

Abstract
The bacterial melibiose permease (MelB) belongs to the glycoside–pentoside–hexuronide:cation symporter family, a part of the major facilitator superfamily (MFS). Structural information regarding glycoside–pentoside–hexuronide:cation symporter family transporters and other Na+-coupled permeases within MFS has been lacking, although a wealth of biochemical and biophysical data are available. Here we present the three-dimensional crystal structures of Salmonella typhimurium MelBSt in two conformations, representing an outward partially occluded and an outward inactive state of MelBSt. MelB adopts a typical MFS fold and contains a previously unidentified cation-binding motif. Three conserved acidic residues form a pyramidal-shaped cation-binding site for Na+, Li+ or H+, which is in close proximity to the sugar-binding site. Both cosubstrate-binding sites are mainly contributed by the residues from the amino-terminal domain. These two structures and the functional data presented here provide mechanistic insights into Na+/melibiose symport. We also postulate a structural foundation for the conformational cycling necessary for transport catalysed by MFS permeases in general.
 The bacterial symporter MelB transports galactosides with Na+, Li+ or H+ ions. Ethayathulla et al. present crystal structures of MelB in two distinct conformations, providing the first structural insights into the transport mechanism of a sodium-coupled permease of the major facilitator superfamily.
The bacterial symporter MelB transports galactosides with Na+, Li+ or H+ ions. Ethayathulla et al. present crystal structures of MelB in two distinct conformations, providing the first structural insights into the transport mechanism of a sodium-coupled permease of the major facilitator superfamily.
Membrane transport proteins from the glycoside–pentoside–hexuronide:cation symporter family1,2 (TC 2.A.2; Supplementary Fig. S1a,b) are widely found in all life forms, and Escherichia coli melibiose permease (MelBEc) is the best-studied representative. However, our understanding of transport mechanisms with these important permeases is limited by lack of high-resolution structures. Salmonella typhimurium MelB (MelBSt) has >85% primary sequence identity with MelBEc3,4, and both proteins exhibit ~54% similarity with the human major facilitator superfamily (MFS) domain-containing protein 2A; moreover, the residues essential for transport in MelB are functionally conserved in MFS domain-containing protein 2A5. Remarkably different from other MFS transporters6, MelB catalyses electrogenic symport of galactosides with Na+, Li+ or H+ (ref. 7); the broad cation selectivity is a prominent feature. Some MelB orthologues selectively couple sugar symport with one or two out of the three cations, but the anomeric configuration of the sugar is important with respect to specific coupling cations7. For instance, sugars in the α-configuration (melibiose and raffinose) utilize all three cations, but β-anomers of galactopyranosides couple only to Na+ and Li+ (ref. 8).
MelBSt shares similarity with regard to cation-coupling specificity and other transport features with the well-studied MelBEc4,7,9,10,11,12. All three coupling cations compete for a single binding site with a protein/sugar/cation stoichiometry of unity4,13,14. Electrogenic sugar symport is driven by Δ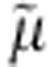 H+, Na+ or Li+, depending on the coupling cation4. Thermodynamically, MelB transduces the free energy from the downhill translocation of a cation to drive translocation of sugar against a concentration gradient and vice versa4,12.
H+, Na+ or Li+, depending on the coupling cation4. Thermodynamically, MelB transduces the free energy from the downhill translocation of a cation to drive translocation of sugar against a concentration gradient and vice versa4,12.
Here we present three-dimensional X-ray crystal structures of MelBSt in two distinct conformations, refined to 3.35 Å. The structure is in general agreement with the predicted model of MelB15. Notably, most MFS members are either H+ symporters or uniporters. Among the members with solved structures16,17,18,19,20,21,22,23, MelBSt is the sole member that utilizes Na+ primarily as a coupling cation for symport and uses a previously uncharacterized coupling mechanism.
Results
Functional characterization
Purified MelBSt is monodisperse, stable (Supplementary Fig. S2a,b) and binds melibiose, 2′-(N-dansyl)aminoalkyl-1-thio-β-D-galactopyranoside (D2G) or methyl-β-D-thiogalactoside, but has no affinity for sugars without a galactopyranosyl moiety (Fig. 1a). A single sugar-binding site was determined by isothermal titration calorimetry (ITC; Fig. 1b,c). Melibiose binding is exothermic with a Kd of ~1 mM in the presence of Na+ or Li+, which is consistent with previous results4. Furthermore, sugar binding is driven energetically by a small favourable enthalpy change (ΔH) and a bigger increase in entropy (TΔS).
Figure 1. Functional characterization.

Purified MelBSt containing 100 mM NaCl or LiCl were subjected to D2G FRET and ITC measurements. (a) D2G FRET measurement. With an excitation wavelength of 290, emission spectra were collected between 415 and 575 nm. Difference spectra (ΔI) were calculated before and after addition of a given sugar (50 mM). Grey curves, melibiose; purple curves, methyl-β-D-thiogalactoside (TMG); green curves, sucrose; blue curves, glucose. (b) ITC measurement. Melibiose titration thermograms (insets) were recorded at 25 °C. Cumulative heat change (ΔQ) is plotted as a function of the molar ratio of melibiose/MelBSt, and fitted with the one-site independent binding model. (c) Melibiose-binding energetics. Enthalpy change (ΔH) and the association constant (Ka) were measured directly; dissociation constant Kd=1/Ka; free-energy change ΔG=−RT ln Ka; entropy change ΔTS=ΔH−ΔG; n, stoichiometry. Error bar, s.d., n=2.
Overall fold of MelBSt
The structure of MelBSt was solved by molecular replacement and refined to a resolution of 3.35 Å (Table 1). The asymmetric unit contains four molecules (Mol-ABCD; Supplementary Fig. S3a,b) with twinning and pseudotranslation symmetries. Surprisingly, twinned Mol-B and Mol-D have a conformation different from Mol-A and Mol-C (Fig. 2 and Supplementary Fig. S4a,b). We describe only Mol-A and Mol-B here as representative structures.
