

Abstract
Activation of AMP-activated protein kinase (AMPK) α2 protects the heart against pressure overload-induced heart failure in mice. Although metformin is a known activator of AMPK, it is unclear whether its cardio-protection acts independently of an AMPKα2-dependent pathway. Because the role of AMPKα1 stimulation on remodeling of failing hearts is poorly defined, we first studied the effects of disruption of both the AMPKα1 and AMPKα2 genes on the response to transverse aortic constriction (TAC)-induced left ventricular (LV) hypertrophy and dysfunction in mice. AMPKα2 gene knockout (KO) significantly exacerbated the degree of TAC-induced LV hypertrophy and dysfunction, whereas AMPKα1 gene KO had no effect on the degree of TAC-induced LV hypertrophy and dysfunction. Administration of metformin to both wild type (WT) and AMPKa2 KO mice attenuated the degree of TAC-induced LV remodeling, as evidenced by reduced LV and lung weights, a more favorable body weight to tibia length ratio, preserved LV ejection fraction, and lower levels of p-mTORser2481 and p-p70S6KThr389. These data support the notion that activation of AMPKα1 plays a negligible role in protecting the heart against the adverse effects of chronic pressure overload, and that metformin protects against adverse remodeling through a pathway that appears independent of AMPKα2.
Keywords: metformin, heart failure
Introduction
AMP-activated protein kinase (AMPK) integrates signals related to energy expenditure and energy production to regulate cellular metabolic processes. In response to metabolic stress, AMPK is rapidly activated to preserve energy homeostasis. AMPK is involved in the hypertrophic response to hemodynamic overload 1 and both AMPKα1 and AMPKα2 activities are elevated in response to pressure overload in rats.2 AMPK activity is also increased in pacing-induced heart failure in dogs.3 Using AMPKα2 deficient mice we previously demonstrated that AMPKα2 protects against Transverse Aortic Constriction (TAC) induced ventricular hypertrophy and dysfunction in part by repressing mTOR signaling1. Recently, we found that AMPKα2 plays an important role in regulating the expression of myocardial ERRα (and its downstream mitochondrial enzymes),4 an important transcriptional factor related to heart failure development. Most recently, we demonstrated that AMPKα2 reduces cardiomyocyte microtubule proliferation, a cytoskeletal abnormality often associated with the development of heart failure.5 Taken together, these observations suggest an important role for AMPKα2 in favorable adaptations of the heart in response to stress. However, the impact of AMPKα1 on ventricular structure and function under stress conditions remains unclear.
Metformin, a drug with insulin-sensitizing properties, is used widely in patients with diabetes mellitus and is known to activate AMPK. In large-scale clinical trials of diabetic patients, metformin decreases the long-term risk of cardiovascular death and non-fatal myocardial infarction.6,7 Metformin has also been shown to attenuate the degree of heart failure in dogs in response to rapid ventricular pacing3, reduce the degree of hypertrophy and fibrosis in mice subjected to a chronic pressure overload8, and preserve LV function and survival in mice following myocardial infarction.9 Because AMPKα2 protects against pressure overload induced LV hypertrophy and heart failure, we tested the hypothesis that metformin exerts its protective effect through activation of AMPKα2. To our surprise, we found that metformin was equally effective in attenuating TAC-induced LV hypertrophy and dysfunction in both wild type and AMPKα2 gene knockout (KO) mice, indicating that metformin can protect the heart in response to chronic pressure overload through an AMPKα2-independent pathway. We also found that AMPKα1 KO had no detectable effect on LV structure and function in mice in response to TAC.
Material and Methods
An extended Material and Methods section can be found in the online-only Data Supplement.
Animals and experimental protocol
All animal studies were performed according to a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee. Adult (12–15 weeks) male AMPKα1 KO mice (mixed background of C57B6J and 129) and AMPKα2 KO mice (background of C57B6J)10,11 and their wild type littermates were used in this study. TAC was performed as previously described.12 To avoid the unwanted influence of cardiac death occurring shortly after the TAC procedure,13 mice were randomly assigned to two groups 3 days after TAC and then treated with either metformin (100mg/kg/day, gavage) or vehicle for 3 weeks. Echocardiography and hemodynamic measurements were performed during anesthesia with 1.5% isoflurane.
