

Abstract
Estrogen receptor α (ERα) is a well-validated drug target for a majority of breast cancers. But the target sites on this receptor are far from exhaustively defined. Almost all ER antagonists in clinical use function by binding to the ligand-binding pocket to occlude agonist access. Resistance to this type of drugs may develop over time, not caused by the change of ERα itself, but by changes in ER associated proteins. This observation is fueling the development of reagents that downregulate ER activity through novel binding sites. However, it is challenging to find general ER antagonists that act independently from other known ER ligands. In this report, we describe the utility of RNA aptamers in the search for new drug target sites on ERα. We have identified three high affinity aptamers and characterized one of them in detail. This aptamer interacted with ERα in a way not affected by the presence or absence of either the steroidal ligands or the estrogen response DNA elements, and effectively inhibited ER-mediated transcriptional activation in a breast cancer cell line. Serving as a novel drug lead, it may also be used to guide the rational chemical synthesis of small molecule drugs or to perform screens of small molecule libraries for those that are able to displace the aptamer from its binding site.
Introduction
Estrogen plays a prominent role in the etiology of various cancers. Its effect on the target tissue is primarily mediated through binding to specific intracellular estrogen receptors, ERα and ERβ. At least 70% of breast cancers are classified as ER-positive, and interfering with estrogen action has been the first and most successful targeted cancer therapy in history (Liang and Shang, 2013). An early implementation of this strategy was surgical oophorectomy to eliminate estrogen production in premenopausal breast cancer patients. A more sophisticated approach is to modulate ER function through molecular mimicry by small molecules structurally related to estrogen. Representing this category of antiestrogen drug therapies, tamoxifen, the first drug developed to target ER function, acts as an ER antagonist in breast cancer cells (Cole et al., 1971; WARD, 1973). While tamoxifen remains the preferred choice for treating hormone-sensitive breast cancers, there has been rapid development of other selective estrogen receptor modulators and aromatase inhibitors (aromatase is a critical enzyme in estrogen biosynthesis in postmenopausal women) for the treatment of breast cancer and other estrogenopathies (Shelly et al., 2008; Litton et al., 2012). Unfortunately, although more than 65% of breast tumors express ERα, fewer than half of them respond favorably to conventional antiestrogen therapy. And tumors initially sensitive to tamoxifen become resistant over time. Overcoming endocrine resistance has been the main motivation driving the research of estrogen signaling, which revealed the molecular mechanism underlying ER pharmacology (Droog et al., 2013).
Estrogen receptors are members of the large conserved nuclear receptor superfamily of transcriptional activators, which share conserved structural and functional organization comprising multiple domains responsible for DNA binding, ligand binding, or transcriptional activation. The ligand-binding domain (LBD) of ER serves as the densely connected hub of a regulatory network for the coordinated recruitment of factors to the promoters of specific genes in the chromatin environment of the nucleus. The binding of a ligand triggers the association of ER with various coactivators or corepressors, which determines the response of the target gene (Merrell et al., 2011; Cirillo et al., 2013). As a result, ER activity is affected by the relative and absolute levels of these receptor-associated proteins in different cells. This mechanistic insight prompted a new strategy of antagonizing ER function by directly or indirectly interfering with receptor-coregulator interaction downstream of ligand binding (Carraz et al., 2009). However, more than 300 proteins have been shown to interact with one or more nuclear receptors, and many of these coregulators interact with ER (Manavathi et al., 2013). This daunting complexity gradually brought the attention back to the well-validated target, ERα itself (McDonnell and Wardell, 2010). Although not the effector, ERα is a nucleating point whose mere presence makes it possible to engage the various coregulators. Therefore, even after tamoxifen resistance, ERα is still a legitimate target as long as the cancer is ER positive.
For historical reasons, when the term “ligand” is used in the ER-related literature, it often designates a small lipophilic molecule that recognizes the ligand-binding pocket on the LBD of ER. But in a broader sense, the DNA estrogen response elements (ERE; Helsen et al., 2012) and the coregulators are also ligands of the receptor. Currently, almost all ER modulators in clinical use interact with the classical ligand-binding pocket (Dai et al., 2008), which is well characterized (Eiler et al., 2001). But therapeutics that target ER by means other than those currently available may be useful in the treatment of endocrine resistant breast cancers (Moore et al., 2010; Shapiro et al., 2011). In particular, we are interested in finding new ligands whose interaction with ER is not affected by the presence or absence of other known ligands (i.e., estrogens, DNA, or other factors). For this purpose, we sought after aptamers that bind and inhibit ER activity in a way indifferent to the binding of estrogen and DNA. In this report, we describe RNA aptamers identified for apo-ERα by in vitro selection, which recognize a novel but undefined site. We demonstrated that their binding to ER was not affected by either 17β-estradiol (E2) or ERE. We further refined one of the aptamers and proved its specific interaction with ER in living cells. When expressed in the ERα-positive breast cancer cell line MCF7, this aptamer significantly inhibited ERα mediated transcriptional activation. We believe that these novel RNA antagonists will serve as useful tools to define new functional sites on ERα with therapeutic utility.
Materials and Methods
Proteins, plasmids, and chemicals
The recombinant human ERα and the glutathione S-transferase (GST)-tagged ERα ligand-binding domain (ERα-LBD) proteins were purchased from Invitrogen; the recombinant human ERβ was purchased from EMD4Biosciences. According to the manufacturers, they were all expressed in and purified from recombinant baculovirus-infected insect cells. The GST-tagged ERα-LBD contains amino acids 282–595 of human ERα and a GST tag at its N-terminus. The activity of these purchased constructs was certified by the manufacturers using their capability to bind radiolabeled estradiol. The His6-tagged ERα DNA binding domain (ERα-DBD) expression construct was a gift from Dr. W. Lee Kraus (University of Texas Southwestern Medical Center). It contains amino acids 180–268 of human ERα and a hexa-histidine tag at its N-terminus. The protein was expressed in E.coli BL21(DE3) strain, and purified using ProBond™ Resin (Invitrogen) according to the manufacturer's instructions. The human ERα coding gene in pSNAP-hERα vector was lifted from pJ3-FLAG-hERα (a gift from Dr. Carolyn L. Smith, Baylor College of Medicine). It was cloned immediately downstream of the SNAP coding sequence between the SbfI (5′ end) and XhoI (3′ end) sites in the pSNAP-tag®(m) vector (New England Biolabs). pSUPER.retro.puro was obtained from OligoEngine. pSHAG-MAGIC has been described previously (Paddison et al., 2004). Reporter vectors 3×ERE TATA luc (Addgene plasmid 11354), 2×PRE TK luc (Addgene plasmid 11350), and pGL3 luc (Promega) all contained a firefly (Photinus pyralis) luciferase gene. Vector 3×ERE TATA luc had three copies of Xenopus laevis vitellogenin A2 ERE in the reporter gene promoter (Hall and McDonnell, 1999); 2×PRE TK luc contained two copies of a consensus progesterone response element (PRE) upstream of the thymidine kinase promoter (Giangrande et al., 2000). The pRL vector expresses Renilla (Renilla reniformis) luciferase. 17β-estradiol (E2) and 4-hydroxytamoxifen (OHT) were purchased from Sigma. The ERE used in binding assays was derived from the Xenopus vitellogenin A2 promoter and synthesized by Integrated DNA Technologies (IDT), with the two strands linked by a GAAA loop in the following sequence: 5′-AGT CAG GTC ACA GTG ACC TGA TCG AAA GAT CAG GTC ACT GGT ACC TGA CT-3′.
In vitro selection
The initial RNA pool contained∼1.8×1015 different sequences, each having a 50-nt randomized region in the middle flanked by 25-nt constant regions on either side (Fan et al., 2004). A procedure described previously (Fan et al., 2004) was followed with minor modifications. The 1×binding buffer contained 12 mM HEPES/pH 7.6, 150 mM NaCl, and 10 mM MgCl2. Bound and unbound RNA fractions were separated by nitrocellulose filters. After seven cycles of selection and amplification, the final pool in the form of DNA was cloned into pSTBlue-1 blunt vector (EMD4Bioscienses). Fifty-four clones were isolated by colony polymerase chain reaction (PCR) to have the insert of correct size, and divided into seven groups to screen for binding activity. Groups showing binding activity were sequenced to identify the aptamers.