Table 1. Data collection and refinement statistics (molecular replacement).
| MelBSt | |
|---|---|
| Data collection | ALS 5.0.2 |
| Space group | P32 |
| Cell dimensions | |
| a=b, c (Å) | 127.20, 206.30 |
| γ (°) | 120.00 |
| Resolution (Å) | 40.00–3.35 (3.47–3.35)* |
| Rsym or Rmerge | 0.092 (0.77) |
| I/σI | 6.0 (1.52) |
| Completeness (%) | 99.4 (99.4) |
| Redundancy | 2.8 (2.7) |
| Refinement | |
| Resolution (Å) | 38.0–3.35 (3.44–3.35) |
| No. of reflections | 50,526 |
| Rwork/Rfree | 0.30/0.35 |
| No. of atoms | |
| Protein | 12,963 |
| B-factors | |
| Protein | 86.0 |
| Root mean squared deviations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.7 |
MelBSt, Salmonella typhimurium melibiose permease.
*Values in parentheses are for highest-resolution shell.
Figure 2. Crystal structures of MelBSt in two outward conformations.

(a) Surface electropotential maps with side chains forming the outer gate and the internal cavity shown in yellow and green (Mol-A), respectively, or cyan (Mol-B) sticks. (b) Overall structure of MelBSt. The N-terminal domains of Mol-A or Mol-B are shown in green and cyan, respectively, and the central loops are shown in yellow. The helices are labelled with Roman numerals. CH1–3 denote the helices in the cytoplasmic loops and the C-terminal tail. All figures showing MelB structures were prepared by Cα1–430 superposition of Mol-B on Mol-A.
As predicted15, MelBSt adopts a typical MFS fold, organized in amino- and carboxy-terminal six-transmembrane α-helix bundles (Fig. 2), with a connecting cytoplasmic central loop containing two short helices (CH1 and CH2), and the cytoplasmic C-terminal tail with another short α-helix (CH3). Both the N and C termini are on the cytoplasmic side of the membrane and the overall shape is consistent with the electron-microscopic map of MelBEc24. The N- and C-terminal domains are related by a pseudo two-fold symmetry axis perpendicular to the membrane plane and separated from each other by an internal cavity facing the periplasm. Within each domain, there is a two-fold inverted pseudosymmetry between helices I–III and IV–VI, as well as between VII–IX and X–XII, forming repeats A–D (Supplementary Fig. S4c). Similar to other MFS members16,18,25, MelB may have evolved from triple-helix repeats with a similar genetic origin.
Most MelBSt helices are irregular with kink(s) and tilts (Fig. 2b and Supplementary Figs S3 and S4). Two to three broken helices are observed in both molecules: helices IV, X and XI in Mol-A and helices II, VII and X in Mol-B. Mol-A exhibits a periplasmic-facing conformation with a partially occluded internal cavity due to the interactions between helices I and VII in the N- and C-terminal domains, respectively. This structure is described as an outward partially occluded conformation (Figs 2a and 5c). The cavity is closed on the cytoplasmic side by interdomain contacts between inner helices IV/X, V/VIII and II/XI; solvent is accessible from the periplasmic side. Mol-B has a partial outward opening compared with fully opened FucP20, and is described as a partial outward conformation (Figs 2a and 5c). The root mean squared deviation between Mol-A and Mol-B is 2.7 Å, and the major differences are found in the first two helices of each repeat (Supplementary Fig. S4c), indicating that the inner helices move relatively independently between the two conformations.
Viewed from the periplasmic side, the internal cavity in Mol-A has an asymmetric charge distribution (Fig. 3a). Except for Lys377 in the C-terminal domain, all other charged residues, including Asp residues (19, 55, 59 and 124), Lys18 and Arg residues (52 and 149), are in the N-terminal domain. The calculated omit maps do not reveal interpretable densities for either bound sugar or cation.
Figure 3. Cosubstrate-binding sites.

(a) Wall-eyed stereo view from the periplasmic side of the internal cavity in Mol-A with the N- and C-halves in green and blue, respectively. (b) Identical view of Mol-B with N- and C-halves in cyan and blue, respectively. (c) Steady-state levels of [3H]melibiose accumulation by intact cells at 10 min presented as histograms. DW2, cells without MelB. Error bar, s.e.m.; n=2 for all mutants with a single-site mutation and n=10 for the WT and DW2. (d) Trp→D2G FRET with RSO vesicles. The emission signals were collected at wavelength of 490 nm after being excited at wavelength of 290 nm. ⁁, adding 10 μM D2G; pink arrows ↓, adding 120 mM melibiose; grey arrows ↓, adding water; black arrows ↓, adding 20 mM or 50 mM Na+ or Li+. Black double-headed arrows ↔, Na+ or Li+ stimulation; pink double-headed arrows ↔, melibiose displacement of D2G.
The cation-binding site
A cluster of Asp residues at positions 55 and 59 (helix II), and 124 (helix IV), forms a pyramidal-shaped pocket in Mol-A (Fig. 3a and Supplementary Fig. S5a). In close proximity, there are Tyr120, Thr121 (helix IV) and Thr373 (XI). Together these residues could form the trigonal bipyramid geometry required for a metal-binding site26,27. Thr121 forms H-bonds with Asn58 and Asp59, stabilizing the interaction between helices II/IV, and Lys377 H-bonds with Tyr120 separating the negatively charged pocket from the central water-filled cavity (Fig. 3a). Asp124 is buried between Tyr120 and Trp128. Asp59 is buried in a hydrophobic environment (Supplementary Fig. S5c).
