

Abstract
Aims: The human branched-chain aminotransferase proteins (hBCATm and hBCATc) are regulated through oxidation and S-nitrosation. However, it remains unknown whether they share common redox characteristics to enzymes such as protein disulfide isomerase (PDI) in terms of regulating cellular repair and protein misfolding. Results: Here, similar to PDI, the hBCAT proteins showed dithiol-disulfide isomerase activity that was mediated through an S-glutathionylated mechanism. Site-directed mutagenesis of the active thiols of the CXXC motif demonstrates that they are fundamental to optimal protein folding. Far Western analysis indicated that both hBCAT proteins can associate with PDI. Co-immunoprecipitation studies demonstrated that hBCATm directly binds to PDI in IMR-32 cells and the human brain. Electron and confocal microscopy validated the expression of PDI in mitochondria (using Mia40 as a mitochondrial control), where both PDI and Mia40 were found to be co-localized with hBCATm. Under conditions of oxidative stress, this interaction is decreased, suggesting that the proposed chaperone role for hBCATm may be perturbed. Moreover, immunohistochemistry studies show that PDI and hBCAT are expressed in the same neuronal and endothelial cells of the vasculature of the human brain, supporting a physiological role for this binding. Innovation: This study identifies a novel redox role for hBCAT and confirms that hBCATm differentially binds to PDI under cellular stress. Conclusion: These studies indicate that hBCAT may play a role in the stress response of the cell as a novel redox chaperone, which, if compromised, may result in protein misfolding, creating aggregates as a key feature in neurodegenerative conditions such as Alzheimer's disease. Antioxid. Redox Signal. 20, 2497–2513.
Introduction
Optimal cellular function depends on the interplay of numerous repair and chaperone systems. Repair enzymes such as thioredoxin, glutaredoxin, and the disulfide isomerase catalyst, protein disulfide isomerase (PDI) represent the thioredoxin superfamily that mediates a co-ordinated adaptive response to changes in the cellular redox environment (5, 45). These oxidoreductases, characterized by their conserved catalytic recognition sequence, the CXXC motif, function to repair and chaperone proteins through dithiol-disulfide exchange mechanisms (7, 21, 27, 51, 70). PDI (EC 5.3.4.1), largely located in the endoplasmic reticulum (ER), is a versatile protein that can catalyze the formation and breakage of disulfide bonds, facilitating correct refolding of proteins by rearranging the pattern of disulfide bonds (26, 68). In cell models, proteins such as PDI are proposed to act by alleviating the build-up of mis-folded proteins, preventing neurotoxicity (58). Should these protection mechanisms fail, mis-folded proteins accumulate creating aggregates, which can interfere with cell function by contributing to the pathogenesis of neurodegenerative conditions such as Alzheimer's disease (AD) (62, 62, 71).
Innovation.
The physiological role of the redox active CXXC motif of human branched-chain aminotransferase (hBCAT) remains unclear. This report demonstrated a novel functional role of hBCAT in redox protein folding. Moreover, this is the first study which reports that hBCATm can bind to protein disulfide isomerase (PDI) in the human brain and that PDI is co-localized with hBCATm in mitochondria. Overall, this has key implications, as both proteins are modified in Alzheimer's disease (AD) brains, which could impact the regulation of protein folding by possibly contributing to the pathogenesis of AD. Understanding the role of dys-regulated redox mechanisms will assist in the identification of therapeutic targets for the treatment of neurodegeneration.
The human branched-chain aminotransferase proteins (hBCATs) (EC 2.6.1.42) catalyze the reversible transamination of the α-amino group of the branched-chain amino acids to α-ketoglutarate, forming their respective branched chain α-keto acids and glutamate (30, 32). The branched chain α-keto acids are further metabolized through irreversible decarboxylation that is catalyzed by the branched chain keto acid dehydrogenase complex (BCKDH) (25). Although other isoforms exist, the cytosolic (hBCATc) and mitochondrial (hBCATm) form predominate, with hBCATm expressed in most tissues and hBCATc primarily expressed in the brain and peripheral nervous system (22, 29, 30, 57). The hBCAT proteins operate as homodimers with a subunit molecular mass of 41.73 and 43.40 kDa for hBCATm and hBCATc, respectively (20). Transamination by the BCAT proteins in the brain contributes to more than 30% of de novo brain glutamate (30). In human and rat brain, hBCATc is specific to glutamatergic and GABAergic neurons, where expression is predominantly in the soma and proximal dendrites, in keeping with its role in contributing to the storage and metabolic pools of glutamate, which is, in turn, decarboxylated in GABAergic neurons to form GABA (22, 57). The mitochondrial isoform is localized specifically to the human brain vasculature with localized neuronal staining, where a role in brain glutamate metabolism has been suggested (28); whereas it is located in astrocytes of rat models (8, 9, 12).
Unique to the mammalian BCAT proteins is a conserved CXXC motif, which is ∼10 Å from the active site (23, 72). X-ray crystallography and biochemical studies of overexpressed hBCAT have shown that these thiol groups confer redox-linked regulation to the hBCAT proteins and that the active and inactive form results from the reduced and oxidized CXXC motif, respectively (18, 23, 72). In response to oxidation, the thiol at the N-terminal position acts as a redox sensor, whereas the C-terminal residue permits reversible regulation through the formation of a disulfide bond via a sulphenic acid intermediate, resulting in a loss of hBCAT activity (16, 17). X-ray crystallography studies and kinetic analysis demonstrated that the predominant effect of oxidation was on the second half reaction rather than on the first half reaction, where disruption of the CXXC center results in altered substrate orientation and an unprotonated pyridoxal mono-phosphate amino group, thus rendering the enzyme catalytically inactive (73). Both isoforms were differentially inactivated through S-nitrosation; however, prolonged exposure to S-nitrosoglutathione (GSNO) resulted in a shift from S-nitrosation to S-glutathionylation, suggesting that S-glutathionylation plays a protective role in preserving hBCAT function during periods of cellular stress (13). Physiologically, the reduced state of the CXXC motif of the hBCAT proteins is essential for the binding of hBCATm to the E1 subunit of the BCKDH complex, facilitating substrate channeling of the α-keto acids (33). In a separate study, using extracts from neuronal cells, hBCATc was shown to have specific peroxide-related redox associations with several proteins known to be involved in protein cell signaling, indicating a novel role for hBCATc in cellular redox control (15). However, neither the cellular mechanisms that govern this redox sensitivity nor their redox substrates are completely understood.
In these studies, we have found that the hBCAT proteins have dithiol-disulfide isomerase activity that is regulated through S-glutathionylation. We provide in-vitro, ex-vivo, and in-vivo evidence that hBCAT associates with PDI, the interaction of which is altered through oxidative and S-nitrosative stress-induced conditions, where we propose that hBCAT may have a novel chaperone role in mediating protein folding. The localization of these proteins to neuronal and endothelial cells of the vasculature of the human brain along with their co-immunoprecipitation demonstrates that the proposed chaperone role may have physiological relevance. These findings provide the first evidence that the hBCAT proteins have oxidoreductase properties similar to PDI and that these proteins may act as neuro-protectors during the cellular stress response, which may become compromised by contributing to protein misfolding, a key feature of neurodegenerative conditions such as AD.
Results
Endogenous PDI-like activity of the hBCAT proteins in altered redox environments
Both hBCATm and hBCATc catalyzed the refolding of reduced and denatured RNase (rdRNase) through the introduction of disulfide bonds at a rate significantly greater than air oxidation alone, with activity at 55%–60% relative to PDI (Fig. 1A), confirming their oxidase activity. Ovalbumin, which does not have a redox-active CXXC motif but contains free thiols and disulfide bonds, did not exhibit measureable activity over 24 h. The refolding of rdRNase by the hBCAT proteins was found to be both time- and concentration dependent and saturated at concentrations of 12 μM (Fig. 1B, C, respectively). Since protein folding is inherently influenced by the redox environment (31), we assessed the influence of various glutathione (GSH) buffers on hBCAT oxidase activity. Although GSH buffers alone offer partial refolding of rdRNase, the oxidase activity of both hBCAT isoforms was increased two-fold at a 3:1 ratio of GSH/Glutathione disulphide (GSSG) compared with GSH alone (Fig. 2). However, as the environment became more oxidizing, the oxidase activity of hBCATm continued to increase relative to hBCATc, which itself remained unchanged at these concentrations.
FIG. 1.