RNA-protein binding assay
All binding assays were performed in 20 μL of 1×binding buffer. A typical binding assay mixture contained about 20 fmol of 32P-labeled RNA aptamer and different amounts (0.1 to 3 pmol) of ERα. The RNA aptamers were uniformly labeled with [α-32P]CTP (Perkin Elmer) using the MAXIscript™ in vitro transcription kit (Applied Biosystems). Both bovine serum albumin and yeast RNA at 1 μg/20 μL were added to the binding reaction to prevent nonspecific binding. When a ligand, such as E2 or OHT, was involved, 40 nM to 5 μM ligand was pre-incubated with ERα at 37°C for 10 minutes before adding RNA aptamers. When nonradioactive RNA aptamer competitors or estrogen response element (ERE) were added in a competition assay, they were mixed with the 32P-labeled RNA aptamer before the addition of ERα. The binding mixtures were allowed to equilibrate for 45 minutes at 37°C before being subjected to filter binding or electrophoresis. The filter-binding assay was modified from that of Wong and Lohman (Wong and Lohman, 1993), using nitrocellulose membrane (Schleicher and Schuell; pore size 0.45 μm) and Bio-Dot microfiltration apparatus (Bio-Rad). The signal intensity of RNA retained on the nitrocellulose membrane was quantified using the ImageQuant software (GE Healthcare) and the data were fit to the Hill equation using Sigma Plot 9.0.1 (Systat Software Inc.). Electrophoretic mobility shift assay (EMSA) was performed at 4°C. The binding reaction mixtures were run on a 4.8%–6% polyacrylamide gel (PAGE) in 0.25×TBE buffer (22.25 mM Tris base, 22.25 mM borate, 0.5 mM EDTA).
In-line and enzymatic probing
Radiolabeled RNA was produced by in vitro transcription using the MAXIscript™ kit. As AptER-1 starts with a G nucleotide the transcription mix was supplemented with [γ-32P]GTP (6,000 Ci/mmol, 10 mCi/mL from Perkin Elmer) along with all four unlabeled nucleotides. Transcription mix containing RNA and DNA template was treated with DNase 1. Full-length aptamer was extracted using denaturing PAGE (8%). The gel slices were washed and the transcripts concentrated using Millipore's Amicon ultra centrifugal filters.
In-line probing was performed by folding AptER-1 in 1×binding buffer at 65°C for 1–2 minutes followed by incubation at 37°C for 10 minutes. This folded RNA was incubated at 25°C for 40–45 hours. Both untreated and in-line probed AptER-1 was mixed with gel loading buffer (95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% xylene cyanol, 0.025% bromophenol blue in RNase-free solution), heat denatured, and separated on PAGE (12%, 19:1 w/v with 50% urea).
Enzymatic probing was performed according to manufacturer's recommendation (RNases A, V1, and T1 from Ambion Life Technologies) with minor modifications. For digestion with RNases A, V1, or T1, AptER-1 (3–4 K cpm) was folded in 1×binding buffer followed by addition of yeast RNA (2–3 μg), and incubated at 37°C either in the presence or absence of ERα for 15–20 minutes. After incubation, RNases were added (RNase A, 0.1 μg; RNase V1, 0.01U; and RNase T1, 1U) and incubated at room temperature for 15 minutes, followed by inactivation and RNA precipitation. The samples were sequenced using 12% PAGE (19:1) containing urea (50%).
RNA aptamer expression cassette design
Two vectors were used for aptamer expression: pSUPER.retro.puro was purchased from OligoEngine; pSHAG-MAGIC had been described previously (Paddison et al., 2004). A set of aptamer coding genes with BglII and HindIII-compatible overhangs were designed for the pSUPER.retro.puro vector: AptER-1, GGA TCC GAG AGG CAC CGC GAA CAA AAC GCA AGA CAG AGT GCC GAC AAG AGC ACT ACA AGC CTC TCT TTT TTG GAA AAG CTT; AptER-1×2, GGA TCC GCG TGA CGG GCA CCG CGA ACA AAA CGC AAG ACA GAG TGC CGA CAA GAG CAC TAC AAG CCC GTC CAT ACT CCG GCA CCG CGA ACA AAA CGC AAG ACA GAG TGC CGA CAA GAG CAC TAC AAG CCG GAG GCG CTT TTT TGG AAA AGC TT. A second set with identical aptamer coding sequences but different restriction sites, BtsCI and BamHI, were designed for the pSHAG-MAGIC vector: AptER-1, GGA TGC GAG AGG CAC CGC GAA CAA AAC GCA AGA CAG AGT GCC GAC AAG AGC ACT ACA AGC CTC TCT TTT TTG GAT CC; AptER-1×2, GGA TGC GCG TGA CGG GCA CCG CGA ACA AAA CGC AAG ACA GAG TGC CGA CAA GAG CAC TAC AAG CCC GTC CAT ACT CCG GCA CCG CGA ACA AAA CGC AAG ACA GAG TGC CGA CAA GAG CAC TAC AAG CCG GAG GCG CTT TTT TGG ATC C. In both vectors, a tract of six thymidines was included at the 3′ end to terminate RNA polymerase III transcription. As a negative control, an insert encoding a randomized sequence of RNA (Ctrl), 5′- ACU ACC GUU GUU AUA GGU GUU CAA GAG ACA CCU AUA ACA ACG GUA GUU-3′, was cloned into each vector.
Cell culture and transfection
HeLa and MCF7 cells were maintained in Dulbecco's modified Eagle's medium-high glucose (Hyclone) supplemented with 10% fetal bovine serum (FBS; Hyclone), 100 U/mL penicillin, and 100 mg/mL streptomycin (Cellgro). HeLa cells were transfected using FuGENE 6 (Roche); MCF7 cells were transfected using FuGENE HD (Roche). Transfection reagents were used according to manufacturer's instructions. Transfection efficiency was monitored by co-transfecting the murine stem cell virus (MSCV)-Puro/EGFP vector, which expressed green fluorescent protein (EGFP).
Western blot analysis
The cell samples were harvested in 1×SDS loading buffer and run on a precast 12% tris-glycine gel (Invitrogen). The protein was transferred to a 0.45-μm nitrocellulose membrane (Millipore). After blocking, the membrane was probed with rabbit anti-SNAP polyclonal antibody (New England Biolabs) overnight at 4°C. The secondary antibody, anti-rabbit immunoglobin G (IgG) from mouse conjugated to alkaline phosphatase (Cell Signaling Technology) was incubated with the blot for one hour at 4°C. The signal was visualized with LumiGLO and Peroxide (Cell Signaling Technology).
Fluorescence microscopy
HeLa cells were seeded in an 8-well chamber slide (Lab-Tek™) the day before transfection. Cells were transiently transfected with the SNAP-hERα expression vector at 150 or 300 ng per well. Around 16 hours post-transfection, live cells were stained with SNAP-Cell TMR-Star (New England Biolabs) and Hoechst 33342 (Invitrogen), according to the manufacturers' instructions. Cells were imaged under an inverted fluorescent microscope with a 63×oil lens (Zeiss).