Remarkably, Cys replacement at position 55, 59, 124, 58, or 121 differentially alters cation selectivity. Thus, the D55C mutant in MelBSt retains only a low level of H+-coupled melibiose binding and active transport (Fig. 3c,d and Supplementary Fig. S6a), but Na+ or Li+ does not stimulate either binding or transport. The D59C and D124C mutants bind ligand with reduced affinity (Fig. 3d), but do not catalyse active melibiose transport (Fig. 3c and Supplementary Fig. S6a). Reduced rates of melibiose efflux and exchange are observed with mutant D59C, but mutant D124C does not exhibit exchange activity. In contrast to wild type (WT), melibiose binding, efflux and exchange in the D59C mutant are not stimulated by Na+ or Li+ nor is efflux inhibited at acidic pH (Fig. 4); thus, the D59C mutant behaves similar to a sugar uniporter. None of these mutations has an adverse effect on protein expression (Supplementary Fig. S6b). Furthermore, both the N58C and T121C mutations selectively inhibit H+- and Na+-coupled activity, with little effect on Li+-coupled melibiose transport (Fig. 3c and Supplementary Fig. S6a). Combined with the observed geometry and functional data presented here and elsewhere7,28,29,30, we propose that the cation-binding site selectively coordinates Na+, Li+ or H3O+ through a dynamic change in size and shape27 dictated by the orientation of Asp55, Asp59 and Asp124. This supports the conclusion that all coupling cations in MelB compete for a single binding site4,13,14. It is apparent that each Asp residue at these positions has a specific role in cation binding and coupling. All three Asp residues are required for Na+ or Li+ binding, and Asp59 is essential for H+ binding. Asp59 is buried in a hydrophobic environment (Supplementary Fig. S5c); based on the PROPKA prediction31, it has a largely elevated pKa of 9.0. We propose that Mol-A is in a protonated state. As the cation-binding site binds H+, Na+ or Li+ mutually exclusively4,13,14, a bound Na+ is not expected in this configuration.
Figure 4. Melibiose efflux and exchange.

RSO membrane vesicles (28 mg ml−1) containing MelBSt were tested for outwardly directed flow of [3H]melibiose in the absence (efflux, squares) or presence (exchange, red circles) of equimolar concentration of unlabelled melibiose. The same experiment was performed in the presence of 20 mM NaCl or LiCl. 2-(4′-maleimidylanilino)naphthalene-6-sulphonic acid-treated vesicles were used for the negative control (blue triangles). Acidic pH effect on melibiose efflux was tested by decreasing the dilution buffer pH to 5.5 (green squares). Error bar, s.e.m., n=2.
In Mol-B, the pyramidal-shaped cation-binding site is deformed. Displacement of the helix XI kink between helices II and IV places Asp55 and Asp59 in a different environment (Fig. 3b and Supplementary Fig. S5d), distorting the pyramidal arrangement. The deformation of the cation-binding site is probably necessary to release the bound Na+. This conformational change, which is involved in rearrangement of the helices, is expected to contribute to the rate-limiting step of transport4,9,32. In this state, Arg52 holds helix XI by H-bonding with backbone atom of Gly378, indicating a primary structural role. Furthermore, Asp55 also stabilizes packing between helices II and XI.
The known crystal structures of other Na+-coupled symporters26,33,34 have a relatively low-affinity Na+-binding motif, where four to six partial charges coordinate Na+. In contrast, MelB utilizes three Asp residues representing a unique, higher-affinity Na+-binding motif4,29.
The sugar-binding site
The sugar specificity of MelBSt is highly selective for di- or trisaccharides containing a D-galactopyranosyl moiety or the monosaccharide D-galactose, as in MelBEc8 or LacY35,36,37. In close proximity to the cation-binding site, a water-filled cavity is observed between residues Asp19 (helix I), Arg149 (helix V), Tyr120, Asp124 and Trp128 (helix IV), as well as Lys377 (helix XI; Fig. 3a). Asp19 and Arg149 form a salt bridge on one side of the cavity; Tyr120 and Lys377 are H-bonded on the other side. Cys replacement at each of these positions abolishes or markedly decreases binding, as measured by Trp→D2G fluorescence resonance energy transfer (FRET), and melibiose transport (Fig. 3c,d, and Supplementary Figs S6 and S7), although mutant K377C retains 30–40% of WT symport activity with each cation (Fig. 3c and Supplementary Fig. S6a). In MelBEc, single-Cys mutation of Asp19 selectively abolishes sugar binding with little effect on Na+ binding30, mutant W128F has decreased affinity for 4-nitrophenyl-α-D-galactopyranoside38, and mutants Y120F and D124E exhibit decreased sugar- and Na+-binding affinities39. Therefore, these charged or polar side chains, particularly Asp19 and Arg149, play critical roles in sugar selectivity and affinity, and the aromatic residues Trp128 and Tyr120 may contribute to affinity by CH/π-interactions with the pyranosyl ring of the sugar40,41. Clearly, a crystal structure with bound sugar is required to determine the sugar-binding coordinates precisely. In Mol-B, the salt bridge between Asp19 and Arg149 is broken, and Trp128 is also displaced from the cavity, suggesting deformation of the sugar-binding site (Fig. 3b). It is noteworthy that the cation-binding site is also collapsed in this conformation; hence, the Mol-B probably represents an inactive state of MelBSt.
It is interesting that both LacY and MelB have similar sugar specificity and location of the sugar-recognition sites35; however, their cation-binding sites are related by a pseudo two-fold symmetry axis perpendicular to the membrane plane. The location of the Na+-binding site in MelB is away from Arg149, which is involved in sugar binding, thereby allowing the presence of a strongly negatively charged metal-binding site without a negative influence on sugar binding.
