | 0.50 | 0.64 | 0.52 |
| White dish | 0.81 | 0.83 | 0.81 | 0.80 |
| RedDot2 | ||||
| Black dish | ND | −17 | −3.1 | −3.1 |
| White dish | ND | 0.65 | 0.44 | 0.39 |
ND, not determined.
Dual Readout Assay
As several dyes demonstrated excellent separation between positive and negative controls during 3-day incubation, we considered them potentially useful in a real-time HTS format cytotoxicity assay. Their robust performance in opaque white plates that enhance luminescent assay signal was particularly intriguing. Therefore, we sought, as proof principle, to test whether a representative, fluorescent, DNA-binding dye could be used in combination with a luminescence-based readout to set up a screening assay for small molecules that block intracellular growth and therefore virulence of the bacterial pathogen, L. pneumophila.
To allow us to quantify intracellular bacterial replication in HTS format, we made use of a previously described L. pneumophila strain expressing the luxCDABE operon from P. luminescens.10 Legionella is noteworthy for its replication within macrophages, but inability to replicate in tissue culture medium.17 Therefore, any increase in luminescence observed during macrophage infection with this strain should reflect intracellular replication. Furthermore, our own qPCR experiments (data not shown) and prior analysis10 indicated that luminescence was an accurate quantitative indicator of intracellular bacterial growth. Notably, the bacterial lux operon encodes both luciferase and substrate.18 Therefore, in contrast to mammalian luciferase systems, addition of exogenous substrate is not required prior to readout.
Several considerations are important for interpretation of data from a screen seeking to identify compounds that inhibit intracellular growth of L. pneumophila. Importantly, intracellular bacterial growth is dependent on host cell viability. Therefore, compounds that are cytotoxic to host cells will lead to a large class of biologically uninteresting “false positives”—in this case, low luminescence resulting from host cell toxicity, rather than inhibition of intracellular bacteria replication. In addition, bacterial replication itself is toxic to host macrophages at late time points.17 Therefore, we expect true inhibitors of intracellular growth to reduce luminescence and prevent host cell death. A dual-assay readout for both luminescence and cytotoxicity would therefore prove advantageous in simultaneously identifying potential inhibitors of intracellular growth and ruling out a large class of false positives.
We therefore made use of observations with DNA-binding dyes to design an assay with dual readout for both bacterial growth (luminescence) and eukaryotic cell toxicity (fluorescence) in the same screening well. For proof of principle experiments, a cytotoxicity positive control (the permeabilizing agent, saponin) and two antibiotics highly active against intracellular Legionella growth (levofloxacin and azithromycin) were dissolved in DMSO and arrayed in a pin transfer plate. Robotic pin transfer to white 384-well screening plates containing J774 cells was then effected, followed by the combined addition of bacteria and SYTOX Green reagent. Plates were incubated and read daily for 3 days.
As shown in Figure 2, in DMSO-treated negative controls, bacterial luminescence increased roughly three logs during a typical experiment. At later time points (day 2–3), macrophage viability was reduced (increasing fluorescence) consistent with known lysis of host cells by replicating bacteria at later time points. As expected, antibiotics prevented both bacterial growth (increased luminescence) and associated host cell death (increased fluorescence). Lastly, a cytotoxic compound, saponin (specifically toxic to mammalian cells),14 led to host cell death (high fluorescence) and thereby prevented intracellular growth (low luminescence). Saponin is not cytotoxic to bacteria because of absence of cholesterol in bacterial membranes,19 therefore also likely accounting for modestly higher luminescence in saponin-treated relative to antibiotic-treated controls. Of note, bacterial luminescent signal in the DMSO negative control was robustly separated from positive antibiotic- and saponin-treated controls on days 2 to 3 (Z′=0.5–0.7), consistent with limitation of intracellular growth through inhibitory effects on bacteria or viability of the host cell (Table 4). Interestingly, on days 2 and 3, SYTOX Green fluorescent signal was significantly lower in antibiotic-treated positive controls (levofloxacin and azithromycin vs. DMSO; Z′=0.5–0.7), suggesting that compounds that block infection could also be identified simultaneously by their ability to prevent host cell death. Conversely, cytotoxic compounds could be identified by increased fluorescence signal on day 1 (saponin vs. DMSO; Z′=0.4).
Fig. 2.