Statistical Analysis
Results are expressed as mean ± standard error of the mean. For the study of TAC-induced LV hypertrophy and dysfunction, two-way analysis of variance (ANOVA) was used to test each variable for differences among the treatment groups. If the ANOVA demonstrated a significant effect, post hoc comparisons were made pairwise using Fisher’s least significant difference test. A p value ≤ 0.05 was considered significantly different.
Results
AMPKα2 but not AMPKα1 protected the heart from TAC-induced LV hypertrophy and heart failure
Consistent with our previous observation,1 AMPKα2 KO had no effect on LV weight, lung weight and LV ejection fraction under control conditions, but significantly exacerbated TAC-induced LV hypertrophy, pulmonary congestion and LV dysfunction (Figure S1, Table S3, Table S4). Western blots showed that AMPKα2 KO also caused significant reductions of the protein expressions of myocardial ERRα and its downstream targets (such as MCAD, CPT1b, CD36, FATP1, cytochorome-C oxidase subunit-3, cytochrome C and UCP3) under control conditions or after TAC (Figure S2). AMPKα2 KO also significantly reduced mRNAs of ERRα and its downstream target genes (Figure S3).
Interestingly, AMPKα1 KO had no detectable effect on LV weight, the ratio of LV weight to body weight or to tibia length, or LV function either under basal conditions or after TAC (Figure S4, Table S5). Thus, after 3 weeks of TAC, the ratio of LV weight to tibia length and the ratio of lung weight to tibia length were increased to a similar extent in both wild type and AMPKα1 KO mice. The degree of LV dilation and dysfunction in the AMPKα1 KO mice exposed to TAC were also comparable to those in the wild type littermates (Figure S4, Table S6). The heart rates were similar between wild type and AMPKα1 KO mice under control conditions and after TAC (Table S6). After TAC, LV dp/dtmax, LVdP/dtmin and systolic aortic pressure (proximal to the TAC site) were not different between wild type and AMPKα1 KO mice (Table S7). AMPKα2 expression was not altered in the AMPKα1 KO mice under control conditions (Figure S5). Myocardial AMPKα1 expression was significantly increased in wild type mice after TAC, while AMPKα2 expression was significantly reduced in the AMPKα1 KO mice after TAC (Figure S5). In addition, AMPKα1 KO had no detectable effect on expression of cardiac ERRα and its downstream targets (such as MCAD, CPT1b, CD36, FATP1, cytochorome C oxidase 3, cytochrome C and UCP3) under control conditions or after TAC (Figure S6).
Metformin attenuated TAC-induced LV hypertrophy and heart failure in wild type mice
In order to study the cardioprotective effect of metformin, we treated TAC-operated wild type mice with metformin or vehicle (normal saline) for 3 weeks. Under control conditions, metformin had no detectable effect on LV mass, structure and function (Table S8, S9). TAC caused significant increases of LV weight, lung weight, and their ratio to body weight (Figure 1). TAC also caused a significant decrease in LV fractional shorting, and a significant increase in LV end-systolic and end-diastolic diameters (Figure 1D–E). Metformin significantly attenuated the TAC-induced LV hypertrophy, increase of lung weight, decrease of LV ejection fraction, and increase of LV end-diastolic and end-systolic diameters as shown in Figure 1 and Table S10. Based on histological analysis, metformin did not affect the TAC-induced increase of LV fibrosis (Figure S7), indicating that the reduced LV hypertrophy in the metformin treated mice is likely a result of reduced cardiac myocyte hypertrophy. Metformin also did not affect the TAC-induced increase of LV TGF1β expression but significantly attenuated the TAC-induced increase of the myocardial oxidative stress marker 4-Hydroxynonenal (4-HNE) (Figure S8). Metformin attenuated expression of myocardial ANP (Figure 2), a biochemical marker associated with LV hypertrophy and dysfunction. Metformin had no detectable effect on blood glucose (186.1±14.2mg/dL in vehicle treated mice vs 184.3±21.4mg/dL in metformin treated mice).
Figure 1. Metformin treatment protects wild type mice from TAC-induced ventricular hypertrophy and heart failure.