Positive cooperative binding between the cosubstrates is well established in MelB4,38,39,42. Helix IV, in the middle of the N-terminal domain, physically connects both cosubstrate sites. In addition to Tyr120 and Asp124, Lys18 (Fig. 3c and Supplementary Fig. S6) H-bonds with the backbone atom of Met123 (Fig. 3a,b), which links helices I–IV, underscoring the crucial role of helix IV in cooperative binding and transport. On the basis of the structures, we anticipate that Na+ binding leads to movement of helix IV, which recruits Asp124, optimizing the pyramidal shape of the cavity with Asp55 and Asp59, and aligning Tyr120 and Trp128 for aromatic stacking with the sugar, thereby increasing affinity. It is well documented that sugar affinity is increased by more than threefold in the presence of Na+ or Li+ (refs 4, 13, 29, 30, 43), and Asp124 is required for completion for the Na+-binding site30.
Ionic locks and conformational cycling
Both conformers are present in the same crystal, indicating that MelBSt has multiple conformations. Available crystal structures show MFS permeases in inward or outward conformations with occluded or partial-to-fully open cavities (Fig. 5), implying that an individual transporter may favour few conformers within a conformational cluster depending on the lowest free-energy state. The energy barrier for altering conformational equilibrium between inward and outward clusters may be the formation of an occluded intermediate state. Here we show that extramembrane electrostatic interactions play a crucial role in this regard.
Figure 5. Clusters of MFS permeases in different conformations.

Structures were categorized into clusters according to their conformational state. *, Disordered region in structure; −, at an unlocked state; 1/2/3, the presence of lock-1, lock-2 or lock-3. PDB ID is shown for each structure.
Both inside-closed structures of MelBSt exhibit multiple hydrophobic interactions (hydrophobic patches) on the cytoplasmic side of the transmembrane helices sealing the cavity between N- and C-domains (Fig. 6a, left). This sealed dynamic domain is also stabilized by interdomain electrostatic interactions, designated as ionic locks, involving three Arg residues: Arg295 (cytoplasmic end of helix IX), Arg141 (cytoplasmic end of the helix V) and Arg363 (flexible loop10–11; Fig. 6a,b). At the first ionic lock (L-1), Arg295 forms multiple H-bonds with Gln143 (helix V) and Pro287 (helix VIII) to hold helix V close to the C-terminal domain. At L-2, Arg141 forms four H-bonded ion pairs with Asp351 and Asp354 (helix X), stabilizing helix X. At L-3, Arg363 (loop10–11) forms one ion pair and two H-bonding interactions with the backbone atoms of Val204, Asp208 (N-terminal side of central loop6–7) and Gly74 (loop2–3), holding the N-terminal domain in an outward-facing conformation. It is likely to be that by stabilizing helix X, ionic locks L-1 and L-2 facilitate interaction of Arg363 with its partners. Replacing Arg363, Arg141 or Arg295 individually with Cys yields conformationally compromised mutants that fail to transport but retain affinity for both cosubstrates (Fig. 6c,d and Table 2). As indicated from a thread model of MelBSt at an inward-facing conformation (Fig. 6a, right), all three cytoplasmic interdomain locks are at an unlocked state, as the residues involved in these electrostatic interactions are 20 Å far apart. Therefore, these three ionic locks play important role in facilitating the formation of outward-facing conformation. On the periplasmic side, Asp35 (helix I) organizes L-4 (Fig. 6a,b) by forming a salt bridge and an H-bond with Arg175 (helix VI) and stabilizes the outward-facing conformation.
Figure 6. Ionic locks and conformational cycling.

N- and C-terminal domains are shown in green and blue, respectively, and hydrophobic patches are in yellow. Arg141–Arg149 stretch is coloured as black. ‘L’ denotes positions of ionic locks. (a) Left, outward partially occluded conformation (Mol-A); right, threading model for inward conformation. (b) Residues forming ionic locks are shown in sticks connected by broken lines. Grey spheres represent the residues involved in sugar binding. (c) Cys mutant of Arg residue at position 295, 141 or 363. Intact E. coli DW2 cells containing WT MelBSt or a given mutant were assayed for [3H]melibiose transport (0.4 mM, 10 mCi mmol−1) in the absence or presence of 20 mM NaCl or LiCl. Intracellular melibiose expressed as nmol mg−1 of total proteins is plotted as a function of time. Each single-site mutation is in the WT background. Error bar, s.e.m., n=2 for the mutants and n=3 for the WT. (d) Western blotting. Twenty-five micrograms of RSO vesicles was loaded on each well and detected by anti-His-tag antibody.
Table 2. Affinity for melibiose and Na+.
| Melibiose displacement of D2G (IC50, mM) | Na+ activation constant for D2G FRET (K0.5, mM) | |
|---|---|---|
| WT MelBSt | 2.65±0.80* | 1.92±0.78 |
| R295C | 4.04±0.42 | 3.22±1.07 |
| R141C | 5.93±1.61 | 4.77±0.39 |
| R363C | 2.83±0.57 | 5.51±1.07 |
D2G, 2′-(N-dansyl)aminoalkyl-1-thio-β-D-galactopyranoside; FRET, fluorescence resonance energy transfer; MelBSt, Salmonella typhimurium melibiose permease; WT, wild type.
*s.e.m., n=2.
Discussion
Similar ionic lock interactions are present in other MFS structures (Fig. 5 and Supplementary Fig. S8). From top to bottom in the Fig. 5, conformations are depicted from the full-inward to the full-outward states17,18,20,21,22,23,44,45,46. None of the predicted locks L-1, L-2 and L-3 exists in cluster-1, which includes the full and partially inward states. L-1 is formed in cluster-2, the inward partially occluded state. When the conformation reaches a more occluded state (cluster-3), both L-1 and L-2 are observed. L-3 is not observed in any of the inward conformers, but exists in most outward structures (cluster-4). Thus, formation of an occluded state probably requires L-1 and L-2, and the outward state requires all three ionic locks. Reverse order of lock formation is expected for the conformational changes to proceed in the opposite direction. The formation and deformation of these ionic interactions are the structural foundation for the function-required conformational switching between inward and outward conformers.