Intracellular binding assay
A dish (15 cm in diameter) of transiently transfected HeLa cells were treated with 1% formaldehyde for 10 minutes at 37°C to crosslink RNA with associated proteins. Cells were resuspended in the lysis buffer (50 mM Tris, 100 mM NaCl, 1 mM DTT, 0.1% NP-40, 1×protease inhibitor (Roche) and 100 U/mL of SUPERaseIn (Applied Biosystems), and then sonicated on ice until no intact cells could be detected under the microscope. The RNA pull-down assay was performed by incubating half of the soluble protein extracts with SNAP-Capture Pull Down Resin (New England Biolabs). Beads were washed five times with 1 ml of high-stringency washing buffer containing 1×RIPA (Cell Signaling Technology) supplemented to reach 1 M NaCl, 1 M urea and 0.1% SDS. The RNA co-immunoprecipitation (IP) was performed by incubating the other half of the soluble protein extracts with rabbit anti-SNAP polyclonal antibody (New England Biolabs)-coated protein G beads slurry (Cell Signaling Technology). Beads were washed four times with NT-2 buffer (20 mM EDTA, 50 mM NaCl, 1 mM DTT, 100 U/mL of SUPERaseIn). To release RNA, both types of beads were incubated with proteinase K buffer (20% glycerol, 100 mM KCl, 0.2 mM EDTA, 0.5 mM DTT) containing 200 μg/mL of proteinase K at 42°C for 1 hour. RNA was isolated by phenol/chloroform extraction and ethanol precipitation, and then quantified using quantitative reverse transcription–PCR.
Quantitative reverse transcription-PCR
MCF7 cells were transiently transfected with DNA plasmids of interest and harvested between 12 and 96 hours post-transfection. Total RNA was extracted using TRizol (Invitrogen), and complementary DNA was synthesized in a 20 μL reverse transcription consisting of 1 μg of RNA, 1 μg of Oligo(dT)15 primer (Promega), 4 μM of the AptER-1×2 reverse primer, and 200 units of SuperScript III reverse transcriptase (Invitrogen), carried out at 53°C for 1 hour. Real-time PCR was performed on ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) using DyNAmo™ HS SYBR® Green qPCR Kit (Finnzymes Oy.). The primer pairs used have the following sequence: AptER-1×2 forward, GCG TGA CGG GCA CCG C; reverse, AAG CGC CTC CGG CTT GT. After initial incubation at 95°C for 10 minutes, the amplification protocol consisted of 40 cycles of a 95°C, 15-second step and a 60°C, 1-minute step. Product levels were calculated after normalization with a β-actin control.
Dual-luciferase assay
MCF7 cells were co-transfected with the two reporter constructs expressing either firefly or Renilla luciferase, together with the aptamer expression vectors of interest. Experiments were performed in 96-well plates with ∼5,000 cells per well. Each transfection reaction is 5 μL and contains 0.45 μL of Fugene, 4.55 μL of serum-free medium, and 150 ng of DNA. The ratio of Fugene to DNA is 3:1. The combination of DNA is 75 ng of aptamer plasmid, 15 ng of Renilla luciferase plasmid, and 60 ng of firefly luciferase plasmid. A randomized RNA sequence described above was expressed to replace the aptamer and serve as a control. As the media contained 10% FBS, estrogen from FBS was present throughout the culture. Cells were lysed 72 hours post-transfection and firefly (reporter) and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay Kit (Promega) on a BioTek HT Synergy plate reader. The ratio of firefly to Renilla luciferase activity was calculated and presented as percentage of control.
Results
RNA aptamers bind ER with high affinity
When a target molecule bears multiple potential sites for aptamer recognition, the in vitro selection procedure does not direct aptamers to any particular one. Therefore, a rational approach to discovering novel sites is to use existing ligands to mask known sites in the process of selection. But either E2 or ERE would change the surface topography of ER, and the aptamer thus isolated may be dependent on a third party for binding to ER. Because a general antagonist of ER should be able to inhibit ER activity independently, we decided to perform a selection with apo-ERα as the target molecule. The isolated aptamers were then tested to find out whether their binding would be affected by other existing ligands and whether they could perturb ER function in the complex milieu of living cells. This procedure yielded three aptamers after 7 cycles of selection and a post-selection screening for binding. One of them, AptER-1, was the most abundant and occurred 9 times in a sample of 54 individuals, while the other two, AptER-2 and AptER-3, were only isolated once (Fig. 1A). There was neither shared consensus sequence among these three sequences, nor an obvious common secondary structure as predicted by the computer program mfold (ZUKER, 2003) (Fig. 1B).
Fig. 1.

RNA aptamers and their affinity for the unliganded estrogen receptor α (apo-ERα). (A) Aptamer sequences in the randomized region and their occurrences in the final sequenced pool. The names of the individual aptamers are given to the left of each sequence. The underlined lower case letters at the 3′ end are sequences derived from the first 3 positions (AGU, underlined) of the 3′ constant region (the primer covers 22 bases of the 25-nt sequence). The frequency of occurrence of each aptamer is given on the right. The sequences of the flanking constant region are shown at the bottom. (B) Predicted secondary structures of aptamers by mfold. The sugar-phosphate backbone is represented by bold lines, while hydrogen bonds between paired bases by faint lines. Each base is represented by a dot. (C) Binding curves of the aptamers for ERα determined using filter-binding assays. The apparent Kd determined from binding curves is shown next to the title of each curve. Each experiment was repeated at least three times and the standard deviation was shown as error bars. (D) Similar mobilities of the three aptamer-ERα complexes in electrophoretic mobility shift assay (EMSA). Binding mixtures are resolved on 4.8% polyacrylamide gel (acrylamide:bis-acrylamide=37.5:1) in 1/4× TBE buffer. ERα concentrations are 0, 10, 20, and 40 nM. The asterisks signify the radiolabel. (E) Affinity of the three aptamers for apo-ERα (0, 20, and 40 nM) and apo-ERβ (0, 20, and 40 nM). Conditions are the same as in (D).
The affinity of the three ERα aptamers was measured using nitrocellulose filter binding assays, and the data were fit to the Hill equation to determine the dissociation constant (Kd) of the aptamers. As shown in Fig. 1C, the apparent Kd for AptER-1, 2, and 3 was 16 nM, 21 nM, and 30 nM, respectively. In an electrophoretic mobility shift assay (EMSA), the aptamer-protein complexes formed by AptER-1, -2 or -3 showed similar mobility, though the one formed by AptER-1 moved slightly slower. Interestingly, a very faint second or “supershift” band was visible in the AptER-2 and AptER-3 binding reactions at the highest protein concentration (Fig. 1D). This is consistent with their sigmoidal binding curves (Fig. 1C) and their calculated Hill coefficients (n >1, indicating positive cooperativity). In contrast, AptER-1 has a Hill coefficient approximating 1, and no “supershift” band was observed. Whether ERα in our assay existed as homodimers and allowed the binding of two aptamers in the cases of AptER-2 or -3 remains to be determined.
As the human estrogen receptors α and β share an overall sequence identity of about 47%, we also tested whether the aptamers recognize ERβ. As shown in Fig. 1E, all three aptamers were able to interact with ERβ in its unliganded form, suggesting that they recognized some conserved site or sites on the two ER subtypes. As a negative control, we tested binding of an unrelated aptamer, AptC3-1 (Mallik et al., 2010), which is of similar size to AptER-1, 2, and 3. As expected, it showed binding to the human complement protein C3 but not to ERα or β (data not shown).
Binding of aptamers to ERα is not affected by estrogen or DNA elements
As mentioned above, the three aptamers we identified for ER did not share any similarity in sequence or predicted secondary structure. Therefore, it was interesting to determine whether they bound to the same site or overlapping sites. We performed a cross competition assay as shown in Fig. 2A, and found out that AptER-2 and -3 competed with AptER-1 for binding to ERα, suggesting that the three aptamers interacted with overlapping sites on ERα. As AptER-1 was the predominant aptamer and marginally the tightest binder among the three, it was chosen to represent the three aptamers in subsequent assays to find out whether the binding of the aptamer is affected by other known ligands, namely the estrogen and the ERE element.
Fig. 2.