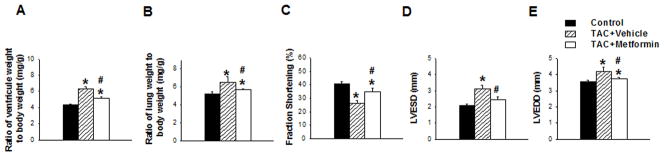
Ventricular weight (A) or lung weight (B) to body weight ratios were measured in WT mice exposed to TAC in the presence or absence of metformin as compared with control condition. Echocardiography was used to measure fractional shortening (C), end systolic diameter (D), and end diastolic diameter (E) in each group. *p<0.05 as compared with SHAM; #p<0.05 as compared with corresponding wild type mice; n=9 to 14/group.
Figure 2. Metformin increases AMPK phosphorylation and reduces ANP expression in Mice exposed to TAC.

Mice were exposed to SHAM conditions, 3 weeks of TAC, or 3 weeks TAC + metformin. Ventricular lysates were collected and analyzed by Western blot for expression of ANP, p-ACCSer79, ACC, total ACC, p-AMPKThe172, total AMPKα1, and total AMPKα2, and GAPDH (loading control) (A) and quantitated by densitometry (B) *p<0.05 vs SHAM; #p<0.05 vs vehicle treatment; n=5/group.
Metformin attenuated TAC-induced mTOR pathway activation in wild type mice
Metformin treatment significantly increased myocardial p-AMPKThr172 and p-ACCSer79 in wild type mice, indicating that metformin increased myocardial AMPK activity in these mice (Figure 2). Metformin did not affect the expression of myocardial AMPKα1 or AMPKα2 (Figure 2). In agreement with previous reports that activation of AMPK inhibits the mTOR signaling pathway, metformin treatment significantly attenuated TAC induced activation of the mTOR signaling pathway as demonstrated by decreases of p-mTORser2448, p-p70S6KThr389, p-S6ser235, and the ratios to their corresponding total protein content (Figure 3). Metformin also significantly attenuated the TAC-induced increase of p-AktSer473 and p-GSK-3βSer9 (Figure 3).
Figure 3. Metformin attenuates TAC-induced activation of mTOR signaling in wild type mice.
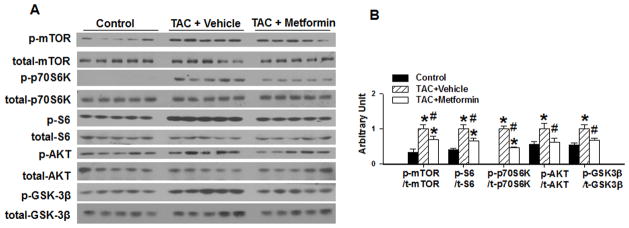
WT mice were exposed to SHAM conditions, 3 weeks of TAC, or 3 weeks TAC + metformin. Ventricular lysates were analyzed by Western blot for p-mTORSer2448, total-mTOR, p-p70S6 kinaseThr389, total-p70S6 kinase, p-S6Ser235/236, total-S6, p-AKTSer473, total-AKT, p-GSK3βSer9, and total-GSK3β (A) and results were quantitated by densitometry (B) *p<0.05 as compared with SHAM; #p<0.05 as compared with corresponding vehicle treated group; n=5/group.
Metformin attenuated TAC induced LV hypertrophy and dysfunction in AMPKα2 KO mice
In order to further investigate the possible mechanism underlying the cardioprotective effect of metformin, we treated sham or TAC-operated AMPKα2 KO mice with metformin or with vehicle for 3 weeks. Under control conditions, metformin had no detectable effect on LV mass, structure and function (Figure 4, Table S11). Metformin significantly attenuated the TAC-induced increase in LV weight, lung weight and their ratios to body weight and tibia length (Figure 4, Table S11). Metformin also significantly attenuated the TAC-induced decrease of LV fractional shortening, and the increase of LV end-diastolic and end-systolic diameters (Figure 4). Metformin did not affect TAC-induced LV fibrosis in AMPKα2 KO mice (Figure S9). Metformin also significantly attenuated the TAC-induced increases of myocardial ANP and 4-HNE in AMPKα2 KO mice (Figure 5, Figure S10). These results indicate that metformin effectively attenuates TAC-induced LV hypertrophy and dysfunction in AMPKα2 KO mice (Figure S11).