What triggers the locking/unlocking processes? In MelB, the salt bridge between Arg149 and Asp19 is involved in sugar binding. It is noteworthy that a short cytoplasmic stretch of helix V (Arg141–Arg149) links the sugar-binding site with two ionic locks (L-1 and L-2; Fig. 6a,b) and also contains two other conformationally important residues, Glu142 (ref. 42) and Pro148 (ref. 47). We propose that sugar binding promotes a dynamic state of the cytoplasmic stretch of helix V (Fig. 6a,b) and triggers locking/unlocking processes associated with a cascade of structural rearrangements for reorientation of the sugar-binding pocket.
We previously proposed a sequential binding kinetic model to explain reversible cation/melibiose symport in MelB based on an alternating-access mechanism (Fig. 7)9,12,42. The structural and functional studies discussed here allow integration of the conformational states with kinetic steps in the proposed model for explaining melibiose/Na+ symport reactions. Although melibiose/Na+ efflux is explained for experimental simplicity4, influx is the reverse reaction starting from the intermediate [6] (Fig. 7) and proceeds via the red arrows around the circle; active melibiose transport driven by Δ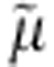 H+, Na+ or Li+ is likely to occur in similar fashion. The polarity of transport is controlled by the direction of the net free energy stored in the form of either the sugar gradient or the electrochemical gradient of Na+, Li+ or H+, and the transport process is reversible.
H+, Na+ or Li+ is likely to occur in similar fashion. The polarity of transport is controlled by the direction of the net free energy stored in the form of either the sugar gradient or the electrochemical gradient of Na+, Li+ or H+, and the transport process is reversible.
Figure 7. Scheme for Na+/melibiose symport.

[1–8], Kinetic steps in the overall transport cycle. The green colour-filled cycle represents the cell inner membrane. Na+, blue circles; melibiose, black squares. N, the N-terminal domain in green colour; C, the C-terminal domain in blue colour. Melibiose influx down a sugar concentration gradient starts at step [6] and proceeds via the red arrows around the circle, and melibiose efflux down a sugar concentration gradient starts at [1] and proceeds via the black arrows around the circle. Active transport of melibiose against a concentration gradient proceeds from step [6] via the red arrows as the melibiose influx.
This simplified scheme for melibiose efflux involves eight states (Fig. 7, black arrows): [1] inward Na+-bound state. Binding of a Na+ to the cation-binding site induces helix IV movement with an increase in sugar affinity. [2] Inward Na+- and melibiose-bound state. Sugar binding from the cytoplasm initiates the locking process (Fig. 6a,b). [3] Occluded Na+- and melibiose-bound state. Closure of the cytoplasmic cavity by the hydrophobic patch is locked by L-1 and L-2. [4] Outward Na+- and melibiose-bound state. Interactions at L-3 and L-4 facilitate a conformational transition from the occluded to an outward state. [5] Outward melibiose-released state. Bound melibiose is released to the periplasmic side from the open outer gate. The crystal structures of MelBSt suggest that the cation exits via the internal cavity that contains the sugar-binding site and that a bound sugar should restrain the dynamics of the bound cation; therefore, release of sugar is a prerequisite for releasing Na+. The structural basis for the ordered release is supported by kinetic studies4,11 and the behaviour of mutant G117R9, which catalyses melibiose exchange but no reaction involving net Na+ transport, that is, involving only steps [1] ⇔ [5]. [6] Outward Na+-released state. Deformation of the cation-binding site facilitates Na+ release as suggested by the Mol-B structure (Fig. 3b). [7] Occluded-empty state. Formation of the unloaded intermediate state occluded on both sides. [8] Inward-empty state. Once the cavity is open to the cytoplasm, the next transport cycle begins. Melibiose efflux involves the whole cycle with one sugar and one charge moving across the membrane. In contrast, melibiose exchange involves only states [1–5].
Interestingly, it appears that the kinetic model is similar for different MFS permeases regardless of the coupling cation35,48,49. In the solely H+-coupled LacY50,51,52,53, it is well established that at physiological pH, LacY is protonated52. Sugar binds only to protonated LacY50,53 and dissociates before the release of H+ during symport50. The inward-facing crystal structures of LacY16,17 indicate that the sugar-binding site is coupled to the H+-binding site through an electrostatic network; as a result, the affinity for the sugar binding is optimized in a protonated state. A similar coupling phenomenon is observed in MelB. The comparison of the structures of Mol-A and Mol-B reveals that a protonated cation-binding site is coupled with a properly formed sugar-binding pocket, and the deformation of the cation-binding site induces deformation of the sugar-binding site. In LacY, the H+ may involve in a single side chain (for example, Glu325)16,17,35,54; in MelB, the H+ may be bound in different forms, a protonated side chain (for example, Asp59) or as H3O+ that is shared by Asp55, Asp59 and Asp124 in the metal-binding cavity. Possibly, these two protonation forms represent different states of bound H+ in MelB.
What is the effect of Na+ or Li+ on MelB? In general, binding of Na+ or Li+ requires four to six coordination numbers from two to three helices, and the small ionic radius of Na+ and Li+ require the coordination ligands in closer distance. Thus, metal binding should induce a relatively large change in movement of helices. On the basis of the structures of MelBSt, it is anticipated that Na+ binding recruits Asp124 to complete Na+ binding, resulting in movement of helix IV with Tyr120 and Trp128 for aromatic stacking with the bound sugar. It is well known that sugar affinity is largely increased by aromatic stacking in LacY40. According to this notion, Na+ and Li+ are more effective ‘activators’ than H+. This structural interpretation is consistent with the previous conclusion derived from functional studies of MelBEc30,32,38. Therefore, it is likely to be that an essential aspect of coupling is that binding of the cation activates the substrate-binding site, a property that is conserved in both MelB and LacY.