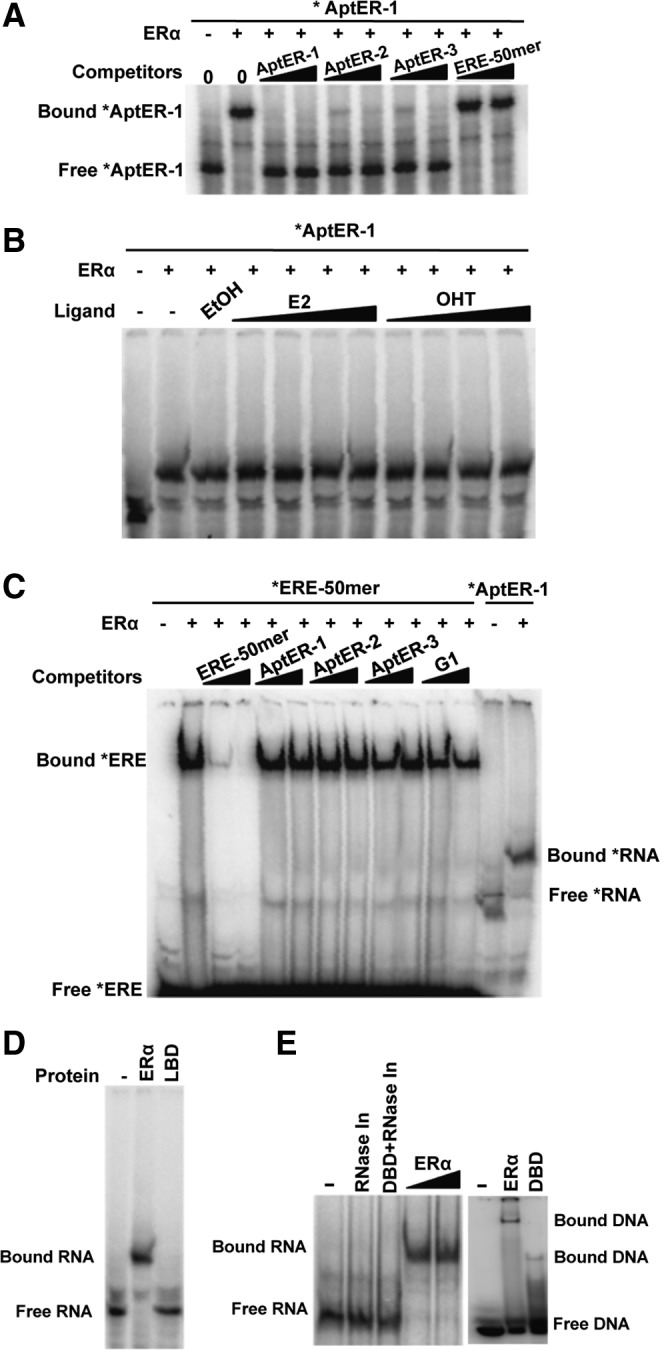
Aptamer binding in the presence or absence of other ligands. (A) A common binding site shared by the three aptamers. In this aptamer cross competition assay, radiolabeled AptER-1 RNA (0.1 nM) is mixed with 2000× or 8000× more concentrated nonradioactive competitors AptER-1, -2, -3 RNA or estrogen response elements (ERE)-50mer before addition of apo-ERα (+, 20 nM) in the binding reactions. (B) Binding assay in the presence of 17β-estradiol (E2) or hydroxy-tamoxifen (OHT). ERα (+, 40 nM) is incubated with ligands (40 nM to 5 μM of E2 or OHT) or the vehicle control (ethanol) at 37°C for 10 minutes before addition of RNA. (C) Cross competition assay with ERE. End-radiolabeled ERE-50mer at 5 nM is mixed with 100× or 400× more concentrated unlabeled competitors ERE-50mer, AptER-1, -2, -3, or an early generation pool of the in vitro selection (G1), before addition of ERα (+, 40 nM). (D) EMSA with radiolabeled AptER-1 and GST-tagged ERα–ligand-binding domain (ERα-LBD) (100 nM) or ERα (100 nM). (E) EMSA with radiolabeled AptER-1 and His6-tagged ERα–DNA binding domain (ERα-DBD) (100 nM) or ERα (50 nM and 100 nM). The activity of the His6-tagged DBD is verified by EMSA with radiolabeled ERE shown in the right panel.
ERα belongs to the nuclear receptor superfamily of ligand-inducible transcription factors, whose members share a common structural architecture (i.e., six domains, A to F, from N-terminus to C-terminus). Among them, the DNA-binding domain (DBD, in domain C) and the ligand-binding domain (LBD, in domain E) are best characterized. In the classic ER signaling pathway, estrogen binds the ligand-binding pocket in LBD, which induces a conformational change promoting the dimerization of the receptor. The ER dimer then translocates from the cytoplasm to the nucleus, binds to specific estrogen response elements (EREs, 5′-GGTCAnnnTGACC-3′) in the target promoters to modulate transcription. If an aptamer could act as a general antagonist, its binding to ER should not be affected by either estrogen or the ERE.
Aptamer affinity to ERα was measured in the presence or absence of E2 or OHT to find out whether these small molecule ligands would interfere with aptamer binding (E2 and OHT bind to the same site but cause different conformational change in LBD). As shown in Fig. 2B, increasing the amount of either E2 or OHT up to 5 μM showed no effect on the binding of AptER-1. Additional binding assays were performed to further investigate whether the aptamer could bind to the LBD using a GST-tagged ERα-LBD construct in EMSA (Fig. 2D). Consistent with the results of ligand interference described above, no aptamer-LBD complex was detected in this assay. The activity of the LBD construct used in this assay was confirmed by the manufacturer with its capability to bind radiolabeled estradiol. Like the full-length protein, the LBD was produced in insect cells and should have similar post-translational modifications. Therefore, the aptamer might bind a site outside the LBD or required other portion of the ER in addition to the LBD to bind, which needs further structural investigation beyond the scope of this study.
Several aptamers isolated previously for transcription factors were shown to interact with their target protein through the DNA binding site or domain and compete with the cognate DNA for binding (Lebruska and Maher, 1999; Fan et al., 2004; Zhao et al., 2006). But this was not the case for AptER-1, whose binding to ERα was not affected by the presence of 8,000-fold of ERE (Fig. 2A, lanes 9 and 10). This result was confirmed in a reciprocal assay, in which excess amounts (100- or 400-fold) of all three aptamers was used to compete with ERE for binding to ERα. As a control, ERE competed with itself effectively (Fig. 2C). This somewhat surprising result prompted an independent binding assay using a recombinant His6-tagged ERα-DBD. Although it was produced in bacteria, the recombinant DBD interacted well with the ERE (Fig. 2E, right panel). In contrast, as shown in the left panel of Fig. 2E, AptER-1 did not exhibit any specific binding activity to this isolated domain.
AptER-1 can be minimized and augmented
AptER-1 was able to bind to the full-length ERα but not to LBD or DBD as isolated recombinant constructs, and its binding was not affected by E2 or ERE. These interesting behaviors indicated the existence of a novel site defined by the aptamer, which may have functional implications. Further functional analyses would have to be performed in a physiological context, which required the assurance of correctly folded aptamer being delivered or produced inside living cells. Towards this goal, we characterized the secondary structure of AptER-1 to find a portable form. Using the structure predicted by the mfold program as a reference model, we performed in-line probing and enzymatic probing using RNases A, V1, and T1. Footprinting of AptER-1 in the presence or absence of ERα showed that the protein interacted with RNA at various positions as highlighted in Fig. 3A. The aptamer cleavage pattern when mapped to the mfold predicted structure agreed with known cleaving properties of the RNases. This structural data was further supported by in-line probing experiments, where the majority of the breaks on RNA were observed in the predicted loop regions, suggesting the dynamic nature of the loops as opposed to stable stems (Fig. 3A).
Fig. 3.