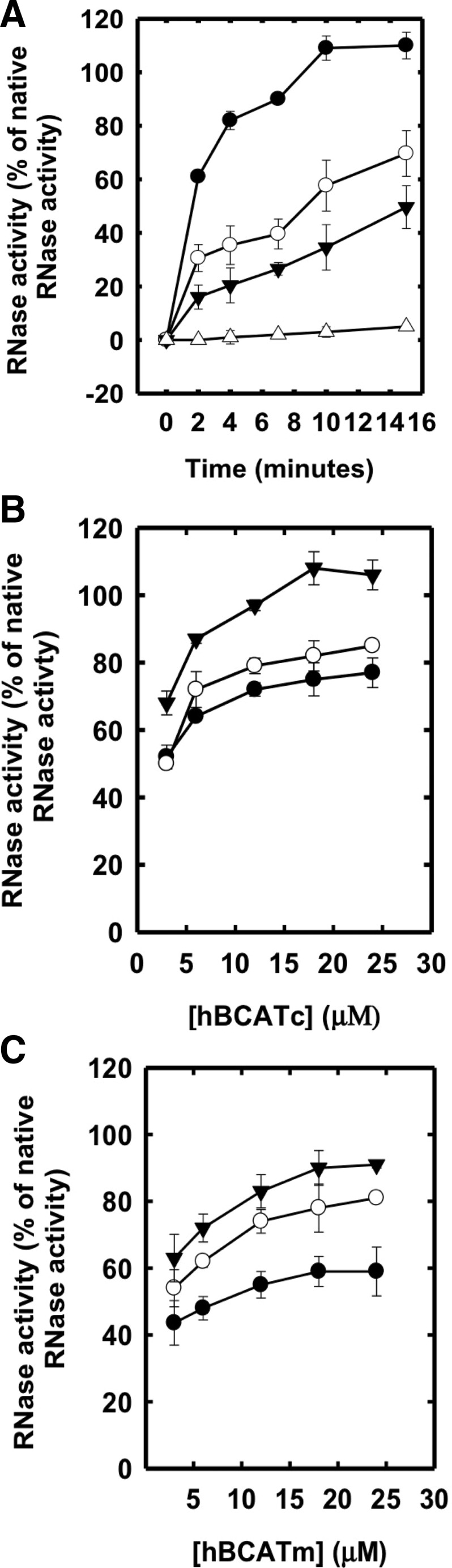
The human branched-chain aminotransferase (hBCAT) proteins catalyze the refolding of reduced denatured ribonuclease A (rdRNase). rdRNase (30 μM) was incubated with protein disulfide isomerase (PDI), hBCATm, or hBCATc (5 μM), respectively, in 0.1 M Tris-HCl, pH 7.4, and 1 mM EDTA over 24 h (A) and then monitored for the effect of both time (12, 24 and 48 h) and concentration (4–24 μM) of each isoform with rdRNase (B, C, respectively). Refolding was determined using the RNase activity assay, monitoring the increase in absorbance at 284 nm. Results were expressed as a % of the activity observed in native RNase preparations. (A) Dithiol-disulfide exchange activity for PDI (●), hBCATc (○), hBCATm (▾), and rdRNase alone (Δ). (B, C) Thiol-disulfide exchange in hBCATc and hBCATm, respectively, over concentrations, 4, 6, 12, 18, and 24 μM at 12 (●), 24 (○), and 48 (▾) h, respectively. Data are the mean±SEM, n=6.
FIG. 2.
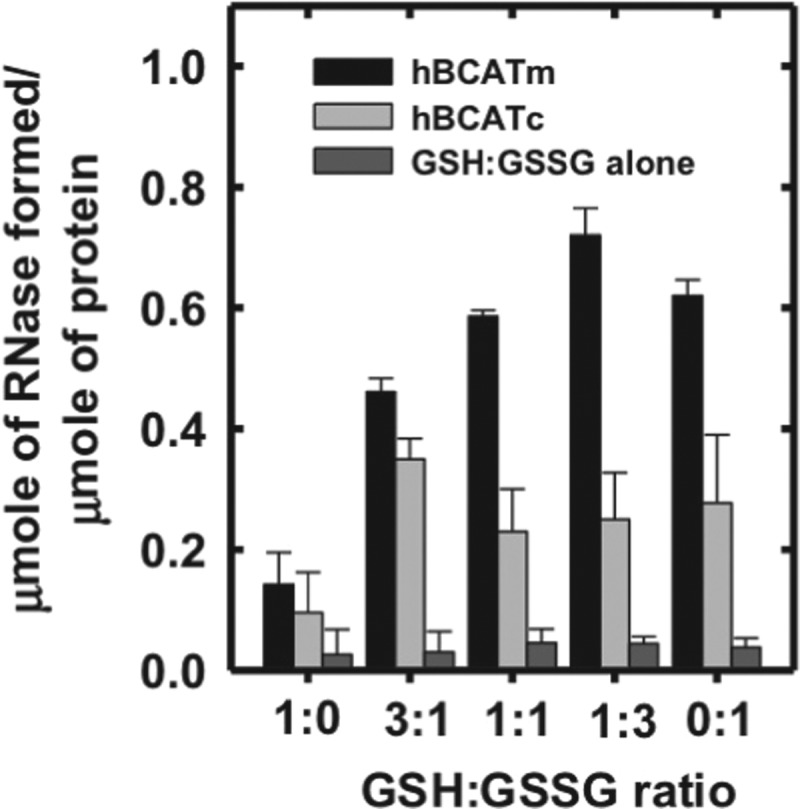
Refolding of rdRNase is influenced by the redox environment. RNase (30 μM) was reduced and denatured as described in the “Materials and Methods” section and incubated with hBCATm or hBCATc at 10 μM +/− varying glutathione (GSH)/Glutathione disulphide (GSSG) ratios (1:0, 3:1, 1:1, 1:3, and 0:1) in 0.1 M Tris-HCl, pH 7.4, and 1 mM EDTA over 24 h. Controls included incubating the unfolded rdRNase with the same ratios of GSH/GSSG with an exception to the hBCAT proteins and rdRNase with hBCAT alone. After 48 h, the contribution of the GSH:GSSG buffer alone was considerable. Refolding was determined using the RNase activity assay, which monitored the change in absorbance at 284 nm. Results were expressed as the μmoles of active RNase formed/μmole of hBCAT. Data are the mean±SEM, n=3.
In the absence of a GSH buffer, the apparent Vmax was 4.4- and 2.4-fold less for hBCATm and hBCATc, respectively (Fig. 3). To investigate the ability of hBCAT proteins to correctly align mis-paired disulfides, we used scrambled RNase (scRNase) as a substrate, which is rdRNase with random disulfide bond formation. Titration of scRNase with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) before and after oxidation resulted in a loss of thiols to 0.04 μmoles/μmole of protein with <1% RNase activity. Although in the presence of GSH/GSSG the apparent Vmax for refolding of scRNase was 3.4- and 2.4-fold higher relative to the reaction without the redox buffer, the dithiol isomerase activity was ∼50% less than that reported for their oxidase activity with rdRNase (Fig. 3). These results demonstrate that the hBCAT proteins have oxidase and thiol isomerase activity, the kinetics of which is dependent on the redox environment.
FIG. 3.
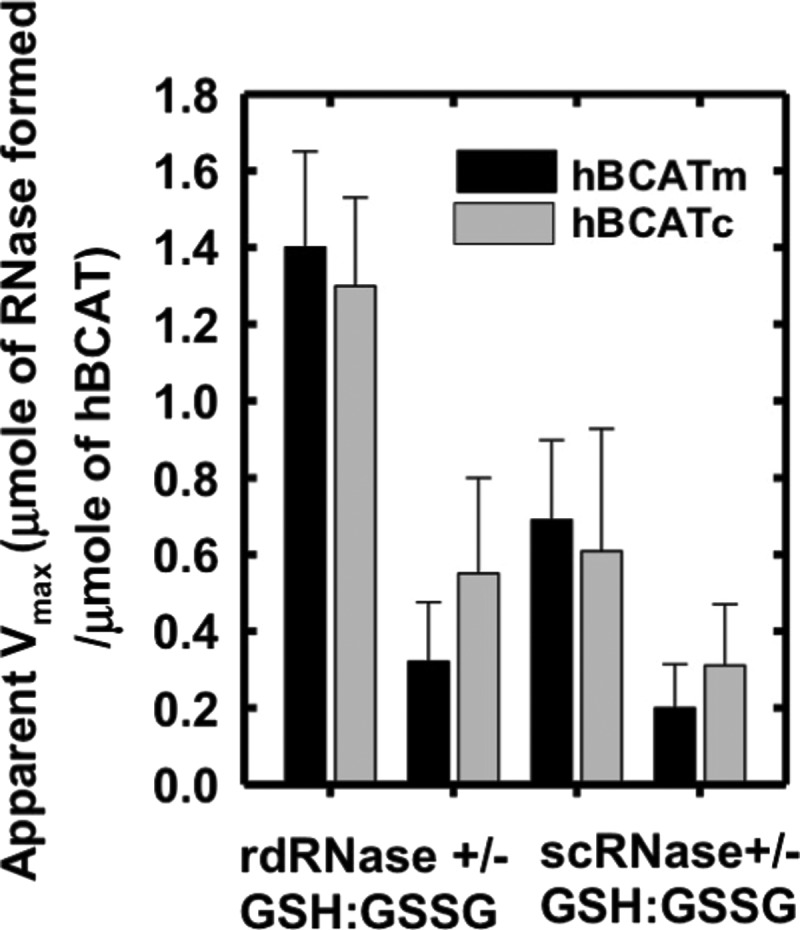
The apparent Vmax of rdRNase and scrambled ribonuclease A (scRNase) with hBCATm and hBCATc. Varying concentrations of rdRNase or scRNase (5–60 μM) were incubated with hBCATc and hBCATm at 10 μM, respectively, in 0.1 M Tris-HCl, pH 7.4, with 1 mM EDTA +/− a GSH buffer (3:1 GSH:GSSG) over 24 h. Refolding was determined using the RNase activity assay, which monitored the change in absorbance at 284 nm. Lineweaver–Burke plots were used to determine the apparent Vmax in μmoles min−1, which was converted to μmoles of active RNase formed/μmole of each respective hBCAT protein. Data are the mean±SEM, n=3.