What prevents cation leakage in MelB? The opening and closing of the cytoplasmic side to form the substrate exit pathway involves dynamics of cytoplasmic helix V in MelB (Fig. 6a,b), which is initiated only by sugar binding. In the absence of sugar, ‘the leader’, MelB/Na+ complexes should preferentially populate outward conformations. This structural arrangement can prevent bound Na+ from futile cycling and also offers an explanation for how the extramembrane ionic locks, as ‘the follower’, respond to the primary effect of sugar binding in the middle of the molecule.
Methods
Large-scale protein production
Full-length MelBSt encoded by plasmid pK95 ΔAH/MelBSt/CHis10 (refs 4, 55) with Leu5→Met and a His10 tag at the C terminus (namely the WT) was expressed in E. coli DW2 strain (melA+, ΔmelB and ΔlacZY)55. In a 10-l fermenter, cells were grown in Luria–Bertani (LB) broth supplemented with 50 mM KPi (pH 7.0), 45 mM (NH4)H2PO4, 0.5% glycerol and 100 mg l−1 ampicillin at 30 °C until they approached stationary phase at A600 of ~3.3. Cells with constitutively overexpressed MelBSt were then harvested, resuspended in 20 mM Tris–HCl (pH 7.5), 200 mM NaCl and 10% glycerol, broken with an EmulsiFlex instrument and centrifuged at 20,000 g for 15 min to remove unbroken cells56. Membranes were harvested from the supernatant by ultracentrifugation at 144,651 g for 3 h in a Beckman rotor, type 45 Ti. The pellets were resuspended in the same buffer, frozen in liquid N2 and stored at −80 °C. The protein concentration of the membrane samples was assayed using Micro BCA Protein Assay Kit (Pierce).
Protein purification
Membranes (14 mg ml−1) were extracted with 1.5% n-undecyl-β-D-maltoside (UDM) followed by ultracentrifugation at 265,000 g for 30 min. Purification was performed by cobalt-affinity chromatography (TALON resins). The column was pre-equilibrated with 50 mM NaPi (pH 7.6), 200 mM NaCl, 10% glycerol, 0.035% UDM, 5 mM imidazole. After loading and washing the column with 35 mM imidazole buffer, MelBSt was eluted with buffer containing 200 mM imidazole, concentrated with VIVASPIN 20 (50 kDa cutoff value), dialysed twice against 1 litre of 20 mM Tris–HCl (pH 7.5), 100 mM NaCl, 10% glycerol, 0.035% UDM, further concentrated to >20 mg ml−1, and finally ultracentrifuged at >360,000 g for 45 min. The protein was flash-frozen in liquid nitrogen and stored at −80 °C. From a 10-l culture, highly purified MelBSt at 30–40 mg was obtained routinely.
Isothermal titration calorimetry
ITC measurements were performed in a nano isothermal titration calorimeter (TA Instruments). The purified MelBSt in the dialysis buffer was injected into the ITC sample cell and 6 mM melibiose dissolved in the same dialysis buffer was titrated incrementally into the protein sample, and the heat rate was recorded at 25 °C. Melibiose binding was also measured in the presence of 100 mM LiCl. The cumulative heat change (ΔQ) was plotted against the molar ratio of melibiose to MelBSt and fitted with the one-site independent binding model (NanoAnalyse software).
Protein expression for functional studies
E. coli DW2 cells containing a given plasmid were grown in LB broth with 100 mg l−1 of ampicillin at 37 °C. The overnight cultures were diluted by 5% to LB broth supplemented with 0.5% glycerol and 100 mg l−1 of ampicillin, and the constitutive overexpression was obtained by shaking at 30 °C for another 5 h.
Preparation of right-side-out membrane vesicles
RSO membrane vesicles were prepared from E. coli DW2 cells by osmotic lysis4,57,58. Cells were incubated with 50 μg ml−1 lysozyme in the presence of 10 mM EDTA (pH 7.5), 5 mM dithiothreitol (DTT) and 30% (w/v) sucrose for 45 min; the yielded spheroplasts were harvested by centrifugation at 14,000 g for 40 min at 4 °C and suspended with 20-ml cocktail containing 100 mM KPi (pH 7.5), 20 mM MgSO4, 30% sucrose, 5 mM DTT, 1 mg ml−1 deoxyribonuclease and 1 mg ml−1 ribonuclease. The resuspended spheroplasts were lysed by rapid dilution into 1-litre 50 mM KPi (pH 7.5) buffer with 5 mM DTT, and the solution was incubated at 37 °C for 10 min. After further incubation with 10 mM EDTA (pH 7.0) for 10 min and 15 mM MgSO4 for another 10 min, the total membrane vesicles were harvested by centrifugation at 14,000 g for 25 min at 4 °C. The RSO vesicles were collected from the supernatant after centrifugation at 800g for 12 min at 4 °C, then extensively washed and resuspended with 100 mM KPi (pH 7.5) at a protein concentration of ~20–28 mg ml−1, frozen in liquid N2 and stored at −80 °C.