Minimization and augmentation of AptER-1. (A) Structural analysis of 5′-[γ-32P]-labeled AptER-1 using in-line probing and footprinting. RNA is sequenced on denaturing gel (12%, acrylamide:bis-acrylamide=19:1) after treatment as follows: −OH, partial alkaline hydrolysis; NR, no reaction; T1, partial digest of denatured AptER-1 with RNase T1 (cleaves after G residues); A, V1, and T1, folded aptamer treated with RNase A, V1, and T1 in the absence (−) or presence (+) of ERα (400 nM); IP, in-line probed sample. Note that NR and IP lanes look very much alike, indicting the hyperactive regions on the folded aptamer. The RNA cleavage by different RNases and in-line probed regions are mapped on the predicted secondary structure of AptER-1. (B) Predicted secondary structures of the truncated AptER-1 and AptER-1 dimer by mfold. Sequences in the variable regions are represented as capital letters, sequences in the constant regions or additional GC pairs in the end-most stem are shown in lower case, and sequences in the three-way junction in AptER-1×2 are enclosed in a box. The nucleotide substitutions in the 65-nt-long truncation B30 are circled in AptER-1(65nt). (C) EMSA showing different binding affinity of the truncated radiolabeled B30, sB30 (65 nt in length), and AptER-1(65nt). ERα concentration is 25 nM. (D) Comparison of binding affinity of radiolabeled AptER-1(65nt) and AptER-1×2(138nt) to ERα. Left panel: shifted band is labeled with asterisk (*). Right panel: band single-astericked (*) represents protein occupying one aptamer unit; band double-astericked (**) represents protein occupying both aptamer units.
Based on these observations, a 65-nt-long truncation AptER-1(65nt) containing the central stem loop region was made (Fig. 3B). It preserved the three-way junction with two apical loops in the predicted AptER-1 structure and retained full binding activity as the full-length aptamer (Fig. 3D). Although the bands representing C18, G31 (RNase V1), and U21 (RNase A) showed reduced intensity in the footprinting assay, indicating their involvement in binding, the 5′ segment up to A30 was not required in the minimized version. This discrepancy may be partly explained by the fact that two-thirds of this region (C18 to C25) was part of the 5′ constant region of the aptamer candidates. So C18 and its vicinity were not “selected” by ERα. G31 was preserved in the 65-nt version as G1, while U32 and U33 were both changed to Gs in the truncated version to facilitate in vitro transcription. More truncation in the stem formed by the 5′ and 3′ ends, as in AptER-1(53nt) (Fig. 3B), decreased the affinity, and a further deletion of 17 nt in the 3′ end of AptER-1 (65 nt) totally abrogated the three-way junction structure, as predicted by mfold, and also its binding activity. Thus, the two stem-loops connected by the three-way junction in AptER-1 (53 nt) may be the true aptamer moiety required for binding. Consistent with this conclusion, sequence B30 (one of the nine isolates of AptER-1) showed two nucleotide substitution in comparison with the sequence of the representative AptER-1 sequence (Fig. 3B) (i.e., substitutions A51C and C68A). The 65-nt truncated B30 (sB30), corresponding to AptER-1(65nt), showed lower affinity to ERα than the canonical AptER-1(65nt) (Fig. 3C).
Constructing a composite molecule containing more than one copy of an aptamer would enhance its avidity. We designed a divalent AptER-1, named AptER-1×2(138nt), emulating the bivalent form of natural antibodies. To avoid incorrect base-pairing between the identical stem sequences of the two units, the two minimized AptER-1were grafted to two stems of a three-way junction of the Haloarcula marismortui 5S RNA, a well-characterized structural element described before (Ban et al., 2000; Xu and Shi 2009). The secondary structure of this dimeric construct was predicted by mfold (Fig. 3B), and the avidity of AptER-1×2(138nt) to apo-ERα, in comparison to that of the monovalent AptER-1(65nt), was assayed by EMSA. As shown in Fig. 3D, the divalent aptamer bound to ERα more avidly than the monovalent aptamer, and both aptameric sites were occupied by the target protein.
AptER-1 recognizes and binds ERα in living cells
The successful construction of the minimized monomeric and stable dimeric versions of AptER-1 made it possible to deliver them into living cells as synthetic genes. Two AptER-1 coding genes were designed, a 61-nt monomeric AptER-1, AptER-1×1, and a 132-nt dimeric AptER-1 with minor modification to AptER-1×2(138nt), AptER-1×2. To introduce the aptamers into the mammalian cell lines, two RNA expression vectors were adopted, which utilized two different RNA polymerase III promoters to drive RNA expression [i.e., a H1 gene promoter in pSUPER.retro.puro (OligoEngine) and a U6 gene promoter in pSHAG-MAGIC (Paddison et al., 2004)]. In these constructs, RNA transcription would start at the initial purine of inserted sequences and terminate with the addition of two uridines (U) at the 3′ end. Before examining the effect of AptER-1 on the activity of ERα, we used two intracellular binding assays to confirm the physical association of aptamers with ERα in living cells.
As the location of the aptamer binding site on ERα remained undetermined, a SNAP tag affixed to the N-terminus of ERα was chosen to be an epitope, to avoid aptamer interfering with ERα immunoprecipitation by an anti-ERα antibody. The SNAP tag is a 20-kDa mutant of a human DNA repair protein, O6-alkylguanine-DNA-alkyltransferase. It reacts with a benzylguanine group on SNAP substrates, such as SNAP-Cell TMR Star and SNAP-Capture pulldown resin, forming a covalent bond with the benzyl group. HeLa cells were chosen for the intracellular binding assays, because ERα was expected to be absent in this cell line (Maminta et al., 1991; Zhai et al., 2010), thus the expressed AptER-1×2 would only be captured by SNAP-ERα. An additional binding assay for AptER-1 was carried out using the HeLa nuclear extract. No specific binding activity was observed (data not shown).
HeLa cells were co-transfected with SNAP-ERα expression vector and pSUPER/AptER-1×2 expression vector. In a group of control cells, a SNAP expression vector replaced the SNAP-ERα. The expression of the recombinant SNAP-ERα was confirmed by fluorescent microscopy. Twenty-four hours post-transfection, live HeLa cells were labeled with SNAP-Cell TMR-Star and Hoechst 33342. As shown in Fig. 4A, the SNAP fusion protein was located in both the cytoplasm and nucleus of transfected cells, but mainly in the nucleus. This pattern agreed with previously described distribution of ERα in the literature (Liang and Shang, 2013). The cell samples were subjected to a western blotting analysis to examine whether the SNAP or SNAP-ERα were expressed at comparable levels. As shown in Fig. 4B, the rabbit anti-SNAP polyclonal antibody detected similar expression level of SNAP and SNAP-ERα fusion protein.
Fig. 4.

Physical association of the RNA aptamer with ERα in living cells. (A) Localization of SNAP-ERα in HeLa cells. HeLa cells are transfected with the SNAP-hERα expression vector. Live cells are labeled with SNAP-Cell TMR-Star and Hoechst 33342, and visualized under an inverted fluorescent microscope. (B) Comparable protein expression levels in the experimental and the control cells. An aliquot of the cell samples are subjected to western blotting, and the SNAP-tagged protein is detected by rabbit anti-SNAP polyclonal antibody. (C) Quantification of SNAP-hERα or SNAP pulled down by the aptamer or immunoprecipitated by anti-SNAP antibody. HeLa cells are co-transfected with the SNAP-hERα expression vector and the pSUPER/AptER-1×2 expression vector. In the control, SNAP expression vector is used to replace the SNAP-hERα expression vector. The same cell samples are used for both pull-down assay and RNA co-IP assay. The amount of RNA in association with the SNAP proteins is quantified using real-time RT-PCR and expressed as “fold of enrichment” over the amount of RNA isolated in the control. Each experiment was repeated at least three times and the standard deviation was shown as error bars.
After a small fraction was used in the western blot to compare protein expression levels, the remaining cell samples were aliquoted and used in two parallel assays: an RNA pull-down assay and an RNA co-immunoprecipitation assay. Considering the different strengths of bonding formed between the SNAP tag and the two kinds of beads used in the two systems, more stringent washing conditions were employed in the pull-down assay. RNA recovered from the beads was quantified by real-time RT-PCR. As shown in Fig. 4C, SNAP-ERα pulled down 292-fold more AptER-1×2 than the SNAP control; SNAP-ERα immunoprecipitated 67 fold more aptamer than the SNAP tag. These results demonstrated that ERα and AptER-1×2 interacted inside HeLa cells. The difference in “fold of enrichment” (SNAP-ERα vs. SNAP) from these two assays might be caused by the higher stringency of buffers used in the pull-down assay, resulting in less nonspecific interaction in the SNAP control.