Target site of dithiol-disulfide exchange
To determine the target site of these refolding mechanisms, active-site cysteine mutants of the hBCAT proteins were assessed for their ability to catalyze the refolding of rdRNase. At 24 h, the oxidase activity of cysteine active-site mutants was negligible (Fig. 4A), but was recovered by the addition of a GSH buffer or GSNO, similar to native hBCAT under these conditions (Fig. 4B, C, respectively). Western blot analysis confirmed that native hBCAT associates and forms a complex with RNase (estimated M.Wt.=53 kDa), which is not evident in the N-terminal mutant proteins unless S-glutathionylated (Fig. 4D, E, respectively, arrow). Also observed were multimers when hBCATc was incubated with GSNO or the GSH buffer (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars), which disappeared when RNase was introduced. Multimers were not reported for hBCATm, suggesting different mechanisms of action between isoforms and rdRNase. Dimers of rdRNase were also noted (*). These findings indicate that the thiols of the CXXC motif are central to their oxidase activity and that S-glutathionylation is fundamental to this mechanism.
FIG. 4.
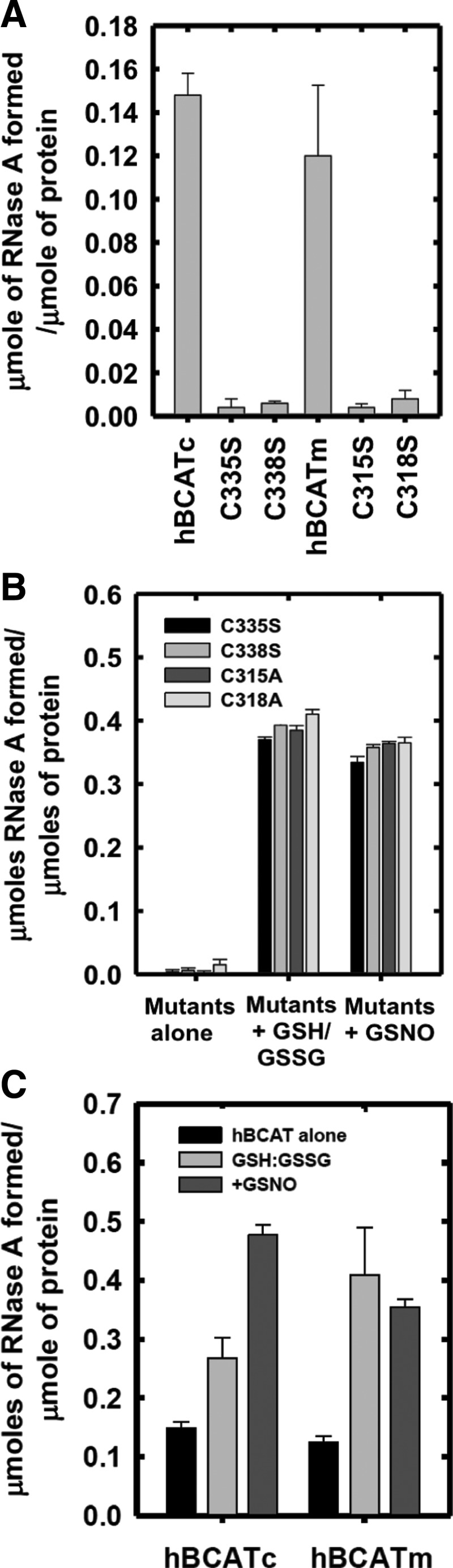

Thiols of the CXXC motif are integral to the oxidase activity of hBCAT, which is mediated by S-glutathionylation. The hBCAT proteins or their respective CXXC mutants, C315A, C318A (hBCATm), C335S, and C338S (hBCATc) (12 μM), in 0.1 M Tris-HCl, pH 7.4, and 1 mM EDTA were incubated with rdRNase (30 μM) for 24 h at room temperature +/− 1 mM S-nitroso-glutathione (GSNO) or GSH buffer (3:1 GSH:GSSG). Refolding was determined using the RNase activity assay, which monitored the change in absorbance over time at 284 nm, converted to μmoles of active RNase formed/μmole of protein. Aliquots of each reaction were separated on a 4%–12% nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) system, and hBCAT was detected by Western blot analysis (antibody dilution 1/5000). (A) Active RNase formed by native hBCAT relative to their mutant proteins. (B) Active RNase formed by mutant proteins +/− a GSH redox buffer or +/− 1 mM GSNO, respectively. (C) Active RNase formed of native hBCAT +/− a GSH redox buffer or +/− 1 mM GSNO. (D, E) Coomassie stain and Western blot analysis of hBCATm and hBCATc with rdRNase under S-glutathionylation conditions. Data are the mean±SEM, n=3. Arrows in the box show that hBCAT has formed a complex with rdRNase.
Association of the hBCAT proteins with human PDI
Far Western blot analysis demonstrated that there is a dose-dependent direct binding of hBCATm and hBCATc with hPDI (Fig. 5A [i, ii]). Binding is also demonstrated using a reverse dot blot for PDI with hBCATm and hBCATc, respectively (Fig. 5B [i, ii], varying concentrations of PDI and Fig. 5C, PDI constant at 7 nmol). Although PDI did not significantly influence hBCAT transaminase activity (Fig. 5D [i]), incubation of hBCAT with PDI resulted in an increase in oxidase activity (Fig. 5D [ii]). Since the initial phase of the reaction occurs earlier relative to the proteins in their independent reactions, we conclude that hBCATm acts in a synergistic manner along with PDI in the initial stages of the refolding reaction but has less influence during the linear phase (Supplementary Fig. S2). Thus, these proteins can associate with each other in a dose-dependent manner, and protein folding is influenced by this binding.
FIG. 5.

The in vitro association of the hBCAT proteins with hPDI. (A) Human PDI (hPDI) (0.1–0.5 μg) was applied to a nitrocellulose membrane. After blocking, 5 μg/ml of hBCATm (i) and hBCATc (ii), respectively, were incubated with hPDI. Bound hBCAT was detected by using anti-hBCAT (antibody dilution 1/1000). (B) Each respective hBCAT protein [hBCATm (i) and hBCATc (ii)] (1.0–3.5 μg) was applied to a nitrocellulose membrane. After blocking, 5 μg/ml of hPDI was incubated with each respective hBCAT isoform. Bound PDI was detected by using anti-PDI-2 (antibody dilution 1/1000). (C) hPDI at 0.4 μg was incubated with hBCATm (0.3–25 μg) (i) and hBCATc (10–30 μg) (ii). Bound hBCAT was detected using anti-hBCAT (antibody dilution 1/1000). (D)(i) The hBCAT transaminase activity (Units/mg) of both isoforms incubated with PDI+/− a GSH buffer (3:1 GSH:GSSG). (D)(ii) PDI and each respective hBCAT isoform were incubated with rdRNase (30 μM), and aliquots were removed at 24 h to determine the effect on RNase folding. Active RNase formed/μmole of protein catalyzed by PDI, hBCAT, and PDI+hBCAT, respectively. Data are the mean±SEM, n=3.
Co-localization of hBCATm with PDI and Mia40 in IMR-32 neuronal cells using confocal microscopy and electron microscopy
Confocal microscopy established that both isoforms differentially co-localize with PDI, suggesting that they occupy the same space within the cell and are interacting (Fig. 6A, B, merged image). The co-localization of hBCATm/hBCATc (red channel) and PDI (green channel) is shown as yellow (merged image) (Fig. 6A, B, respectively). Mander's correlation coefficients (Mx and My) were derived using Volocity (Perkin-Elmer) from an average of 15 individual cell images (6). Mx signifies the correlation of hBCATm/hBCATc co-localizing with PDI, and the My value reflects the correlation of overlap between PDI and hBCAT. This method quantifies the signal intensity and can be measured in a range of 0–1, with 0 indicating no co-localization and 1.0 indicating complete co-localization. The average Mx values for hBCATm with PDI were 0.675 (Fig. 6A) and 0.648 (Supplementary Fig. S3), respectively, validated using antibodies specific for PDI from two independent manufacturers, demonstrating a strong co-localization between hBCATm and PDI; whereas the My value, a measure of PDI co-localizing with hBCATm, was recorded at 0.31. The co-localization of hBCATc with PDI was not as strong with an Mx value of 0.29 recorded (Fig. 6B, merged image). In addition, hBCATm showed co-localization with Mia40 (Mx=0.849, Fig. 6C), a mitochondrial-specific redox protein that also co-localized with PDI with an Mx value of 0.96, confirming that this co-localization is specific to the mitochondria (Fig. 6D). The My correlation for PDI with Mia40 was 0.23. Together, these data indicate that in the mitochondria, the co-localization of hBCATm with PDI is strongly correlated but relative to the whole cell, co-localization of PDI with these mitochondrial proteins (that will account for ER expression of PDI); the signal is weak, illustrating that the co-localization is clearly specific to the mitochondria.
FIG. 6.