Melibiose transport assay with intact cells
Melibiose active transport with intact cells were carried out with [1-3H]melibiose4, which was custom synthesized by PerkinElmer (Boston, MA). E. coli DW2 cells expressing MelBSt were washed with 50 ml of 100 mM KPi (pH 7.5), so-called Na+-free buffer. The cell pellets were resuspended with 100 mM KPi (pH 7.5) and 10 mM MgSO4, adjusted to an A420=10 (~0.7 mg protein ml−1) and aliquoted with 50 μl. Transport was initiated by adding 2 μl of [3H]melibiose with a specific activity of 10 mCi mmol−1 at a final concentration of 0.4 mM in the absence or presence of either 20 mM NaCl or LiCl, and stopped at a given incubation time. Intracellular melibiose was assayed by a fast filtration.
Melibiose efflux and exchange assays
RSO membrane vesicles containing the WT or a mutant MelBSt in 100 mM KPi (pH 7.5) and 10 mM MgSO4 were concentrated to ~28 mg ml−1 and pre-equilibrated overnight on ice with 20 mM [3H]melibiose (10 mCi mmol−1), 0.75 μM monensin and 10 μM carbonylcyanide m-chlorophenylhydrazone4,9,11, without or with 20 mM NaCl or LiCl. Aliquots (2 μl) were diluted 200-fold into a given buffer in the absence (efflux) or presence (exchange) of 20 mM unlabelled melibiose50, and reactions were terminated by dilution and rapid filtration at a given time.
Western blotting
Twenty-five micrograms of RSO membranes were loaded onto each well of SDS–16% PAGE. After transfer onto the polyvinylidene difluoride membrane, MelBSt proteins were detected with anti-His-tag antibody and imaged by the ImageQuant LAS 4000 Biomolecular Imager (GE Health Care Life Science)47.
Trp→D2G FRET
D2G (refs 4, 9, 43, 47, 59) was used to test affinity for the cosubstrate via FRET from Trp residues to the dansyl moiety of the D2G bound with MelBSt. Steady-state fluorescence measurements were performed in a 3-mm quartz cuvette (Hitachi F-7000 Fluorescence Spectrophotomer or AMINCO-Bowman Series 2 Spectrometer).
The purified sugar-free MelBSt (1 μM) in the dialysis buffer or the buffer with 100 mM LiCl instead of NaCl was excited at wavelength of 290 nm and the emission fluorescence was recorded between 415 and 575 nm. With 10 μM D2G (the KD value for the WT4), displacement of D2G bound by MelBSt was carried out by adding 50 mM melibiose, methyl-β-D-thiogalactoside, glucose or sucrose, respectively. The difference spectra before and after the addition of a given sugar for displacement (ΔI) is calculated and plotted.
The RSO membrane vesicles prepared from DW2 cells expressing MelBSt (1 mg ml−1)4,9 in 100 mM KPi (pH 7.5) were excited at wavelength of 290 nm; the emission fluorescence at wavelength of 490 nm was recorded for 1 min after each of the following addition unless specified: (1) 10 μM D2G; (2) 20 mM NaCl or LiCl; (3) 50 mM NaCl or LiCl; (4) 120 mM melibiose or same volume of water.
Half-maximal displacement of bound D2G (IC50)
Melibiose was added stepwise to the samples containing the RSO vesicles with D2G (10 μM) and 20 mM NaCl until no change in fluorescence emission occurred. The decrease in fluorescent intensity after each addition was corrected by the water dilution and was plotted as a function of melibiose concentration. The IC50 value was determined by fitting a hyperbolic function to the data (OriginPro).
Na+-stimulation constant (K 0.5) for D2G FRET
Applying the same experimental setup, NaCl was consecutively added to the sample containing the RSO vesicles and D2G (10 μM)9. An identical volume of water was used for the control. Fluorescent increase (ΔI, the difference before (I0) and after the addition of NaCl) was expressed as the percentage of the I0, corrected by a dilution effect, and then plotted as a function of Na+ concentration. The K0.5 for Na+ simulation was determined by fitting a hyperbolic function to the data (OriginPro).
Mutagenesis
All MelBSt mutants were constructed with the QuickChange Site-Directed Mutagenesis Kits (Stratagene) using the plasmid pK95 ΔAH/MelBSt/CHis10 as the template4.
Gel filtration chromatography
Gel filtration was performed in a Superdex 200 10/300 GL column on a Fast Protein Liquid Chromatography instrument. For the stability studies, the protein sample was placed at 23 °C for 5 days and analysed by the gel filtration chromatography and the D2G FRET assay.
Precrystallization phospholipid treatment
Before setting up crystallization trials, the protein samples were thawed out from −80 °C, ultracentrifuged at 384,492 g for 45 min at 4 °C (Beckman Coulter Optima MAX, TLA-100 rotor), diluted to a final protein concentration of 7 mg ml−1 and incubated with phospholipids at a concentration of 3.3 mM (E. coli Extract Polar, Avanti, 100600) from a 15-mM stock dissolved with the dialysis buffer.
Crystallization, data collection and processing
Crystallization of MelBSt was carried out by the hanging-drop vapour-diffusion method at 23 °C by mixing 2 μl of phospholipid-treated protein samples60 containing 5 mM αNPG (4-nitrophenyl-α-D-galactopyranoside) with 2 μl reservoir containing 100 mM 2-(N-morpholino) ethanesulfonic acid (pH 6.5), 100 mM NaCl, 50 mM CaCl2, 35–37% PEG 400 and 0.08% octyl-β-D-galactopyranoside. Crystals were frozen with liquid nitrogen and tested for X-ray diffraction at the Lawrence Berkeley National Laboratory, ALS BL 8.2.1, 8.2.2, 5.0.1, 5.0.2 or at the Stanford Synchrotron Radiation Light Source BL 7-1 via remote data collection. The complete diffraction data set was collected at 100 K from a single cryocooled crystal at wavelength of 1.004 Å with an ADSC QUANTUM 315 detector at ALS BL 5.0.2; image data were processed with HKL 2000 (ref. 61) to a resolution of 3.35 Å with 98% completeness (Table 1).