AptER-1 inhibits ER function in cancer cells
A most important function of ERα is to recruit cofactors and the basal transcription machinery to form the transcription pre-initiation complex. We hypothesized that in a transcription system regulated by ERα, addition of aptamers would restrain ERα transcriptional activation at an ERE-containing promoter. A transcription factor–reporter assay in an ERα-positive breast cancer line, MCF7, was employed to investigate this mechanism. To determine whether the aptamer expression vectors could produce the RNA aptamers, the pSUPER/AptER-1×2 expression vectors was introduced into the ERα-positive breast cancer cell line MCF7 using transient transfection, and accumulated AptER-1×2 between 12 and 96 hours post-transfection was quantified using quantitative RT-PCR. As shown in Fig. 5A, AptER-1×2 accumulation level, expressed as “fold of β-actin mRNA level,” peaked between 48 and 72 hours post-transfection. Thus, the pSUPER vector could successfully generate relatively high level of RNA aptamers in MCF7 cells.
Fig. 5.
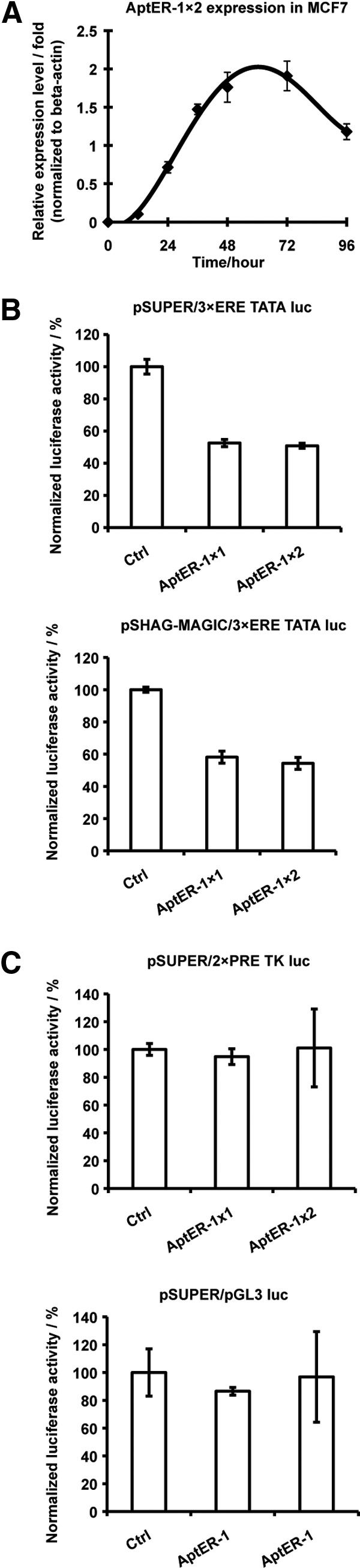
Inhibitory effect of the aptamer to ER-mediated transcriptional activation in MCF7 cells. (A) Accumulation level in MCF7 of AptER-1×2 expressed from the pSUPER vector. Cells are harvested at several time points within 96 hours post-transfection of the pSUPER/AptER-1×2 plasmid. The level of AptER-1×2 is quantified using real-time RT-PCR and expressed as relative expression level (fold) of β-actin. (B) Dual-luciferase assays for aptamer expressed using two different vectors. MCF7 cells are co-transfected with the firefly luciferase vector (3×ERE TATA luc), Renilla luciferase vector and an aptamer expression vector of interest. “Ctrl” is a scrambled RNA expression vector used in place of the aptamer expression vectors. All experiments are carried out in triplicate and the average is used to make the plot. (C) Specificity of the inhibitory effect. 3×ERE TATA luc is replaced with firefly luciferase reporter gene 2×PRE TK luc or pGL3 luc. The pSUPER vectors are used to express aptamers or the control RNA. Each experiment was repeated at least three times and the standard deviation was shown as error bars.
We used dual-luciferase assays to examine the efficacy of RNA aptamers on inhibiting transcription activation mediated by ERα. Two luciferase reporter vectors and an aptamer expression vector or an expression vector for a randomized control RNA were co-transfected into MCF7 cells. The firefly luciferase reporter vector 3×ERE TATA luc carries three copies of Xenopus vitellogenin A2 ERE in the luciferase promoter region, which allows transcriptional regulation by the endogenous ERα in MCF7 cells. The amount of firefly luciferase enzyme assayed is proportional to the activity of the receptor, whereas Renilla luciferase constitutively expressed from the other reporter vector served as an internal control to normalize transfection efficiency among different transfection reactions. Both luciferase levels were measured sequentially in one dual-luciferase assay.
The ratio of firefly luciferase activity to Renilla luciferase activity was determined, and the value in the control was considered to be 100%, which represents full transcriptional activation. The ratios of firefly luciferase activity to Renilla luciferase activity in all aptamer-expressing samples were then normalized against that of the control and expressed as normalized firefly luciferase activity (%) of the control. As shown in Fig. 5B, expression of the aptamers from pSUPER or pSHAG-MAGIC resulted in 42%–50% reduction in normalized firefly luciferase activity. The AptER-1 dimer was always more efficient than the monomer, but only by 5%–10%. The same aptamer when expressed from either the pSUPER vector or the pSHAG-MAGIC vector showed comparable effects, although the pSUPER vector performed slightly better.
Two additional firefly luciferase reporter genes were used to replace the vector 3× ERE TATA luc in two control assays in order to investigate the specificity of the aptamer. The 2× PRE TK luc contained the DNA motif recognized by progesterone receptor (PR), another member of the nuclear receptor family, which is also expressed in MCF7 (Neve et al., 2006). As shown in Fig. 5C, the expressed aptamers had no inhibitory effect on the PR-dependent transcriptional activation. Similar negative results were observed with the pGL3 luc reporter vector, which is a firefly reporter vector backbone without specific element for transcriptional activation. Taken together, these results demonstrated that the expressed aptamers could specifically inhibit transcriptional activation by ER, but had no apparent effect on the closely related nuclear receptor PR.
Discussion
Although ERα is a well-validated drug target for treating estrogenopathies including breast cancers, it is far from being fully exploited (Nilsson et al., 2011; Shapiro et al., 2011). In particular, the target sites on this protein are far from exhaustively defined. Almost all ER antagonists currently in clinical use act through binding to the site reserved for its steroidal agonists on the LBD. As demonstrated by tamoxifen, the pharmacological properties of this type of ER ligands may change over time, making them unsuitable for chronic treatment of cancer (Musgrove and Sutherland, 2009). In the search for new classes of ER modulators, it might be a fruitful approach to target additional, non-classic regulatory surfaces on the receptor. However, crystal structures are only available currently for the LBD and DBD (Schwabe et al., 1993; Shiau et al., 1998; Eiler et al., 2001), and thus structure-based rational drug design can only be applied to these two isolated domains. This bias is eliminated in our scheme, as the entire solvent accessible surface of the full-length ERα was presented to the aptamer candidate pool. A similar approach was used to identify peptide aptamers (McDonnell et al., 2000). Many of these “peptamers” have a core LXXLL motif and interact with the coactivator binding site (Chang et al., 1999); some are conformation-specific and only recognize the ER after its binding by E2 or tamoxifen (Norris et al., 1999). Although they are useful structural and functional probes for mechanistic studies, the requirement of coexistence with E2 or tamoxifen would limit their pharmaceutical utility.
A critical finding of the tamoxifen study revealed the cause of the reverse in its pharmacological activity: endocrine resistance is the result of changes in ER's cellular environment rather than change in ER itself (Osborne and Schiff, 2011). The mere presence of the receptor may be sufficient for downstream transcriptional activity that supports malignancy, as cells can be selected to produce a coactivator compatible with the ER-tamoxifen complex. Therefore, a more desirable ER antagonist should be able to act on ER without the help of other ligands. However, in addition to the well-known ligand-induced conformational changes, binding to the ERE has also been demonstrated to exert allosteric modulation to the receptor (Wood et al., 2001). In this work, we identified RNA aptamers that function as general antagonists of ER. The representative aptamer AptER-1, was able to bind ERα avidly without being affected by either E2 (and tamoxifen) or ERE, and yet it was capable of inhibiting ER function as a transcription activator in living cancer cells. We believe that these novel RNA antagonists will serve not only as useful tools in mechanistic dissection of ERα biology but also as drug leads for the treatment of estrogenopathies.