Figure 4. Metformin attenuates TAC induced hypertrophy and LV dysfunction in AMPKα2 KO mice.
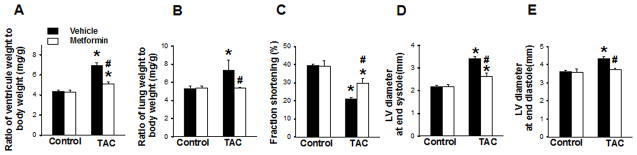
AMPKα2 KO mice were exposed to 3 weeks of SHAM or TAC conditions in the absence or presence of metformin. Heart weight (A) and lung weight (B) to body weight ratios. Echocardiography was used to measure fractional shortening (C), LV end systolic diameter (D), and LV end diastolic diameter (E). *p<0.05 as compared with SHAM; #p<0.05 as compared with corresponding vehicle treated group; n=5 to 9/group.
Figure 5. Metformin attenuates TAC induced expression of ANP in AMPKα2 KO mice.
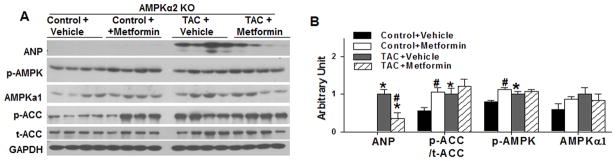
AMPKα2 KO mice were exposed to 3 weeks of SHAM or TAC conditions in the absence or presence of metformin. Ventricular lysates were analyzed by western blot for ANP, phospho-AMPKThr172, total AMPK, phospho-ACCSer79, total ACC and GAPDH (A) and results were quantitated by densitometry (B); n=4/group.
Metformin attenuated TAC-induced mTOR activation in AMPK α2 KO mice
Under sham conditions, metformin treatment significantly increased myocardial p-AMPKThr172 and p-ACCSer79, indicating increased activation of AMPKα1. However, this increase was not significantly different from the slightly elevated levels of p-AMPKThr172 observed after TAC (Figure 5). Metformin did not affect myocardial AMPKα1 expression (Figure 5). TAC caused a significant increase of p-mTORser2448, p-p70S6KThr389 and p-S6ser235, indicating activation of the mTOR signaling pathway. Interestingly, the addition of metformin to the AMPKα2 KO mice led to a significant attenuation of the TAC-induced increases of p-mTORser2481, p-p70S6KThr389 and p-S6ser235 (Figure 6) as well as a significant reduction in the TAC-induced increase of p-AktSer473 and p-GSK-3βSer9.
Figure 6. Metformin attenuates TAC induced mTOR signaling in AMPKα2 KO mice.
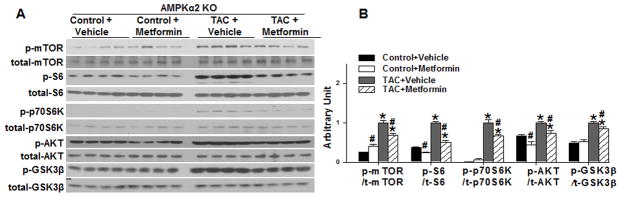
AMPKα2 KO mice were exposed to 3 weeks of SHAM or TAC conditions in the absence or presence of metformin. Ventricular lysates were analyzed by western blot for p-mTORSer2448, total mTOR, p-p70S6 kinaseThr389, total p70S6 kinase, p-S6Ser235/236, total S6, p-AKTSer473, total AKT, p-GSK3βSer9, and total GSK3β, (A) and results were quantitated by densitometry (B) *p<0.05 as compared with SHAM; #p<0.05 as compared with corresponding vehicle treated group; n=4/group.
Discussion
The present study reveals two major new findings. First, we demonstrated that the AMPK activator metformin was effective in attenuating TAC-induced LV hypertrophy and heart failure in both wild type and AMPKα2 KO mice, indicating that metformin protects mice from chronic pressure overload in part, through an AMPKα2-independent molecular pathway. Second, we demonstrated that AMPKα1 KO has no detectable effect on LV structure and function in mice either under control conditions or following TAC.