The mitochondrial hBCAT co-localizes with PDI and Mia40. IMR-32 cells were fixed for immunostaining as described in the “Materials and Methods” section using primary antibodies: (A, B) anti-hBCATm-1, anti-hBCATc-1 (1/500 dilution), and anti-PDI-1 (1/150 dilution). (C) anti-hBCATm-2 (1/250 dilution) and anti-Mia40 (1/100). (D) anti-PDI-1 (1/150 dilution) and anti-Mia40 (1/100 dilution). Secondary antibodies: goat anti-rabbit Alex Fluor® 568 (hBCATc-1, hBCATm-1, Mia40 [1/500 dilution]) and goat anti-mouse Alex Fluor 488 (PDI-1 and hBCATm-2, [1/500 dilution]). Slides were washed and mounted in 300 nM 4′,6 diamino-2-phenylindole (DAPI) in glycerol. Mander's correlation coefficient (Mx) was used to statistically assess the co-localization of hBCAT proteins with PDI and Mia40, respectively and also between PDI and Mia40. The Mander's co-localization coefficients were derived using Volocity (Perkin-Elmer) from an average of 15 individual cell images. (A) PDI (Green)+hBCATm (red). (B) PDI (Green)+hBCATc (red). (C) hBCATm (red)+Mia40 (Green). (D) PDI (Green)+Mia40 (Red). Merged images show yellow, where co-localization is observed as indicated by the arrows in each image. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Electron microscopy independently validated that hBCATm can co-localize with PDI in the mitochondria as determined by calculating distances between cellular proteins, where a distance of approximately 50 nm apart is considered co-localization, which factors the distance of the antibodies and gold particles (64). IMR-32 cells showed a predicted distribution of hBCATm and Mia40 within the mitochondria (distance apart=9.46–11.59 nm), suggesting co-localization (Fig. 7A). Validation that PDI was also detected in the mitochondria was determined using Mia40 as a mitochondrial control protein (Fig. 7B). In a separate section, PDI was observed in the mitochondria and in close proximity with hBCATm (distance apart 11.16–15.85 nm), validating our confocal microscopy data that indicated these two proteins were occupying the same space, suggesting an interaction (Fig. 7C). It is also important to note the detection of hBCATm in the intermembrane space (Fig. 7C). These studies were validated using antibodies from several suppliers (Supplementary Fig. S4), demonstrating that PDI is found in the mitochondria of neuronal cell cultures and is co-localizing with hBCATm.
FIG. 7.

Localization of hBCATm, Mia40, and PDI in the mitochondria of IMR-32 cells. Electron micrograph of IMR-32 cells expressing, (A) hBCATm and Mia40 labeled with anti-mouse-10 nm (hBCATm-2–black arrowheads) and anti-rabbit-6 nm (Mia40–white arrowheads) gold-conjugated secondary antibodies. (B) PDI and Mia40 labeled with anti-mouse-10 nm (PDI-1–white arrowheads) and anti-rabbit-6 nm (Mia40–black arrowheads) gold-conjugated secondary antibodies. (C) hBCATm and PDI labeled with anti-mouse-10 nm (hBCATm-2–black arrowheads) and anti-rabbit-6 nm (PDI-2–white arrowheads) gold-conjugated secondary antibodies. Scale bar: A–C (50 nm).
Co-immunoprecipitation of hBCATm and PDI from IMR-32 cells
Direct binding of hBCATm with PDI was demonstrated using immunoprecipitation (IP) analysis, where antibodies specific for hBCATm and HIS-tagged hBCATm isolated PDI from neuronal cell lysates transfected with hBCATm-pDEST™26 Gateway® destination vector (HIS-tagged) and homogenates from human brain (Fig. 8). The expected 57 kDa PDI protein was precipitated with anti-hBCATm and detected using Western blot analysis using anti-PDI (Fig. 8A [i]). This band corresponded to the bands observed in whole cell lysates and pure overexpressed hPDI protein. The absence of a band in the control IP samples confirms that the proteins eluted are specific to anti-hBCATm. Moreover, using the same conditions, PDI was co-immunoprecipitated from human brain samples using anti-hBCATm (Fig. 8A [ii]). To further validate the specificity of this interaction, neuronal cells were transfected with HIS-tagged hBCATm and shown to specifically interact with PDI, where the concentration of hBCATm in the purified complex is ∼31 nM with 16 nM of PDI (as estimated from pure hBCATm and PDI, respectively), indicating a 2:1 ratio of hBCATm to PDI (Fig. 8B). These studies demonstrate that PDI was co-purified with hBCATm, suggesting that hBCATm directly binds to PDI in neuronal cells and, more importantly, in the human brain.
FIG. 8.
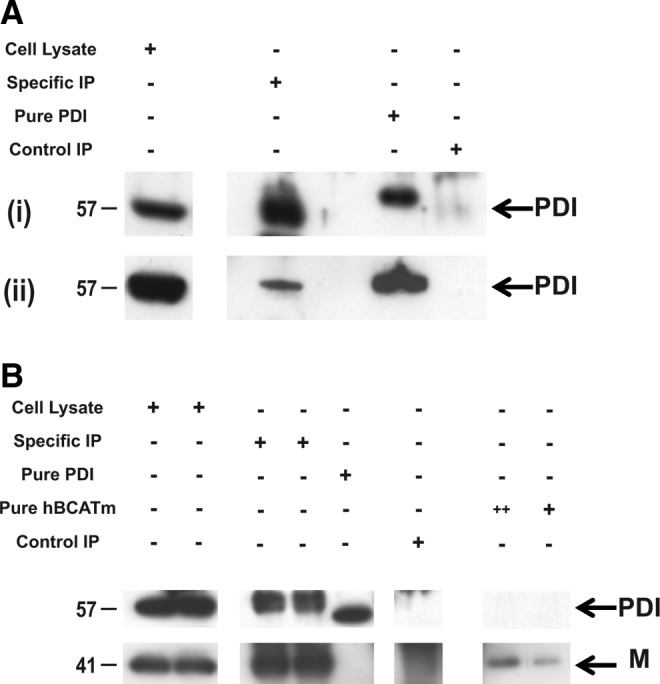
Immunoprecipitation of hBCATm with PDI from IMR-32 cell lysates and human brain homogenates. (A) (i) IMR-32 cell lysates (800 μg) and (ii) 800 μg of human brain tissue homogenate were immunoprecipitated with hBCATm-2 raised in mouse (specific immunoprecipitation [IP]) and rabbit IgG (control IP) using protein G MagSepaharose beads. (B) IMR-32 cells (800 μg) transfected with hBCATm-pDEST26 Gateway destination vector (HIS tagged) (Two standard concentrations of hBCATm were included (+= 5 ng and ++=10 ng of hBCATm)). PDI was detected using anti-mouse HRP (1/2000). Co-purification of PDI was assessed by immunoblotting with anti-PDI-2 (1/1000). These images are representative of three independent repeat experiments.
Co-localization of hBCATm with PDI in IMR-32 neuronal cells is reduced under increased oxidative stress
The co-localization of hBCATm with PDI decreased (Mx=0.675 to 0.159) when the cells were incubated with increasing concentrations of hydrogen peroxide or GSNO, respectively (Fig. 9A). Incubation of IMR-32 cells with GSNO did not impact the association between hBCATc and PDI; however, with hydrogen peroxide, the Mx value of hBCATc with PDI increased from 0.29 to 0.635 (Fig. 9B). Electron microscopy also shows that hBCATm is no longer observed in close proximity to PDI when the cells are treated with 100 μM of hydrogen peroxide (Fig. 9C). The increased association of hBCATm with the mitochondrial membrane under these conditions should be noted. These studies taken together provide evidence that hBCAT can associate with other redox-sensitive proteins such as PDI and that these associations are altered when the cells are exposed to increased oxidative stress.
FIG. 9.

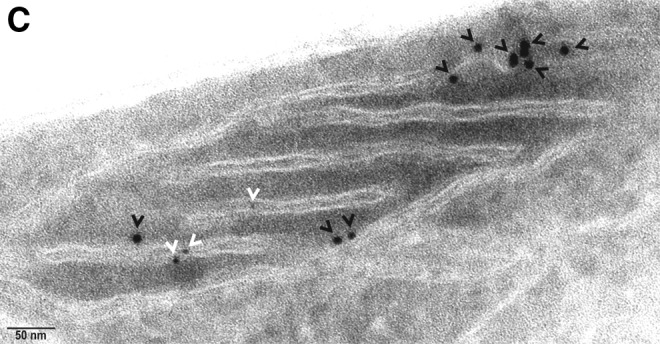
Co-localization of hBCATm with PDI in IMR-32 neuronal cells is reduced under increased oxidative stress. IMR-32 cells were treated with 50 and 100 μM of hydrogen peroxide and 100 and 500 μM GSNO for 30 min, respectively. Cells were subsequently fixed for immunostaining as described in the “Materials and Methods” section using primary antibodies: (A, B) anti-hBCATm-1, anti-hBCATc-1 (1/250 dilution), and anti-PDI-1 (1/150 dilution). Secondary antibodies: goat anti-rabbit Alex Fluor 568 (hBCATc-1, hBCATm-1, [1/500 dilution]) and goat anti-mouse Alex Fluor 488 (PDI-1 [1/500 dilution]). Slides were washed and mounted in 300 nM DAPI in glycerol. Mx was used to statistically assess the co-localization of hBCAT proteins with PDI under altered redox conditions. Mander's co-localization coefficients were derived using Volocity (Perkin-Elmer) from an average of 15 cell images. (A) hBCATm with PDI under oxidizing and S-nitrosylating conditions. (B) hBCATc with PDI under oxidizing and S-nitrosylating conditions. PDI (Green)+hBCATm/hBCATc (red). Merged images show yellow where co-localization is observed. (C) Electron micrograph of IMR-32 cells treated with 100 μM hydrogen peroxide for 30 min expressing hBCATm and PDI labeled with anti-mouse-10 nm (hBCATm-2–black arrowheads) and anti-rabbit-6 nm (PDI-2–white arrowheads) gold-conjugated secondary antibodies. Scale bar: C (50 nm). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Expression of hBCAT proteins and PDI in the human brain
To ascertain whether the hBCAT proteins were expressed in the same human brain, cells as PDI serial sections from the temporal and cerebellum of human brains were analyzed using antibodies that were specific for each respective protein. In agreement with previous studies (28), hBCATc is neuronal specific (Fig. 10A, B) and here using serial sectioning, we show that PDI is expressed in the same neuronal cells with a similar pattern of distribution (Fig. 10C, D). Interestingly, strong staining of PDI was also indicated in the endothelial cells of the vasculature, where hBCATm is predominantly expressed, in addition to some neuronal staining (Fig. 10E, F and G, H, respectively). These studies confirm that PDI is expressed in the same brain cells as the hBCAT isoforms and along with the co-immunoprecipitation studies, indicate that these proteins have the potential to interact in vivo.