Intracellular bacterial growth and cytotoxicity measured in a dual readout high throughput screening (HTS) format assay. J774 cells were infected with Legionella pneumophila Lp02:flaA:lux in the presence of SYTOX Green. Luminescent (A) and fluorescent signals (B) of representative conditions: negative DMSO control (●); positive cytotoxicity control, saponin (■); and bacterial inhibition controls, azithromycin (◆) and levofloxacin (▲). Data points represent the mean and standard deviation of 192 replicates from a representative experiment with the exception that in the data shown for azithromycin luminescence, two outlier wells were excluded from analysis. As expected, bacterial luminescence (displayed in log scale) increases over time as a result of intracellular growth in macrophages. At later time points, bacterial growth kills host cells leading to increase in SYTOX Green fluorescence. We envision use of this dual readout assay to identify specific inhibitors of intracellular bacterial growth. The dual readout will allow us to easily identify and exclude false positive hits (e.g., saponin) that limit intracellular bacterial growth through destruction of the host cell (low luminescence, high fluorescence).
Table 4.
Z′ for Dual Real-Time Fluorescence and Luminescence HTS Readout
| Day 0 | Day 1 | Day 2 | Day 3 | |
|---|---|---|---|---|
| Fluorescence | ||||
| Saponin vs. DMSO | 0.29 | 0.38 | −0.13 | −2.20 |
| Azithromycin vs. DMSO | −19.4 | −4.69 | 0.53 | 0.78 |
| Levofloxacin vs. DMSO | −19.3 | −3.82 | 0.58 | 0.82 |
| Saponin vs. levofloxacin | 0.58 | 0.60 | 0.60 | 0.60 |
| Luminescence | ||||
| Saponin vs. DMSO | −24.7 | 0.36 | 0.51 | 0.71 |
| Azithromycin vs. DMSO | −37.9 | 0.35 | 0.51 | 0.71 |
| Levofloxacin vs. DMSO | −67.4 | 0.38 | 0.51 | 0.71 |
| Saponin vs. levofloxacin | −16.7 | −0.47 | −4.05 | −4.35 |
These predictions were further supported by dose–response analysis performed for a representative antibiotic, azithromycin (Fig. 3). Here, the IC50 for luminescence and fluorescence were almost identical, and also similar to the minimal inhibitory concentration previously reported for macrophage grown Legionella.20,21 Therefore, based on these similar dose–response profiles, bacterial replication, as assessed by luminescence, and cytotoxicity, as assessed by fluorescence, also appear linked.
Fig. 3.

Dose–response for azithromycin. Luminescence (A) and SYTOX Green fluorescent (B) signals on days 0 (○), 1 (■), 2 (△), and 3 (▼) after infection and treatment with twofold serial dilutions of antibiotic. Note that luminescence was plotted in log10 scale to best illustrate the logarithmic growth pattern of Legionella and differentiate patterns of antibiotic effect. The data presented are the mean and standard deviation for 16 test wells per condition, 8 wells each from two separate 384 well plates. IC50 for non-log transformed luminescence data on days 1, 2, and 3 were 189, 221, and 328 nM, respectively, and for fluorescence were 223, 303, and 407 nM, respectively.
As this dual real-time assay appeared to meet goals for identification of inhibitors of intracellular bacterial replication, we therefore set up a pilot screen with libraries containing 5,956 known bioactive compounds to examine performance of the assay during an actual HTS. The real-time assay afforded us the ability to read wells on both days 1 and 2 of incubation, allowing us to choose dish readings that provided the best statistical separation between positive and negative control wells in individual experiments (highest within plate Z′). This was helpful in addressing the biological variability in this cell-based assay; for example, Legionella growth might peak earlier in some experiments than others.
For luminescence, within plate Z′ (levofloxacin vs. untreated controls) were generally slightly better for day 2 than day 1 readings (day 1 median plate Z′ and interquartile range [IQR] of 0.67 and 0.15 respectively; day 2 median and IQR of 0.73 and 0.174 respectively). Day 2 luminescence z scores were therefore almost always used preferentially. For detecting compound cytotoxicity, day 1 fluorescence z score readings were generally used preferentially to help us best differentiate compounds that killed or protected J774 cells, prior to excessive bacterial replication-associated cytotoxicity that might mask direct toxic effects of compounds. Note, the convergence of saponin and DMSO control signal on day 2 versus day 1 in Figure 2B. This choice was also supported by within plate Z′ determinations for saponin versus infected, untreated negative controls. Specifically, for fluorescence, Z′ were almost always better for day one readings (day 1 median and IQR of 0.84 and 0.16; day 2 median and IQR of 0.53 and 1.3 respectively). Overall, Z′ may have improved during this pilot bioactive screening compared to the HTS validation phase (Table 3) as a result of introduction of a room temperature equilibration period before each reading to reduce edge effect.