Our current study supports the notion that AMPKα1 and AMPKα2 exert different physiological and pathological functions in the heart. The findings that only AMPKα2 KO significantly reduced the expressions of myocardial ERRα and its downstream targets such as MCAD, CPT1b, CD36, FATP1, cytochorome-C oxidase subunit-3, cytochrome C, UCP3 and SOD2 under both control conditions and after TAC demonstrates that AMPKα2 is more important than AMPKα1 in maintaining normal LV structure and function. The differences in the influence of AMPKα2 and AMPKα1 on the cardiac adaptation to stress may relate to their different contributions to overall myocardial AMPK activity (AMPKα2 is the dominant isoform in mouse heart), their different cellular distribution (AMPKα2 is mainly expressed in cardiomyoyctes while AMPKα1 is more abundant in other cell types), and their variable subcellular distribution (AMPKα2 is distributed in both nucleus and cytosol while AMPKα1 is expressed only in cytosol). A recent study demonstrated that AMPKα1 and AMPKα2 contribute more than 80% of total AMPK activity, while AMPKα2 contributes ~20% of total AMPK activity in the mouse aorta. However, deletion of AMPKα2 but not AMPKα1 exacerbated neointima formation in mice.14 A study from the same group further demonstrated that activation of AMPKα2 but not AMPKα1 contributed to the nicotine-induced formation of abdominal aortic aneurysms in mice in vivo.15 Thus, different cellular and subcellular distribution or protein interactions of AMPKα2 and AMPKα1 likely contribute to their different cardiac protective effects.
The UK Prospective Diabetes Study has shown that metformin decreased the risk of cardiovascular death and the incidence of myocardial infarction associated with diabetes mellitus.16 Additional clinical studies have demonstrated that metformin decreased the risk of all-cause mortality and cardiovascular death in patients with type-2 diabetes mellitus.6,7 Eurich and colleagues reported the results of a meta-analysis showing that metformin was the only antidiabetic agent to reduce all-cause mortality without causing any harm in patients who had heart failure and diabetes mellitus.17 Moreover, metformin attenuated rapid ventricular pacing-induced heart failure in dogs and pressure overload induced LV hypertrophy in mice.3,8,18 Thus, our finding that metformin attenuated TAC-induced LV hypertrophy and dysfunction in mice is fully anticipated.
The finding that metformin effectively attenuated TAC-induced increases of p-mTORser2448, p-p70S6KThr389 and p-S6ser235 in both wild type and AMPKα2 KO mice suggest that metformin can attenuate myocardial mTOR signaling independent of AMPKα2 activation. mTOR signaling promotes ventricular hypertrophy, while inhibition of mTOR signaling attenuates ventricular hypertrophy. Thus, metformin attenuation of myocardial p-mTORser2448, p-p70S6KThr389 and p-S6ser235 in both wild type and AMPKα2 KO mice may help explain its anti-hypertrophic effects. As metformin did not affect blood glucose in these nondiabetic mice, the beneficial effect of metformin is likely independent of its effect on blood glucose.
We demonstrated that metformin effectively attenuated the TAC-induced increase of LV weight, lung weight and their ratios to body weight in both wild type and AMPKα2 KO mice. Metformin also significantly improved LV fractional shortening, attenuated LV dilation, and limited expression of myocardial ANP in both wild type and AMPKα2 KO mice exposed to TAC. These data indicate that metformin can exert cardiac protective effects through an AMPKα2 independent molecular pathway. Our findings differ from a previous report that metformin attenuation of LV hypertrophy is dependent upon AMPKα2.18 A study from the same group reported that metformin attenuated LV fibrosis in mice,8 a result that is also at variance with our observation. These discrepancies may be the result of technical differences between the studies. Thus, the degree of pressure overload and ventricular hypertrophy produced by TAC was considerably different between studies. Fu et al18 reported a 26% increase in ventricular weight in the vehicle treated AMPKα2 KO mice, as compared with a 58% increase in ventricular mass in the vehicle treated AMPKα2 KO mice in the present study. In addition, the difference in genetic backgrounds of the AMPKα2 KO mice (a 129/C57B6 mixed background for AMPKα2 KO mice was used by Fu18 vs. a C57B6J background for the AMPKα2 KO mice in the present study) could have contributed to the differing results.