FIG. 10.

Co-localization of hBCATc and hBCATm with PDI in the same cell types of the human cerebellum, temporal lobe, and hippocampus. Serial sections that were 7 μm in thickness were taken from each brain region and subsequently immunolabeled as described. Antibodies specific to hBCATc-1 and hBCATm-1 and PDI-2 were used to identifiy each respective protein. A–D show localization of hPDI and hBCATc to the purkinje cells (arrowhead) of the cerebellum. E–H shows the localization of hPDI and hBCATm to the endothelial layer of vessels (arrowhead) on serial sections of the temporal cortex (E+G) and the CA4 region of the hippocampus (F+H). Also shown in E+F is PDI labeling of neurons of the temporal cortex (E) and granule cells (*) of the dentate nucleus (F). Scale bars: A, C (100 μm); B, D, E, F, G, H (20 μM). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Discussion
Accumulation of mis-folded proteins is the hallmark of several neurodegenerative conditions, including Alzheimer's, Parkinson's, Huntington's, and prion diseases (9, 44, 71). However, the mechanisms that underpin this are not clearly defined. Similar to PDI and other oxidoreductases, the mammalian hBCAT proteins have a conserved CXXC motif, where the thiols of the CXXC motif can recycle between an intra-molecular disulfide bond (inactive) and the reduced dithiol (active) (61, 73). The present study demonstrates that hBCAT proteins catalyze the insertion of disulfide bonds into rdRNase-releasing active RNase, with the active cysteines playing a central role in this mechanism. Several other CXXC redox-sensitive proteins such as thioredoxin and platelet integrin alpha IIbbeta 3 have also been reported to have endogenous thiol disulfide isomerase activity (37, 40, 48). Inactivity reported for the N-terminal mutant proteins can be explained by their inability to form an intermediate with RNase, suggesting that the N-terminal cysteine is essential for the first step in forming a mixed disulfide with RNase. The N-terminal cysteine residues of both isoforms are central to their transaminase activity, as mutation of the reactive cysteine results in a complete loss in hBCAT activity (16). A number of the PDI family members, namely, ERp27, ERp28, PDILT, and TMX2, are missing in the surface exposed to N-terminal cysteine and similar to our N-terminal mutants, lack thiol disulfide isomerase activity, demonstrating the importance of this residue for catalysis (21, 65).
Even though the C-terminal mutant can potentially take part in the first step of the reaction, this intermediate may get trapped, as the reaction cannot subsequently progress to complete oxidation, as evidenced by the inability of these mutants to form active RNase, suggesting that this residue is required for substrate escape. This concept has previously been described for other systems, where stable intermediates result when the C-terminal residue is absent, such as that reported for ERp44, which results in the retention of these proteins in the ER, impacting protein function (3, 4). However, here, when S-glutathionylated, the single cysteine mutants have oxidase activity and form intermediates that are comparable to native proteins. This shows that either cysteine residue when forming a mixed disulfide with GSH can catalyze disulfide bond formation and implies that reactive thiols in other proteins subject to S-glutathionylation may also participate in oxidase activity, even if the CXXC motif is not present. Single active-site thiol disulfide exchange has also been explored and found to have effective activity for single mutants of the bacterial isomerase equivalent DsbA (69).
The increased oxidase activity of hBCATm relative to hBCATc reported as the environment became less reducing is consistent with their measured Em values (−310 and −225 mV, for hBCATc and hBCATm, respectively) (14). This indicates that while both proteins are reducing in nature, hBCATm is the less so of the two, and has a similar Em value to those reported for the human mitochondrial isoforms of the repair enzymes, thioredoxin and glutaredoxin (52, 66, 67). Taken together, we propose that hBCAT oxidoreductase activity is initiated through the S-glutathionylation of the hBCAT protein at the N-terminal cysteine position. Under nucleophilic attack, the reactive thiolate anion of RNase forms an inter-disulfide bond with S-glutathionylated hBCAT. Along with the C-terminal cysteine, the second thiol of RNase forms a disulfide bond, releasing RNase (S-S). Without the thiol at the C-terminal position, the mixed disulfide intermediate cannot be released and remains fixed to the N-terminal residue. Although S-glutathionylation is clearly involved in maximizing oxidase turnover, it is likely that other chaperones are involved in this mechanism in cells. A plausible candidate is human Ero1, a flavin adenine dinucleotide-binding oxidase, which has been proposed to act in part to provide oxidizing equivalents to PDI (24, 54). However, recent studies using knock-out Ero-1 have shown little physiological consequence, leading to debate as to whether Ero-1 is the primary donor of oxidizing equivalents or whether other mechanisms exist that substitute for its absence in these models (74). Lappi and Ruddock have recently revaluated the role of GSH buffers in this system and strongly advocate their physiological role in protein folding, in particular calling for further investigations into the physiological role of GSSG in the ER (38). The pathways for dithiol-disulfide exchange are complex and have not been entirely elucidated. Single thioredoxin-like domains of PDI have redox activity but poor isomerase activity, suggesting that a second CXXC motif is required for maximal dithiol-disulfide isomerase activity (19). This is possibly the case seen here for hBCAT, which shows that although they can undergo cycles of reduction followed by correct alignment of disulfide bonds, their isomerase activity is significantly less than their oxidase activity, and likely to be less significant than its oxidoreductase activity. However, in cells, the direction of the reaction to oxidation or reduction will depend on the overall Eh, the redox state of the CXXC motif of the hBCAT proteins, its microenvironment, and also other chaperone and redox repair systems present, which requires further investigation.
Co-localization of hBCATm with PDI in neuronal cells was curious, as PDI is considered to be largely associated with the ER; however, it has also been localized to mitochondria and the cytosol. Although the cytosolic and mitochondrial proteins are the two key mammalian isoforms, other spliced variants of hBCAT homologous to hBCATm have been described, namely a novel alternative splice variant found in placental tissue and a splice variant that acts as a co-repressor of thyroid hormone nuclear receptors (39, 59). It remains unknown whether another hBCAT isoform could exist and reside in the ER or whether the predominant isoforms participate in their PDI-like activity elsewhere in the cell, as disulfide bond formation can also occur in mitochondria and outside the cell in the blood (1, 36). However, here, using Mia40 as a control mitochondrial protein, PDI was located to the mitochondria using electron microscopy and in close proximity (≤50 nm apart) to hBCATm, suggesting co-localization. These findings along with confocal microscopy confirm that the interaction between PDI and hBCATm is specific to the mitochondria. Co-immunoprecipitation assays validated direct binding, not only in neuronal cell cultures but also from human brain tissue. Moreover, the fact that PDI was identified in neuronal and endothelial cells, where both hBCATs reside, suggests the clear potential for this interaction to be physiologically relevant. We hypothesize that hBCATm and PDI could operate through dithiol-disulfide exchange, and hBCATm could potentially act as a chaperone to assist PDI in facilitating protein folding or in its own capacity act as a dithiol-disulfide oxidoreductase.
The importance of molecular chaperones and PDI is reflected in their role for the correct folding of proteins and the elimination of misfolded or irreversibly damaged proteins from the cell (44). Accumulation of aggregates such as α-synuclein and synphillin-1 in Parkinson's disease and amyloid-β and hyper-phosphorylated tau in AD along with inclusion bodies associated with amylotrophic lateral sclerosis (ALS), and prion diseases are common features of these neurodegenerative conditions (35, 44). Molecular chaperones such as the heat shock proteins (HSP70 and HSP90, in particular) and PDI are up-regulated in response to cellular stress and believed to function as neuroprotectors in these diseases [reviewed in Andreu et al. (2)]. These systems operate as a network with the unfolded protein response, which is directed to the clearance of modified or incorrectly folded proteins. However, although these systems are up-regulated, evidence suggests that oxidative and S-nitrosative stress may impact protein function, thus compromising their neuroprotective role (46, 47, 56, 63). One such example is a study by Uehara et al., which showed that PDI was S-nitrosylated in the brains of patients with Parkinson's disease and AD (63). Modeling of this mechanism in SH-SY5Y cells resulted in the accumulation of protein aggregates, supporting their hypothesis that S-nitrosylated PDI contributes to protein misfolding and has the potential to play a role in the pathogenesis of AD. In a separate study, PDI-SNO was also detected in the spinal cord of ALS patients.