A summary of the screening results for day 2 luminescence and day 1 fluorescence data are shown in the scatterplots in Figure 4. Controls performed as expected, specifically nearly complete suppression of luminescent signal in the presence of antibiotic or saponin, and high luminescent signal in negative controls (Fig. 4A). As predicted, fluorescent signal (percent normalized cytotoxicity) was high in saponin-treated controls. In contrast, test compound fluorescent signal was centered around the negative control (Fig. 4B and data not shown). In addition, antibiotic-treated controls demonstrated modestly lower cytotoxicity than negative controls, consistent with suppression of the very low level of cytotoxicity associated with limited bacterial growth on day 1 (Fig. 2B). Of note, screening was performed in duplicate in separate 384-well plates. There was excellent correlation between these replicates with an R2 of 0.91 for luminescent z scores and an R2 of 0.98 for fluorescent z scores (data not shown), suggesting high intra-assay reproducibility.
Fig. 4.

Scatter plots for high throughput screening data. (A) Average luminescence signal from duplicate screening wells on day 2. (B) SYTOX Green fluorescence signal on day 1 normalized to mean negative control (0%) and mean saponin positive control (100%) values within each screening dish. Key: test compounds (X, cyan); negative controls (N, blue); levofloxacin, antibiotic positive controls (P, red); and saponin, cytotoxicity positive controls (O, yellow). Note, in (A), red, levofloxacin-positive control symbols are hidden behind yellow, saponin-positive control symbols and therefore not visible, as both demonstrated close to zero luminescence.
During the pilot HTS screen, we identified 316 strong screening hits defined by a luminescent z score >6.75 in both of the duplicate screening plates. We reasoned that true hits should in general demonstrate less cytotoxicity that the negative control (i.e., a negative z score). Therefore, a fluorescence z of ≥1 was set as a conservative threshold to identify false positive, eukaryotic cell cytotoxic hits. After excluding cytotoxic compounds in this way, 213 screening hits representing 135 unique compounds remained. These included 74 known antimicrobials that were represented often repeatedly in different bioactive libraries, the majority of which were traditional antibiotics in fluoroquinolone, tetracycline, sulfa, rifampin, and macrolide classes. There were also 61 other compounds that may have either unexpected antimicrobial activity or may act in some other way, whether on the host macrophage or bacterial cell, to limit intracellular infection. Cytotoxic compounds with a fluorescence z score ≥1 included those such as actinomycin D, gramicidin, dehydrocholic acid, mercurics, the blistering agent cantharidin, and chemotherapeutics such as idarubicin, camptothecin, and mithramycin that are presumed to limit intracellular bacterial growth through destruction of host cells. The use of a dual readout real-time assay allowed us to exclude these compounds as true hits based on eukaryotic cell toxicity. Therefore, during a pilot HTS, the dual real-time screen appeared to identify compounds with known eukaryotic cytotoxic and/or antimicrobial properties in accordance with our predictions.
Discussion
Membrane-impermeant, DNA-binding dyes have long been used in endpoint assays to ascertain eukaryotic cell membrane integrity and thereby serve as a marker for cell viability. Here, we explored whether such dyes could be used as real-time monitors of cell death through inclusion during the entire assay incubation. From a theoretical standpoint, this appeared practical if dyes remained nonpenetrant and nontoxic over prolonged periods. Indeed, the fluorescent DNA-binding dyes, CellTox Green and DraQ7, have either been promoted by their manufacturer or used in this fashion for microplate screening22 and flow cytometry-based analysis23,24 respectively. However, to the best of our knowledge, their performance in microplate format has not been evaluated in the peer-reviewed literature, nor compared to the large number of “impermeant” DNA-binding dyes that are commercially available.