Several studies have demonstrated that metformin exerts biological and physiological effects through a number of AMPK-independent molecular pathways, and these effects may also influence cardiac hypertrophy. A previous study demonstrated that metformin inhibits hepatic gluconeogenesis in an AMPK-independent manner via a decrease in hepatic energy state.19,20 There is also indirect evidence that metformin can increase adenosine production in the heart.21 Adenosine protects against pressure overload hypertrophy and heart failure and, like AMPK activation, also attenuates mTOR signaling.22,23 In addition, metformin has been shown to decrease mTOR signaling independent of AMPK through either activation of RAG2 GTPase24 or through activation of REDD.25 It is possible that RAG2 or REDD also play a role in the metformin attenuation of cardiac mTOR signaling and hypertrophy observed in AMPKα2 KO mice. Additional studies have demonstrated that metformin alters Stat3 activity independent of AMPK.26 There is evidence that Stat3 plays a role in promoting cardiac hypertrophy,27 so Stat3 suppression may also play a role in the anti-hypertrophic effects of metformin. The extent to which these actions of metformin attenuate cardiac hypertrophy independent of AMPKα2 is not clear.
AMPKα1 KO had no observable influence on TAC induced hypertrophy and heart failure; however, since AMPKα1 is still expressed in AMPKα2 KO mice, we cannot exclude the possibility that metformin might exert its cardiac protective effect through a slight compensatory increase in the activation of AMPKα1. Another limitation of the present study is that because AMPKα1 KO and AMPKα2 KO mice are global KO strains, the phenotypes observed in these KO mice may not be due to effects on cardiomyocytes or myocardial tissues alone. Similarly, metformin may have effects on multiple organ systems or other cell types that also contribute to its beneficial effects in attenuating TAC-induced LV hypertrophy and heart failure.
Perspectives
Although metformin is a known activator of AMPK, it is unclear whether its cardio-protection acts through an AMPKα2-dependent pathway. We demonstrated that metformin attenuates pressure overload-induced heart failure through an AMPKα2-independent mechanism. We also demonstrated that AMPKα1 plays a negligible role in protecting the heart against heart failure development.
Supplementary Material
Novelty and Significance.
What Is New?
AMPK activator metformin is equally effective in attenuating pressure overload-induced heart failure in both wild type and AMPKα2 KO mice.
AMPKα1 KO has no detectable effect on LV structure and function in mice following pressure overload.
What Is Relevant?
This study suggests that metformin attenuated pressure overload-induced LV hypertrophy and dysfunction in mice independent to the activation of AMPKα2.
Summary
Metformin protects heart against LV hypertrophy and dysfunction through a novel molecular pathway that appears independent of AMPKα2 activation.
Acknowledgments
Sources of funding: This study was supported by U.S. Public Health Service Grants HL098669, HL098719, HL102597, HL089249, HL021872 and R01HL105406 from the National Institutes of Health, and Grant 81270319 from Chinese National Natural Science Foundation. Zhongbing Lu is a recipient of Chinese Academy of Sciences Hundred Talents Program.
Footnotes
Disclosures: None.