In rat models, up-regulation of hBCATc has been related to cell survival. A report by Kholodilov et al. showed an increase in mRNA expression of BCATc in the substantia nigra of rats after developmental striatal target injury (34). Although this correlated with cell death, the neurons that were immunopositive for BCATc had normal physiology and rarely contained apoptotic chromatin, suggesting that BCATc played a neuroprotective role. Furthermore, administration of brain-derived neurotrophic factor (role in neuronal survival, growth, and differentiation) after a lesion of the visual cortex in newborn rats resulted in the up-regulation of BCATc (10). Increased expression was also observed in the brain of transgenic rats overexpressing BDNF in a time- and region-dependent manner (43). Collectively, these studies support the role of the BCAT proteins in maintaining neuronal survival. Recent studies by our group have demonstrated that both hBCAT isoforms show increased protein expression (up to 140% for hBCATm) in the brain of patients with AD relative to matched control patients (n=30, frontal and temporal region) (Conway, unpublished data). Based on previous studies, we suggest that the expression of hBCAT is increased in response to changes in the cellular redox environment, to assist in neuronal protection. We speculate that since the hBCAT proteins have oxidoreductase activity and can refold proteins, they may play a role in facilitating thiol-disulfide exchange either independently or as a redox chaperone to PDI or Mia40. However, similar to PDI, the hBCAT proteins are regulated through oxidation and S-nitrosylation (13, 15, 16), such that under increased cellular stress, their proposed neuroprotective role, and their association with these proteins would be affected. Therefore, knowing that the function of these proteins is compromised under cellular stress highlights the impact during pathogenic conditions where protein misfolding features. Future studies will investigate and characterize the role of redox-modified hBCAT with potential redox partners and protein folding in cells.
In conclusion, we have identified that hBCAT proteins have novel dithiol-disulfide isomerase activity that plays a role in protein folding. We demonstrate that S-glutathionylation is not only present to protect the function of a redox-sensitive protein, a concept held in high regard by most groups, but it has also shown value mechanistically and was shown here to be integral to the refolding of proteins catalyzed by hBCAT. Moreover, we have demonstrated that hBCATm is localized with other redox-sensitive proteins such as PDI and Mia40 in the mitochondria. These findings may have physiologically relevant implications in the human brain, but more importantly in protein misfolding, a key mechanism that is associated with AD pathology.
Materials and Methods
Materials
Cytidine 2′,3′–cyclic monophosphate (cCMP), RNase, bovine PDI, nonessential amino acids, fetal calf serum, 100× penicillin/streptomycin solution, glutaraldehyde, sodium borohydride, sodium cacodylate, Tween®20, and Triton X-100 were obtained from Sigma-Aldrich (Dorset, UK). RPMI 1640 was obtained from Lonza (Gloucestershire, UK). Human PDI (hPDI) was obtained from Cambridge Bioscience (Cambridge, UK). IMR-32, neuroblastoma cell line (ATCC® CCL-127™) was obtained from LGC Promochem (Middlesex, UK). NuPAGE® LDS sample buffer (4×), NuPAGE Novex® 4%–12% Bis-Tris 1 mm gel, 20× NuPAGE MES SDS running buffer, and 20× NuPAGE Transfer Buffer were obtained from Invitrogen (Paisley, UK). Electron microscopy (EM) grade paraformaldehyde was purchased from BDH (Leicestershire, UK). Acetylated bovine serum albumin (BSA) (10%) was purchased from Aurion (Wageningen, The Netherlands). Peroxidase, substrate 3,3′-diaminobenzidine (DAB), donkey serum, 4′,6-diamino-2-phenylindole (DAPI)-containing mounting medium, biotin-labeled secondary antibody raised to IgG, and avidin (Vectastain ABC kit) were purchased from Vector Labs (Peterborough, UK). Hematoxylin, Clearene, and Clearium were purchased from SurgiPath (Peterborough, UK). BM chemiluminescent horse radish peroxidase (HRP) substrate was purchased from Roche (West Sussex, UK). Hyperfilm was obtained from GE Healthcare (Cardiff, UK). All antibodies used in these studies are summarized in Supplementary Table 1. All other materials were purchased from Fisher Scientific (Loughborough, UK).
Preparation and activity of rdRNase and scRNase
Twenty milligrams of RNase was incubated with 150 mM DTT and 6 M guanidine hydrochloride over 18 h at room temperature (RT) in 0.1 M Tris, pH 8.6, desalted into 0.01 N HCl, and stored under argon at −80°C. To prepare scRNase, rdRNase in 6 M guandine-HCl was adjusted to pH 8.6 with solid Tris, sparged with oxygen for 1 h at RT, and allowed to re-oxidise in the dark for 4 days. The thiol concentration of scRNase was assessed before and after oxidation by titration with DTNB (18). RNase activity was determined by the protein refolding assay (detailed in 41, 42, 49, 50). Briefly, a final concentration of 1.4 μM RNase was added to 0.44 mM cCMP that was prepared in 0.1 M 4-morpholinepropanesulfonic acid, pH 7.0 with the increase in absorbance at 284 nm monitored over 10 min.
Reactivation of rdRNase catalyzed by native hBCAT and mutant proteins
All hBCAT and mutant proteins were exchanged into 0.1 M Tris-HCL, pH 7.4 with 1 mM EDTA before incubation with rdRNase. Typically, for each refolding assay, 30 μM rdRNase was incubated at RT with test protein and aliquots were removed to assess RNase activity at 24 and 48 h. The effect of time (12–48 h) and concentration (4–24 μM) on this reaction was assessed under these conditions. The redox environment was also varied using five different GSH/GSSG ratios (1:0, 3:1, 1:1, 1:3, and 0:1). In a separate experiment, the hBCAT proteins were pretreated with 500 μM GSNO for 30 min at RT before incubation with rdRNase. Control samples for the hBCAT and mutant proteins±rdRNase, rdRNase alone, for each GSH/GSSG ratio, and GSNO were included for each time point and each concentration. Subsequently, using a GSH/GSSG ratio of 3:1, the apparent Vmax for rdRNase and scRNase was determined by holding hBCAT at 10 μM with the substrate (either rdRNase or scRNase) concentration between 5 and 60 μM. A standard curve was generated from the initial rates of a range of RNase concentrations (0–4 μM). Using the slope, the concentration of active RNase formed/μM of protein was calculated. Western blot analysis of these samples was carried out as described in Supplementary Data.
Far Western immunoblotting
Human BCAT (1.0–3.5 μg, Fig. 5A) or hPDI (0.1–0.5 μg, Fig. 5B and constant at 0.4 μg, Fig. 5C) was applied to a nitrocellulose membrane. After blocking with 1× Tris buffer saline with tween (TBST) containing 5% BSA for 1 h at RT, the membrane was incubated with 5 μg/ml of PDI, hBCATc, or hBCATm, respectively, for 90 min at 37°C, with shaking at 100 rpm. After washing, bound hBCAT was detected by incubating with anti-hBCAT (1/1000 dilution) or for the hBCATs, anti-PDI-2 (1/1000) shaking, overnight at 4°C. Bands were visualized using BM chemiluminescent HRP substrate and exposed to HyperFilm.
Immunocytochemistry of hBCAT and PDI in neuronal cells
IMR-32 cells were allowed to reach 70%–80% confluence and treated +/− hydrogen peroxide (50–100 μM) and GSNO (100–500 μM) for 30 min, respectively, at 37°C in a humidified incubator that was adjusted to contain 5% carbon dioxide. After serial washes, the cells were fixed with 0.25% glutaraldehyde and permeabilized with 0.2% Triton X-100 for 20 min at RT in a 0.1 M sodium cacodylate buffer, pH 7.4 adjusted to contain 0.1 M sucrose. The cells were further treated by a succession of washes with sodium borohydride (1 mg/ml in PBS) and blocked in 3% BSA overnight before incubating with primary antibodies, anti-hBCATc, anti-hBCATm-1 (1/500), anti-PDI-1 (1/150 dilution), anti-PDI-2, anti-Mia40 (1/100), and anti-hBCATm-2 (1/250), respectively, for 1 h at RT. After serial washes, the cells were incubated with goat anti-rabbit Alexa Fluor® 568 (hBCATc, hBCATm-1, PDI-2, and Mia40, 1/500 dilution) and goat anti-mouse Alexa Fluor 488 (PDI-1 and hBCATm-2, 1/500 dilution) for 1 h at RT. Slides were washed in cacodylate buffer and mounted in 300 nM DAPI in glycerol. The Mander's correlation coefficients (Mx and My) were derived using Volocity (Perkin-Elmer) from an average of 15 individual cell images.
Electron microscopy of hBCAT and PDI in neuronal cells
Cells that had been previously incubated +/−100 μM of hydrogen peroxide for 30 min were fixed in a final concentration of 2% formaldehyde and 0.2% glutaraldehyde in phosphate buffer and processed according to the Tokuyasu method for cryosectioning (55, 60). Seventy nanometre sections were prepared at −120°C with 1% methylcellulose in 1.2 M sucrose and transferred onto carbon-coated copper mesh grids. The sections were labeled with primary antibodies to anti-hBCATm-1-3, anti-Mia40, anti-PDI-1, and anti-PDI-2 (1/10), respectively. Gold-labeled secondary antibodies, 6 and 10 nm (1/20), respectively, were incubated for 1 h at RT. All antibodies were prepared in 0.1% acetylated BSA. The sections were counterstained with 0.3% uranylacetate in 2% methylcellulose on ice and imaged on a CM10 (Philips) electron microscope.
Mammalian expression of HIS-tagged hBCATm using Gateway™ technology
According to the manufacturer's instructions, the LR recombination reaction contained 50–100 ng of purified hBCATm pENTR221™ entry vector (synthesized and prepared by GeneArt, Life Technologies), 150 ng of pDEST26 Gateway destination vector and TE buffer, pH 8.0 was used to adjust the final volume to 8 μl. Positive controls containing 100 ng pENTR-gus with 150 ng pDEST26 and negative controls containing 150 ng of purified BCATm pENTR221 with 150 ng pDEST26 were also included. The reaction was started with 2 μl of LR Clonase® II enzyme mix (with exception to the negative control). The samples were incubated in a PCR hot block at 25°C for 3 h. To stop the LR reaction, 1 μl of proteinase K solution was added and incubated at 37°C for 10 min. The completed LR reaction (1 μl) was subsequently transformed into Library Efficiency® DH5α™ Cells according to the manufacturer's instructions. Plasmids were purified using the PureLink™ Hi-Pure plasmid mini-prep purification according to the manufacturer's instructions, and restriction analysis was used to confirm correct orientation of the insert. IMR-32 cells were subsequently transfected with hBCATm expression plasmid using jetPRIME® transfection reagent (Polyplus) and following the protocol prescribed by the manufacturers. Controls included transfection reagent alone, cell only, and pcDNA3-EGFP to assess transfection efficiency.