Notably, a large number of impermeant fluorescent stains have been marketed for use in applications such as fluorescent microscopy, flow cytometry, agarose gel DNA staining, and real-time PCR detection. Of note, manufacturers have marketed some impermeant DNA-binding dyes as environmentally safe DNA stains, with the idea that impermeant dyes would less likely mutagenize living tissue and as such could therefore also be disposed of as nonhazardous waste. Several such “safe” dyes are now offered as alternatives to ethidium bromide and SYBR Green, used for agarose gel staining and real-time PCR assays respectively.25 An observed utility in high throughput assay format would therefore also make these dyes welcome alternatives for the potentially large hazardous waste stream generated in HTS efforts.
We confirmed that the majority of impermeant DNA-binding dyes failed to penetrate a representative macrophage cell line during prolonged incubation. However, the dyes that performed well in microplate format generally had excitation and emission characteristics similar to FITC. They maintained high Z′ during a 3-day incubation (sustained statistical separation of saponin-treated and untreated J774 cells), consistent with low permeance. In contrast, both red and blue fluorescent dyes were unusable in microplate assays: use of the former limited by low signal-to-noise ratios, and use of the latter potentially limited by filters and dichroic mirrors available to us.
Notably, SYTOX Green, EvaGreen, and GelGreen all performed well in 384-well plate format, and are relatively inexpensive on a per assay basis. CellTox Green performed similarly but was significantly more expensive on a per well basis. We found that each gave good to excellent separation in white dishes (Z′>0.5) and good separation in black dishes (Z′>0.4) when comparing positive and negative controls during a 3-day incubation. Furthermore, in proof of principle HTS simulation experiments, dyes in this group were found to be amenable for use in dual readout assays in combination with a separate luminescence reporter (Fig. 2; data for other dyes not shown). Lastly, in a dual readout HTS screen of 5,956 known bioactive compounds, both known antimicrobials and cytotoxic compounds were identified according to prediction. Importantly, we did not note any interaction or interference of the real-time fluorescence and a bacterial luciferase assay whose output spanned a very large dynamic range (Figs. 2, 3, and 4A). Accordingly, we speculate that our findings should be applicable to use of fluorescent cytotoxicity detection in combination with other mammalian luciferase-based assays more commonly used in HTS readouts. Taken together, we believe that several membrane-impermeant DNA-binding dyes can serve as useful real-time monitors of eukaryotic cell viability, whether used alone or as a secondary readout, in HTS assays.
Abbreviations Used
- ATCC
American Type Culture Collection
- ATP
adenosine triphosphate
- CCD
charged coupled device
- DAPI
4′,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- DNA
deoxyribonucleic acid
- FITC
fluorescein isothiocyanate
- HTS
high throughput screening
- IC50
half maximal inhibitory concentration
- IQR
interquartile range
- ND
not determined
- PCR
polymerase chain reaction
- qPCR
quantitative polymerase chain reaction
- SD
standard deviation
- TRITC
tetramethylrhodamine
Acknowledgments
Research reported in this manuscript was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI099122 to J.E.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Su Chiang, Doug Flood, Sean Johnston, Jennifer Nale, Stewart Rudnicki, Jennifer Smith, David Wrobel, Paul Yan, and the National Screening Laboratory for the New England Regional Centers of Excellence in Biodefense and Emerging Infectious Diseases (supported by U54AI057159) for their assistance.
Disclosure Statement
The authors declare no conflicts of interest.