As demonstrated by us and others, RNA aptamers can be a powerful means to modulate transcription factors (Lebruska and Maher, 1999; Fan et al., 2004; Zhao et al., 2006). While most previously characterized RNA aptamers to DNA-binding proteins were found to recognize the DNA-binding surface of the targeted proteins, the aptamers we have selected and characterized here for ERα did not follow this pattern. They did not compete with ERE for binding to the DNA-binding site, and did not show any affinity for the DNA binding domain (DBD) along. We speculate that a cluster of contact points belonging to different structural domains might be involved, and the aptamer binding site is formed only when these domains are connected together. Interestingly, a natural RNA transcript, steroid receptor RNA activator (SRA), was previously found to bind to ERα and enhances the AF-1 activity of ER and other steroid receptors (Lanz et al., 1999). Thus, ERα has intrinsic dual-specificity for DNA and RNA, and there might be an amenable target site for RNA aptamers outside the DBD and LBD.
Whereas aptamers in general exhibit high specificity to their targets, it is desirable to demonstrate empirically whether a particular aptamer binds to any unintended target. Before an exhaustive proteomic assay is available, we took a heuristic approach to address this issue: because an aptamer binds its target primarily through recognition of molecular surface topography, it is more likely to bind non-targets with high homology to the target. Therefore, we set a priority to test binding of our aptamer to proteins structurally related to ER, such as PR (Ellmann et al., 2009; Yang et al., 2013). The inability of the AptER-1 to affect PR-dependent transcriptional activation in the MCF7 cells suggests that the aptamer is specific to ER and does not recognize other nuclear receptors. However, there are two isoforms of ER and the aptamer showed affinity to both in binding assays. Nonetheless, ERα is the predominant form in ER dependent breast cancer cells (Reid et al., 2002; Nilsson and Gustafsson, 2011), suggesting that the primary inhibitory effect of the aptamer was through this isoform. To further improve specificity, we have now created next generation of aptamers capable of distinguishing ERα from ERβ, which will be published elsewhere (Xu et al, unpublished data).
The aptamers described herein defined a new druggable site on ERα, even though the exact location of the site remains to be delineated by structural studies. To our knowledge, most previously identified small molecular antagonists and peptamers bound to LBD or DBD. As mentioned above, peptide aptamers were isolated for either E2- or tamoxifen-activated ER to block the coactivator binding on LBD (McDonnell et al., 2000) and the ERα/cofactor interactions have been explored by other means (Carraz et al., 2009). Small molecule ligands were synthesized to target binding of ERα to ERE (Wang et al., 2004; Mao et al., 2008). In addition, a recent study demonstrated a site on the F domain, which interact with the 14-3-3 proteins, as a drug target interface (De Vries-van Leeuwen et al., 2013). By using a distinct type of probes, the RNA aptamers, we were able to find a novel site on ERα, which should be distinct from those previously identified. Because AptER-1 is capable of inhibiting ERα function in the ERα-positive MCF7 cells, the new site could be another potential drug target to suppress ERα activity in breast cancers. In particular, they can be used to guide the rational chemical synthesis of small molecule drugs or perform screens of small molecule libraries for those that can displace the aptamer from this novel binding site (Hartig et al., 2002; Elowe et al., 2006; Hafner et al., 2006).
Acknowledgments
We thank the other members of the Shi and Conklin Labs for their helpful suggestions and encouragement. This work was supported by a Research Scholar Grant (RSG-09-159-01-CDD) to H.S. from the American Cancer Society.
Author Disclosure Statement
A patent application has been filed for the aptamers.
References
- BAN N., NISSEN P., HANSEN J., MOORE P.B., and STEITZ T.A. (2000). The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 289,905–920 [DOI] [PubMed] [Google Scholar]
- CARRAZ M., ZWART W., PHAN T., MICHALIDES R., and BRUNSVELD L. (2009). Perturbation of estrogen receptor alpha localization with synthetic nona-arginine LXXLL-peptide coactivator binding inhibitors. Chem. Biol. 16,702–711 [DOI] [PubMed] [Google Scholar]
- CHANG C., NORRIS J.D., GRON H., PAIGE L.A., HAMILTON P.T., KENAN D.J., FOWLKES D., and MCDONNELL D.P. (1999). Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol. Cell Biol. 19,8226–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIRILLO F., NASSA G., TARALLO R., STELLATO C., DE FILIPPO M.R., AMBROSINO C., BAUMANN M., NYMAN T.A., and WEISZ A. (2013). Molecular mechanisms of selective estrogen receptor modulator activity in human breast cancer cells: identification of novel nuclear cofactors of antiestrogen-ERalpha complexes by interaction proteomics. J. Proteome Res. 12,421–431 [DOI] [PubMed] [Google Scholar]
- COLE M.P., JONES C.T., and TODD I.D. (1971). A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br. J. Cancer. 25,270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAI S.Y., CHALMERS M.J., BRUNING J., BRAMLETT K.S., OSBORNE H.E., MONTROSE-RAFIZADEH C., BARR R.J., WANG Y., WANG M., BURRIS T.P., DODGE J.A., and GRIFFIN P.R. (2008). Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc. Natl. Acad. Sci. U. S. A. 105,7171–7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE VRIES-VAN LEEUWEN I.J., DA COSTA P.D., FLACH K.D., PIERSMA S.R., HAASE C., BIER D., YALCIN Z., MICHALIDES R., FEENSTRA K.A., JIMÉNEZ C.R., et al. (2013). Interaction of 14-3-3 proteins with the Estrogen Receptor Alpha F domain provides a drug target interface. Proc. Natl. Acad. Sci. U. S. A. 110,8894–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DROOG M., BEELEN K., LINN S., and ZWART W. (2013). Tamoxifen resistance: from bench to bedside. Eur. J. Pharmacol. 717,47–57 [DOI] [PubMed] [Google Scholar]
- EILER S., GANGLOFF M., DUCLAUD S., MORAS D., and RUFF M. (2001). Overexpression, purification, and crystal structure of native ER alpha LBD. Protein Expr. Purif. 22,165–173 [DOI] [PubMed] [Google Scholar]
- ELLMANN S., STICHT H., THIEL F., BECKMANN M.W., STRICK R., and STRISSEL P.L. (2009). Estrogen and progesterone receptors: from molecular structures to clinical targets. Cell Mol. Life Sci. 66,2405–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELOWE N.H., NUTIU R., LALI-HASSANI A., CECHETTO J.D., HUGHES D.W., LI Y., and BROWN E.D. (2006). Small-molecule screening made simple for a difficult target with a signaling nucleic acid aptamer that reports on deaminase activity. Angew Chem. Int. Ed. Engl. 45,5648–5652 [DOI] [PubMed] [Google Scholar]
- FAN X., SHI H., ADELMAN K., and LIS J.T. (2004). Probing TBP interactions in transcription initiation and reinitiation with RNA aptamers that act in distinct modes. Proc. Natl. Acad. Sci. U. S. A. 101,6934–6939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIANGRANDE P.H., KIMBREL E.A., EDWARDS D.P., and MCDONNELL D.P. (2000). The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Mol. Cell Biol. 20,3102–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAFNER M., SCHMITZ A., GRUNE I., SRIVATSAN S.G., PAUL B., KOLANUS W., KREMMER E., BAUER I., and FAMULOK M. (2006). Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature 444,941–944 [DOI] [PubMed] [Google Scholar]