Co-immunoprecipitation of hBCATm with PDI from IMR-32 cell lysates and human brain homogenates
Cell lysates (800 μg) from IMR-32 cells or brain homogenates (800 μg) were incubated with anti-hBCATm-protein G Mag Sepharose beads overnight. Total cell lysates, bound protein, and pure overexpressed hPDI were separated on a 6.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred onto a nitrocellulose membrane for Western blot analysis as described earlier. Primary antibody (1/1000 for hPDI) was prepared in 5% nonfat milk powder in 1× TBST and incubated overnight at 4°C before several washes with 1× TBST. Secondary antibody, mouse anti-rabbit light chain specific (1:2000) was added for 1 h at RT and then washed with 1× TBST followed by a 1 h incubation with anti-mouse HRP (1/2000). The positive bands were visualized using BM chemiluminescent HRP substrate and imaged using hyperfilm.
Immunohistochemistry of the hBCAT isoforms and PDI
The study was approved by the North Somerset and South Bristol Research Ethics Committee. All brain tissue used in this study was from brains donated with consent to the South West Dementia Brain Bank in the University of Bristol, UK. Serial sections (7 μm) were taken from the temporal and cerebellum brain regions, placed in a 60°C oven overnight before immunohistochemical staining, and subsequently dewaxed in clearene (2×5 min) and dehydrated in 100% ethanol (2×3 min). Endogenous peroxidase was quenched in 0.09% hydrogen peroxide/methanol solution for 30 min at RT. The slides were pretreated with citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0), then washed (2×3 min) in PBS containing 0.154 M NaCl, 1.86 mM Na2H2PO4.2H2O, 7.48 mM Na2HPO4.12H2O, pH 7.1, and blocked with 10% horse serum in PBS for 20 min at RT. Sections were incubated at RT overnight with primary antibody (1/6000 for both hBCAT isoforms) in PBS. The sections were washed in PBS (2×3 min) and incubated with biotinylated antibody to IgG for 20 min (Vectastain ABC kit). The slides were washed again in PBS (2×3 min) and incubated with the avidin-biotin complex in PBS for 20 min (Vectastain ABC kit). Slides were developed with DAB/H2O2 in distilled water (DAB substrate kit) for 10 min before immersion in copper sulfate solution (16 mM CuSO4.5H2O, 0.123 M NaCl) for 4 min, and counterstained with Harris' hematoxylin (25% Gill haematoxylin). The slides were dehydrated in 100% ethanol (2×5 min), cleared in 100% Clearene (2×3 min), and mounted in Clearium. Sections were viewed and imaged on a Nikon Eclipse 50i or a Leica DMR microscope.
Supplementary Material
Abbreviations Used
- AD
Alzheimer's disease
- ALS
amylotrophic lateral sclerosis
- BCKDH
branched chain keto acid dehydrogenase complex
- BSA
bovine serum albumin
- cCMP
cytidine 2′,3′–cyclic monophosphate
- DAB
substrate 3,3′-diaminobenzidine
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- ER
endoplasmic reticulum
- EM
electron microscopy
- GSH
glutathione
- GSSG
glutathione disulphide
- GSNO
S-nitroso-glutathione
- HRP
horse radish peroxidase
- hBCATm/c
human mitochondrial/cytosolic branched-chain aminotransferase
- IP
immunoprecipitation
- Mx
Mander's correlation coefficient
- PDI
protein disulphide isomerase
- rdRNase
reduced denatured ribonuclease A
- RT
room temperature
- scRNase
scrambled ribonuclease A
- TBST
Tris buffer saline with tween
Acknowledgments
The authors would like to sincerely thank Leslie Poole, Wake Forest University, USA, for advice on rdRNase kinetics and the discussion of dithiol-disulfide exchange and also Sarah O'Neill, Royal College of Surgeons, Ireland, for technical assistance. They would also like to acknowledge the help of Paul Verkade and the EM technical staff at the Wolfson Bioimaging Facility, University of Bristol, UK. This research was supported by a Research PhD Fellowship grant awarded by Bristol Research to Alzheimer's and Care of the Elderly (BRACE), Bristol, UK, and an HEFCE-funded bursary from the University of the West of England to Myra E. Conway.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, and Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci U S A 103: 13932–13937, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreu CI, Woehlbier U, Torres M, and Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett 586: 2826–2834, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Anelli T, Alessio M, Bachi A, Bergamelli L, Bertoli G, Camerini S, Mezghrani A, Ruffato E, Simmen T, and Sitia R. Thiol-mediated protein retention in the endoplasmic reticulum: the role of ERp44. EMBO J 22: 5015–5022, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A, and Sitia R. ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J 21: 835–44, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appenzeller-Herzog C. and Ellgard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta 86: 535–548, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Barlow AL, Macleod A, Noppen S, Sanderson J, and Guérin CJ. Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of pearson's correlation coefficient. Microsc Microanal 16: 710–724, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Berndt C, Lillig CH, and Holmgren A. Thioredoxins and glutaredoxins facilitators of protein folding. Biochim Biophys Acta 83: 641–650, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Bixel M, Hutson S, and Hamprecht B. Cellular distribution of branched-chain amino acid aminotransferase isoenzymes among rat brain glial cells in culture. J Histochem Cyto 45: 685–694, 1997 [DOI] [PubMed] [Google Scholar]
- 9.Bixel M, Shimomura Y, Hutson S, and Hamprecht B. Distribution of key enzymes of branched-chain amino acid metabolism in glial and neuronal cells in culture. J Histochem Cyto 49: 407–418, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Castellano S, Macchi F, Scali M, Huang JZ, and Bozzi Y. Cytosolic branched chain aminotransferase (BCATc) mRNA is up-regulated in restricted brain areas of BDNF transgenic mice. Brain Res 1108: 12–18, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, and Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304: 1328–1331, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Cole JT, Sweatt AJ, and Hutson SM. Expression of mitochondrial branched-chain aminotransferase and α-keto-acid dehydrogenase in rat brain: implications for neurotransmitter metabolism. Front Neuroanat 6: 18, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coles SJ, Easton P, Sharrod H, Hutson SM, Hancock J, Patel VB, and Conway ME. S-Nitrosoglutathione inactivation of the mitochondrial and cytosolic BCAT proteins: S-nitrosation and S-thiolation. Biochemistry 48: 645–656, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Coles SJ, Hancock JT, and Conway ME. Differential redox potential between the human cytosolic and mitochondrial branched-chain aminotransferase. Acta Biochim Biophys 44: 172–176, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Conway ME, Coles SJ, Islam MM, and Hutson SM. Regulatory control of human cytosolic branched-chain aminotransferase by oxidation and S-glutathionylation and its interactions with redox sensitive neuronal proteins. Biochemistry 47: 5465–5479, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Conway ME, Poole LB, and Hutson SM. Roles for cysteine residues in the regulatory CXXC motif of human mitochondrial branched chain aminotransferase enzyme. Biochemistry 43: 7356–7364, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Conway ME, Yennawar N, Wallin R, Poole LB, and Hutson SM. Human mitochondrial branched chain aminotransferase: structural basis for substrate specificity and role of redox active cysteines. Biochim Biophys Acta 1647: 61–65, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Conway ME, Yennawar N, Wallin R, Poole LB, and Hutson SM. Identification of a peroxide-sensitive redox switch at the CXXC motif in the human mitochondrial branched chain aminotransferase. Biochemistry 41: 9070–9078, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Darby NJ, Penka E, and Vincentelli R. The multi-domain structure of protein disulfide isomerase is essential for high catalytic efficiency. J Mol Biol 276: 239–247, 1998 [DOI] [PubMed] [Google Scholar]
- 20.Davoodi J, Drown PM, Bledsoe RK, Wallin R, Reinhart GD, and Hutson SM. Overexpression and characterization of the human mitochondrial and cytosolic branched-chain aminotransferases. J Biol Chem 273: 4982–4989, 1998 [DOI] [PubMed] [Google Scholar]
- 21.Ellgard L. and Ruddock LW. The human protein disulphide isomerase family:substrate interactions and functional properties. EMBO Rep 6: 28–32, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Espinosa MA, Wallin R, Hutson SM, and Sweatt AJ. Widespread neuronal expression of branched-chain aminotransferase in the CNS: implications for leucine/glutamate metabolism and for signaling by amino acids. J Neurochem 100: 1458–1468, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Goto M, Miyahara I, Hirotsu K, Conway M, Yennawar N, Islam MM, and Hutson SM. Structural determinants for branched-chain aminotransferase isozyme-specific inhibition by the anticonvulsant drug Gabapentin. J Biol Chem 280: 37246–37256, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Gross E, Sevier CS, Heldman N, Vitu E, Bentzur M, Kaiser CA, Thorpe C, and Fass D. Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci U S A 10: 103: 299–304, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris RA, Joshi M, and Jeoung NH. Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem Biophys Res Commun 313: 391–396, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Hatahet F. and Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal 11: 2807–2850, 2009 [DOI] [PubMed] [Google Scholar]