References
- 1.Riss TL, Moravec RA, Niles AL, Benink HA, Worzella TJ; Minor L. (ed.): Cell viability assays. 2013. In Assay Guidance Manual [Internet] (Sittampalam GS, Gal-Edd N, Arkin M, et al., eds.). Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD, 2004–. www.ncbi.nlm.nih.gov/books/NBK144065 [Google Scholar]
- 2.Niles AL, Moravec RA, Riss TL: In vitro viability and cytotoxicity testing and same-well multi-parametric combinations for high throughput screening. Curr Chem Genomics 2009;3:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niles AL, Moravec RA, Riss TL: Update on in vitro cytotoxicity assays for drug development. Expert Opin Drug Discov 2008;3:655–669 [DOI] [PubMed] [Google Scholar]
- 4.Ramirez CN, Antczak C, Djaballah H: Cell viability assessment: toward content-rich platforms. Expert Opin Drug Discov 2010;5:223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Didenko VV: DNA probes using fluorescence resonance energy transfer (FRET): designs and applications. Biotechniques 2001;31:1106–1116, 1118, 1120–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juskowiak B: Nucleic acid-based fluorescent probes and their analytical potential. Analyt Bioanalyt Chem 2011;399:3157–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marras SE: Interactive fluorophore and quencher pairs for labeling fluorescent nucleic acid hybridization probes. Mol Biotechnol 2008;38:247–255 [DOI] [PubMed] [Google Scholar]
- 8.Singer VL, Jones LJ, Yue ST, Haugland RP: Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal Biochem 1997;249:228–238 [DOI] [PubMed] [Google Scholar]
- 9.Furstenberg A, Deligeorgiev TG, Gadjev NI, Vasilev AA, Vauthey E: Structure–fluorescence contrast relationship in cyanine DNA intercalators: toward rational dye design. Chemistry 2007;13:8600–8609 [DOI] [PubMed] [Google Scholar]
- 10.Coers J, Vance RE, Fontana MF, Dietrich WF: Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol 2007;9:2344–2357 [DOI] [PubMed] [Google Scholar]
- 11.ICCB-Longwood Compound Libraries: http://iccb.med.harvard.edu/libraries/compound-libraries (last accessed on March3, 2014)
- 12.Zhang JH, Chung TD, Oldenburg KR: A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67–73 [DOI] [PubMed] [Google Scholar]
- 13.Zhang JH, Chung TD, Oldenburg KR: Confirmation of primary active substances from high throughput screening of chemical and biological populations: a statistical approach and practical considerations. J Comb Chem 2000;2:258–265 [DOI] [PubMed] [Google Scholar]
- 14.Shany S, Bernheimer AW, Grushoff PS, Kim KS: Evidence for membrane cholesterol as the common binding site for cereolysin, streptolysin O and saponin. Mol Cell Biochem 1974;3:179–186 [DOI] [PubMed] [Google Scholar]
- 15.Caron GN, Stephens P, Badley RA: Assessment of bacterial viability status by flow cytometry and single cell sorting. J Appl Microbiol 1998;84:988–998 [DOI] [PubMed] [Google Scholar]
- 16.Darzynkiewicz Z, Bruno S, Del Bino G, et al.: Features of apoptotic cells measured by flow cytometry. Cytometry 1992;13:795–808 [DOI] [PubMed] [Google Scholar]
- 17.Horwitz MA: Characterization of avirulent mutant Legionella pneumophila that survive but do not multiply within human monocytes. J Exp Med 1987;166:1310–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waidmann MS, Bleichrodt FS, Laslo T, Riedel CU: Bacterial luciferase reporters: the Swiss army knife of molecular biology. Bioeng Bugs 2011;2:8–16 [DOI] [PubMed] [Google Scholar]
- 19.van Doorne H, van der Tuuk Adriani WP, van de Ven LI, Bosch EH, de Natris T, Smit Sibinga CT: Saponin, an inhibitory agent of carbon dioxide production by white cells: its use in the microbiologic examination of blood components in an automated bacterial culture system. Transfusion 1998;38:1090–1096 [DOI] [PubMed] [Google Scholar]
- 20.Stout JE, Sens K, Mietzner S, Obman A, Yu VL: Comparative activity of quinolones, macrolides and ketolides against Legionella species using in vitro broth dilution and intracellular susceptibility testing. Int J Antimicrob Agents 2005;25:302–307 [DOI] [PubMed] [Google Scholar]
- 21.Jonas D, Engels I, Daschner FD, Frank U: The effect of azithromycin on intracellular Legionella pneumophila in the Mono Mac 6 cell line at serum concentrations attainable in vivo. J Antimicrob Chemother 2000;46:385–390 [DOI] [PubMed] [Google Scholar]
- 22.Worzella T, Niles A, Hengstl T, Fejtl M, Oberdanner C, Merlino J: Real-time cytotoxicity analysis using CellTox™ green cytotoxicity assay and the Tecan Infinite® 200 PRO with gas control module. www.promega.com/resources/pubhub/real-time-cytotoxicity-analysis-with-celltox-green-cytotoxicity-assay (last accessed on October14, 2013)
- 23.Wlodkowic D, Akagi J, Dobrucki J, et al.: Kinetic viability assays using DRAQ7 probe. Curr Protoc Cytom 2013;Chapter 9:Unit 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akagi J, Kordon M, Zhao H, et al.: Real-time cell viability assays using a new anthracycline derivative DRAQ7(R). Cytometry A 2013;83:227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Q, Baum L, Fu WL: Simple and practical staining of DNA with GelRed in agarose gel electrophoresis. Clin Lab 2010;56:149–152 [PubMed] [Google Scholar]