References
- 1.Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, Xu W, Wiczer B, Bernlohr DA, Bache RJ, Chen Y. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian R, Musi N, D’Agostino J, Hirshman MF, Goodyear LJ. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 3.Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, Ogai A, Asakura M, Kim J, Minamino T, Takashima S, Sanada S, Sugimachi M, Komamura K, Mochizuki N, Kitakaze M. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation. 2009;119:2568–2577. doi: 10.1161/CIRCULATIONAHA.108.798561. [DOI] [PubMed] [Google Scholar]
- 4.Hu X, Xu X, Lu Z, Zhang P, Fassett J, Zhang Y, Xin Y, Hall JL, Viollet B, Bache RJ, Huang Y, Chen Y. AMP activated protein kinase-α2 regulates expression of estrogen-related receptor-α, a metabolic transcription factor related to heart failure development. Hypertension. 2011;58:696–703. doi: 10.1161/HYPERTENSIONAHA.111.174128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fassett JT, Hu X, Xu X, Lu Z, Zhang P, Chen Y, Bache RJ. AMPK attenuates microtubule proliferation in cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013;304:H749–758. doi: 10.1152/ajpheart.00935.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masoudi FA, Wang Y, Inzucchi SE, Setaro JF, Havranek EP, Foody JM, Krumholz HM. Metformin and thiazolidinedione use in Medicare patients with heart failure. JAMA. 2003;290:81–85. doi: 10.1001/jama.290.1.81. [DOI] [PubMed] [Google Scholar]
- 7.Masoudi FA, Inzucchi SE, Wang Y, Havranek EP, Foody JM, Krumholz HM. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation. 2005;111:583–590. doi: 10.1161/01.CIR.0000154542.13412.B1. [DOI] [PubMed] [Google Scholar]
- 8.Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M, Shen Q, Zhu Y, Zhang Y. Metformin attenuates cardiac fibrosis by inhibiting the TGFbeta1-Smad3 signalling pathway. Cardiovasc Res. 2010;87:504–513. doi: 10.1093/cvr/cvq066. [DOI] [PubMed] [Google Scholar]
- 9.Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, Ramachandran A, Anaya-Cisneros M, Tian R, Lefer DJ. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104:403–411. doi: 10.1161/CIRCRESAHA.108.190918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jørgensen SB, Viollet B, Andreelli F, Frøsig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 11.Viollet B, Andreelli F, Jørgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 13.Lu Z, Fassett J, Xu X, Hu X, Zhu G, French J, Zhang P, Schnermann J, Bache RJ, Chen Y. Adenosine A3 receptor deficiency exerts unanticipated protective effects on the pressure-overloaded left ventricle. Circulation. 2008;118:1713–1721. doi: 10.1161/CIRCULATIONAHA.108.788307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song P, Wang S, He C, Wang S, Liang B, Viollet B, Zou MH. AMPKα2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res. 2011;109:1230–1239. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou MH. Activation of AMP-activated protein kinase α2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012;18:902–910. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 17.Eurich DT, McAlister FA, Blackburn DF, Majumdar SR, Tsuyuki RT, Varney J, Johnson JA. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: systematic review. BMJ. 2007;335:497. doi: 10.1136/bmj.39314.620174.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu YN, Xiao H, Ma XW, Jiang SY, Xu M, Zhang YY. Metformin attenuates pressure overload-induced cardiac hypertrophy via AMPK activation. Acta Pharmacol Sin. 2011;32:879–887. doi: 10.1038/aps.2010.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, Viollet B, Guigas B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. 2011;54:3101–3110. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paiva M, Riksen NP, Davidson SM, Hausenloy DJ, Monteiro P, Gonçalves L, Providência L, Rongen GA, Smits P, Mocanu MM, Yellon DM. Metformin prevents myocardial reperfusion injury by activating the adenosine receptor. J Cardiovasc Pharmacol. 2009;53:373–378. doi: 10.1097/FJC.0b013e31819fd4e7. [DOI] [PubMed] [Google Scholar]
- 22.Xu X, Fassett J, Hu X, Zhu G, Lu Z, Li Y, Schnermann J, Bache RJ, Chen Y. Ecto-5′-nucleotidase deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction. Hypertension. 2008;51:1557–1564. doi: 10.1161/HYPERTENSIONAHA.108.110833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fassett JT, Hu X, Xu X, Lu Z, Zhang P, Chen Y, Bache RJ. Adenosine kinase regulation of cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol. 2011;300:H1722–1732. doi: 10.1152/ajpheart.00684.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalender A, Selvaraj A, Kim SY, Gulati P, Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, Thomas G. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 26.Lin CC, Yeh HH, Huang WL, Yan JJ, Lai WW, Su WP, Chen HH, Su WC. Metformin Enhances Cisplatin Cytotoxicity by Suppressing Stat3 Activity Independently of the LKB1-AMPK Pathway. Am J Respir Cell Mol Biol. 2013;49:241–250. doi: 10.1165/rcmb.2012-0244OC. [DOI] [PubMed] [Google Scholar]
- 27.Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, Okabe M, Kishimoto T, Yamauchi-Takihara K. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:315–319. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.