- 27.Hawkins HC, Blackburn EC, and Freedman RB. Comparison of the activities of protein disulphide-isomerase and thioredoxin in catalysing disulphide isomerization in a protein substrate. Biochem J 275 (Pt 2): 349–353, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hull J, Hindy ME, Kehoe PG, Chalmers K, Love S, and Conway ME. Distribution of the branched chain aminotransferase proteins in the human brain and their role in glutamate regulation. J Neurochem 123: 997–1009, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Hutson SM. Subcellular distribution of branched-chain aminotransferase activity in rat tissues. J Nutrition 118: 1475–1481, 1988 [DOI] [PubMed] [Google Scholar]
- 30.Hutson SM, Wallin R, and Hall TR. Identification of mitochondrial branched chain aminotransferase and its isoforms in rat tissues. J Biol Chem 267: 15681–15686, 1992 [PubMed] [Google Scholar]
- 31.Hwang C, Sinskey AJ, and Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257: 1496–1502, 1992 [DOI] [PubMed] [Google Scholar]
- 32.Ichihara A. and Koyama E. Transaminase of branched chain amino acids. J Biochem 59: 160–169, 1966 [DOI] [PubMed] [Google Scholar]
- 33.Islam MM, Wallin R, Wynn RM, Conway M, Fujii H, Mobley JA, Chuang DT, and Hutson SM. A novel branched-chain amino acid metabolon. Protein-protein interactions in a supramolecular complex. J Biol Chem 282: 11893–11903, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Kholodilov N, Neystat M, Oo T, Hutson S, and Burke R. Upregulation of cytosolic branched chain aminotransferase in substantia nigra following developmental striatal target injury. Mol Brain Res 75: 281–286, 2000 [DOI] [PubMed] [Google Scholar]
- 35.Kim HT, Russell RL, Raina AK, Harris PL, Siedlak SL, Zhu X, Petersen RB, Shimohama S, Smith MA, and Perry G. Protein disulfide isomerase in Alzheimer disease. Antioxid Redox Signal 2: 485–489, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Koehler CM, Beverly KN, and Leverich EP. Redox pathways of the mitochondrion. Antioxid Redox Signal 8: 813–822, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Langenbach KJ. and Sottile J. Identification of protein-disulfide isomerase activity in fibronectin. J Biol Chem 274: 7032–7038, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Lappi AK. and Ruddock LW. Reexamination of the role of interplay between glutathione and protein disulfide isomerase. J Mol Biol 409: 238–249, 2011 [DOI] [PubMed] [Google Scholar]
- 39.Lin HM, Kaneshige M, Zhao L, Zhang X, Hanover JA, and Cheng SY. An isoform of branched-chain aminotransferase is a novel co-repressor for thyroid hormone nuclear receptors. J Biol Chem 276: 48196–48205, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Lundström J. and Holmgren A. Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. J Biol Chem 265: 9114–9120, 1990 [PubMed] [Google Scholar]
- 41.Lyles MM. and Gilbert HF. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30: 613–619, 1991 [DOI] [PubMed] [Google Scholar]
- 42.Lyles MM. and Gilbert HF. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: pre-steady-state kinetics and the utilization of the oxidizing equivalents of the isomerase. Biochemistry 30: 619–625, 1991 [DOI] [PubMed] [Google Scholar]
- 43.Madeddu F, Naska S, Menna E, Chiellini C, Sweatt AJ, Hutson SM, Benzi L, Maffei M, Maffei L, and Bozzi Y. Intraocular delivery of BDNF following visual cortex lesion upregulates cytosolic branched chain aminotransferase (BCATc) in the rat dorsal lateral geniculate nucleus. Eur J Neurosci 20: 580–586, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Muchowski PJ. Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 35: 9–12, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Nakamura H. Thioredoxin and its related molecules: update 2005. Antioxid Redox Signal 7: 823–828, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, and Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78: 596–614, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM, and Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer's disease inferior parietal lobule, a proteomics approach. J Neurosci Res 85: 1506–1514, 2007 [DOI] [PubMed] [Google Scholar]
- 48.O'Neill S, Robinson A, Deering A, Ryan M, Fitzgerald DJ, and Moran N. The platelet integrin alpha IIbbeta 3 has an endogenous thiol isomerase activity. J Biol Chem 275: 36984–36990, 2000 [DOI] [PubMed] [Google Scholar]
- 49.Pigiet VP. and Schuster BJ. Thioredoxin -catalyzed refolding of disulfide-containing proteins. Proc Natl Acad Sci U S A 83: 7643–7647, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rancy PC. and Thorpe C. Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry 47: 12047–12056, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruoppolo M, Lundström-Ljung J, Talamo F, Pucci P, and Marino G. Effect of glutaredoxin and protein disulfide isomerase on the glutathione-dependent folding of ribonuclease A. Biochemistry 36: 12259–12267, 1997 [DOI] [PubMed] [Google Scholar]
- 52.Sagemark J, Elgán TH, Bürglin TR, Johansson C, Holmgren A, and Berndt KD. Redox properties and evolution of human glutaredoxins. Proteins 68: 879–892, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Schaffner W. and Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 56: 502–514, 1973 [DOI] [PubMed] [Google Scholar]
- 54.Sevier CS, Qu H, Heldman N, Gross E, Fass D, and Kaiser CA. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 129: 333–344, 2007 [DOI] [PubMed] [Google Scholar]
- 55.Slot JW. and Geuze HJ. Cryosectioning and immunolabelling. Nat Protoc 2: 2480–2491, 2007 [DOI] [PubMed] [Google Scholar]
- 56.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, and Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging 27: 1564–1576, 2006 [DOI] [PubMed] [Google Scholar]
- 57.Sweatt AJ, Garcia-Espinosa MA, Wallin R, and Hutson SM. Branched-chain amino acids and neurotransmitter metabolism: expression of cytosolic branched-chain aminotransferase (BCATc) in the cerebellum and hippocampus. J Comp Neurol 477: 360–370, 2004 [DOI] [PubMed] [Google Scholar]
- 58.Tanaka S, Uehara T, and Nomura Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J Biol Chem 275: 10388–10393, 2000 [DOI] [PubMed] [Google Scholar]
- 59.Than NG, Sumegi B, Than GN, Bellyei S, and Bohn H. Molecular cloning and characterization of placental tissue protein 18 (PP18a)/human mitochondrial branched-chain aminotransferase (BCATm) and its novel alternatively spliced PP18b variant. Placenta 22: 235–243, 2001 [DOI] [PubMed] [Google Scholar]
- 60.Tokuyasu KT. A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol 57: 551–565, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tu BP, Ho-Schleyer SC, Travers KJ, and Weissman JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290: 1571–1574, 2000 [DOI] [PubMed] [Google Scholar]
- 62.Tuite MF. and Melki R. Protein misfolding and aggregation in ageing and disease: molecular processes and therapeutic perspectives. Prion 1: 116–120, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, and and Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513–517, 2006 [DOI] [PubMed] [Google Scholar]
- 64.Verkade P, Schrama LH, Verkleij AJ, Gispen WH, and Oestreicher AB. Ultrastructural co-localization of calmodulin and B-50/growth-associated protein-43 at the plasma membrane of proximal unmyelinated axon shafts studied in the model of the regenerating rat sciatic nerve. Neuroscience 79: 1207–1218, 1997 [DOI] [PubMed] [Google Scholar]
- 65.Walker KW, Lyles MM, and Gilbert HF. Catalysis of oxidative protein folding by mutants of protein disulfide isomerase with a single active-site cysteine. Biochemistry 35: 1972–1980, 1996 [DOI] [PubMed] [Google Scholar]
- 66.Watson WH, Chen Y, and Jones DP. Redox state of glutathione and thioredoxin in differentiation and apoptosis. Biofactors 17: 307–314, 2003 [DOI] [PubMed] [Google Scholar]
- 67.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, and Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem 278: 33408–33415, 2003 [DOI] [PubMed] [Google Scholar]
- 68.Wilkinson B. and Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta 1699: 35–44, 2004 [DOI] [PubMed] [Google Scholar]
- 69.Wunderlich M, Otto A, Maskos K, Mücke M, Seckler R, and Glockshuber R. Efficient catalysis of disulfide formation during protein folding with a single active-site cysteine. J Mol Biol 247: 28–33, 1995 [DOI] [PubMed] [Google Scholar]
- 70.Xiao R, Lundström-Ljung J, Holmgren A, and Gilbert HF. Catalysis of thiol/disulfide exchange. Glutaredoxin 1 and protein-disulfide isomerase use different mechanisms to enhance oxidase and reductase activities. J Biol Chem 280: 21099–21106, 2005 [DOI] [PubMed] [Google Scholar]
- 71.Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, Uehara T, and Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A 101: 10810–10814, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yennawar NH, Conway ME, Yennawar HP, Farber GK, and Hutson SM. Crystal structures of human mitochondrial branched chain aminotransferase reaction intermediates: ketimine and pyridoxamine phosphate forms. Biochemistry 41: 11592–11601, 2002 [DOI] [PubMed] [Google Scholar]
- 73.Yennawar NH, Islam MM, Conway M, Wallin R, and Hutson SM. Human mitochondrial branched chain aminotransferase isozyme: structural role of the CXXC center in catalysis. J Biol Chem 281: 39660–39671, 2006 [DOI] [PubMed] [Google Scholar]
- 74.Zito E, Chin KT, Blais J, Harding HP, and Ron D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J Cell Biol 188: 821–832, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.