- HALL J.M., and MCDONNELL D.P. (1999). The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140,5566–5578 [DOI] [PubMed] [Google Scholar]
- HARTIG J.S., NAJAFI-SHOUSHTARI S.H., GRUNE I., YAN A., ELLINGTON A.D., and FAMULOK M. (2002). Protein-dependent ribozymes report molecular interactions in real time. Nat. Biotechnol. 20,717–722 [DOI] [PubMed] [Google Scholar]
- HELSEN C., KERKHOFS S., CLINCKEMALIE L., SPANS L., LAURENT M., BOONEN S., VANDERSCHUEREN D., and CLAESSENS F. (2012). Structural basis for nuclear hormone receptor DNA binding. Mol. Cell Endocrinol. 348,411–417 [DOI] [PubMed] [Google Scholar]
- LANZ R.B., MCKENNA N.J., ONATE S.A., ALBRECHT U., WONG J., TSAI S.Y., and O'MALLEY B.W. (1999). A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97,17–27 [DOI] [PubMed] [Google Scholar]
- LEBRUSKA L.L., and MAHER L.J., III. (1999). Selection and characterization of an RNA decoy for transcription factor NF-kappa B. Biochemistry 38,3168–3174 [DOI] [PubMed] [Google Scholar]
- LIANG J., and SHANG Y. (2013). Estrogen and cancer. Annu. Rev. Physiol. 75,225–240 [DOI] [PubMed] [Google Scholar]
- LITTON J.K., ARUN B.K., BROWN P.H., and HORTOBAGYI G.N. (2012). Aromatase inhibitors and breast cancer prevention. Expert Opin. Pharmacother. 13,325–331 [DOI] [PubMed] [Google Scholar]
- MALLIK P.K., NISHIKAWA K., MILLIS A.J., and SHI H. (2010). Commandeering a biological pathway using aptamer-derived molecular adaptors. Nucleic Acids Res. 38,e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAMINTA M.L., MOLTENI A., and ROSEN S.T. (1991). Stable expression of the human estrogen receptor in HeLa cells by infection: effect of estrogen on cell proliferation and c-myc expression. Mol. Cell Endocrinol. 78,61–69 [DOI] [PubMed] [Google Scholar]
- MANAVATHI B., DEY O., GAJULAPALLI V.N., BHATIA R.S., BUGIDE S., and KUMAR R. (2013). Derailed estrogen signaling and breast cancer: an authentic couple. Endocr. Rev. 34,1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAO C., PATTERSON N.M., CHERIAN M.T., ANINYE I.O., ZHANG C., MONTOYA J.B., CHENG J., PUTT K.S., HERGENROTHER P.J., WILSON E.M., et al. (2008). A new small molecule inhibitor of estrogen receptor alpha binding to estrogen response elements blocks estrogen-dependent growth of cancer cells. J. Biol. Chem. 283,12819–12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCDONNELL D.P., CHANG C.Y., and NORRIS J.D. (2000). Development of peptide antagonists that target estrogen receptor-cofactor interactions. J. Steroid Biochem. Mol. Biol. 74,327–335 [DOI] [PubMed] [Google Scholar]
- MCDONNELL D.P., and WARDELL S.E. (2010). The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr. Opin. Pharmacol. 10,620–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERRELL K.W., CROFTS J.D., SMITH R.L., SIN J.H., KMETZSCH K.E., MERRELL A., MIGUEL R.O., CANDELARIA N.R., and LIN C.Y. (2011). Differential recruitment of nuclear receptor coregulators in ligand-dependent transcriptional repression by estrogen receptor-alpha. Oncogene 30,1608–1614 [DOI] [PubMed] [Google Scholar]
- MOORE T.W., MAYNE C.G., and KATZENELLENBOGEN J.A. (2010). Minireview: Not picking pockets: nuclear receptor alternate-site modulators (NRAMs). Mol. Endocrinol. 24,683–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUSGROVE E.A., and SUTHERLAND R.L. (2009). Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer. 9,631–643 [DOI] [PubMed] [Google Scholar]
- NEVE R.M., CHIN K., FRIDLYAND J., YEH J., BAEHNER F.L., FEVR T., CLARK L., BAYANI N., COPPE J.P., TONG F., et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 10,515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILSSON S., and GUSTAFSSON J.A. (2011). Estrogen receptors: therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 89,44–55 [DOI] [PubMed] [Google Scholar]
- NILSSON S., KOEHLER K.F., and GUSTAFSSON J.A. (2011). Development of subtype-selective oestrogen receptor-based therapeutics. Nat. Rev. Drug Discov. 10,778–792 [DOI] [PubMed] [Google Scholar]
- NORRIS J.D., PAIGE L.A., CHRISTENSEN D.J., CHANG C.Y., HUACANI M.R., FAN D., HAMILTON P.T., FOWLKES D.M., and MCDONNELL D.P. (1999). Peptide antagonists of the human estrogen receptor. Science 285,744–746 [DOI] [PubMed] [Google Scholar]
- OSBORNE C.K., and SCHIFF R. (2011). Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 62,233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADDISON P.J., SILVA J.M., CONKLIN D.S., SCHLABACH M., LI M., ARULEBA S., BALIJA V., O'SHAUGHNESSY A., GNOJ L., SCOBIE K., et al. (2004). A resource for large-scale RNA-interference-based screens in mammals. Nature 428,427–431 [DOI] [PubMed] [Google Scholar]
- REID G., DENGER S., KOS M., and GANNON F. (2002). Human estrogen receptor-alpha: regulation by synthesis, modification and degradation. Cell Mol. Life Sci. 59,821–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHWABE J.W., CHAPMAN L., FINCH J.T., and RHODES D. (1993). The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 75,567–578 [DOI] [PubMed] [Google Scholar]
- SHAPIRO D.J., MAO C., and CHERIAN M.T. (2011). Small molecule inhibitors as probes for estrogen and androgen receptor action. J. Biol. Chem. 286,4043–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHELLY W., DRAPER M.W., KRISHNAN V., WONG M., and JAFFE R.B. (2008). Selective estrogen receptor modulators: an update on recent clinical findings. Obstet. Gynecol. Surv. 63,163–181 [DOI] [PubMed] [Google Scholar]
- SHIAU A.K., BARSTAD D., LORIA P.M., CHENG L., KUSHNER P.J., AGARD D.A., and GREENE G.L. (1998). The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95,927–937 [DOI] [PubMed] [Google Scholar]
- WANG L.H., YANG X.Y., ZHANG X., MIHALIC K., FAN Y.X., XIAO W., HOWARD O.M., APPELLA E., MAYNARD A.T., and FARRAR W.L. (2004). Suppression of breast cancer by chemical modulation of vulnerable zinc fingers in estrogen receptor. Nat. Med. 10,40–47 [DOI] [PubMed] [Google Scholar]
- WARD H.W. (1973). Anti-oestrogen therapy for breast cancer: a trial of tamoxifen at two dose levels. Br. Med. J. 1,13–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONG I., and LOHMAN T.M. (1993). A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. U. S. A. 90,5428–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOD J.R., LIKHITE V.S., LOVEN M.A., and NARDULLI A.M. (2001). Allosteric modulation of estrogen receptor conformation by different estrogen response elements. Mol. Endocrinol. 15,1114–1126 [DOI] [PubMed] [Google Scholar]
- XU D., and SHI H. (2009). Composite RNA aptamers as functional mimics of proteins. Nucleic Acids Res. 37,e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG X.R., FIGUEROA J.D., HEWITT S.M., FALK R.T., PFEIFFER R.M., LISSOWSKA J., PEPLONSKA B., BRINTON L.A., GARCIA-CLOSAS M., and SHERMAN M.E. (2013). Estrogen receptor and progesterone receptor expression in normal terminal duct lobular units surrounding invasive breast cancer. Breast Cancer Res. Treat. 137,837–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAI Y., BOMMER G.T., FENG Y., WIESE A.B., FEARON E.R., and CHO K.R. (2010). Loss of estrogen receptor 1 enhances cervical cancer invasion. Am. J. Pathol. 177,884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO X., SHI H., SEVILIMEDU A., LIACHKO N., NELSON H.C., and LIS J.T. (2006). An RNA aptamer that interferes with the DNA binding of the HSF transcription activator. Nucleic Acids Res. 34,3755–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZUKER M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31,3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]