Structure solution and refinement
Data were processed in space groups P3221, P3121, P32 and P31. The data set was analysed by Structure Factor Check on CCP4 (ref. 62) suite and Phenix Xtriage63,64, which indicated a strong off-origin peak at 0, 0, 0.5 along c* axis and also revealed merohedral twinning with twin fraction of 0.41 for the data processed in space groups P32 and P31. Search probes were prepared using the server I-TASSER65, using the structures of MFS permeases, the inward LacY (PDB, 2V8N) and GlpT (1PW4), the occluded EmrD (2GFP), as well as the outward FucP (3O7Q). Using Phaser 2.52 programme66 in Phenix suite, a single copy of each threading model with deletion of the longer loop6–7 and loop10–11, and the C-terminal tail was used as the search probe to test the data processed in all space groups. Applying the native Patterson map and omit map to assist the molecular replacement processes, a maximal solution from the FucP-based MelBSt model was identified, and the crystal symmetry was determined to be P32 containing two additional pseudocrystallographic symmetries with 74% solvent content.
The asymmetric unit contains four closely aligned molecules with two types of pseudo two-fold symmetry (twinning) along the b axis, with Mol-A and Mol-B related by 172°, and Mol-C and Mol-D related by 160° (Supplementary Fig. S3). Mol-AB and Mol-CD are related by 0.5 pseudotranslation. Twin refinement was carried out by REFMAC 5.7 programme using the Jelly-Body-Restrained refinement option in CCP4 suite with the twinning operator k, h, -l. Initial refinement yielded R/Rfree of 0.37/0.42. Model verification was done by systematic calculation of omit maps at various regions. With iterative rounds of manual model building followed by simulated annealing and density modification, an interpretable density map to 3.35 Å resolution was obtained allowing assignment of most side chains for Mol-A and Mol-B (Supplementary Figs S3c,d and S5a,b). Final refinement was carried out using the Tangent Least-Square Jelly-Body-Restrained twin refinement, where the R/Rfree values dropped to 0.30/0.35 (Table 1). As pseudosymmetries are present, higher apparent R/Rfree values are expected67. Out of 476 residues, 1–432 in Mol-A, regions 1–210 and 235–450 in Mol-B, 1–431 in Mol-C, as well as 1–100, 109–210, 235–398 and 408–431 in Mol-D, were well resolved. Residues (82.2%) are in most favoured, 17.4% in generously allowed and 0.4% in disallowed regions. Visualization of omit maps and manual model building were performed using Coot 0.7 (ref. 68). Surface electropotential maps were calculated using APBS software69. All crystallographic figures were generated with Pymol 1.5 (ref. 70).
Author contributions
A.S.E., M.S.Y. and L.G. performed protein production, crystallization, X-ray diffraction, data collection and processing. A.S.E. and L.G. performed collection and processing of data for the PDB accessing 4M64 and structure interpretation. L.G., A.S.E. and A.A. designed and A.A. performed all transport assays and FRET measurements. L.G. interpreted functional data and directed the research. H.R.K. and G.L. provided advice and research reagents. All authors contributed to manuscript preparation. L.G. and A.S.E. wrote the manuscript with help from H.R.K.
Additional information
Accession codes: The protein coordinates and structure factor have been deposited in the Protein Data Bank with the accession number 4M64.
How to cite this article: Ethayathulla, A. S. et al. Structure-based mechanism for Na+/melibiose symport by MelB. Nat. Commun. 5:3009 doi: 10.1038/ncomms4009 (2014).
Supplementary Material
Supplementary Figures S1-S8
Acknowledgments
We thank Luis Reuss and R. Bryan Sutton for critical reading of the manuscript, and Shailika Nurva, Hariharan Parameswaran, Elena Tikhonova, Gill Verner and Shushi Nagamori for assistance. We thank Kelsey Markham for constructing most of the mutants. We also thank the staffs at the Advanced Light Source, Lawrence Berkeley National Laboratory and the Stanford Synchrotron Radiation Light Source for assistance with X-ray data collection. The diffraction data set was collected at ALS BL 5.0.2. This work was supported by the National Science Foundation (grant MCB-1158085 to L.G. and grant MCB-0450970 to H.R.K.) and the Norman Hackerman Advanced Research Program (grant 010674-0034-2009 to L.G.), as well as by the National Institutes of Health (grants R21HL087895 and R01GM095538 to L.G. and grants R01DK051131 and R01DK069463 to H.R.K.).
References
- Poolman B. et al. Cation and sugar selectivity determinants in a novel family of transport proteins. Mol. Microbiol. 19, 911–922 (1996). [DOI] [PubMed] [Google Scholar]
- Saier M. H. Jr. Families of transmembrane sugar transport proteins. Mol. Microbiol. 35, 699–710 (2000). [DOI] [PubMed] [Google Scholar]
- Mizushima K. et al. Cloning and sequencing of the melB gene encoding the melibiose permease of Salmonella typhimurium LT2. Mol. Gen. Genet. 234, 74–80 (1992). [DOI] [PubMed] [Google Scholar]
- Guan L., Nurva S. & Ankeshwarapu S. P. Mechanism of melibiose/cation symport of the melibiose permease of Salmonella typhimurium. J. Biol. Chem. 286, 6367–6374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling J. H. et al. A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc. Natl Acad. Sci. USA 108, 11756–11765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier M. H. Jr. et al. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1, 257–279 (1999). [PubMed] [Google Scholar]
- Wilson T. H. & Ding P. Z. Sodium-substrate cotransport in bacteria. Biochim. Biophys. Acta 1505, 121–130 (2001). [DOI] [PubMed] [Google Scholar]
- Wilson D. M. & Wilson T. H. Cation specificity for sugar substrates of the melibiose carrier in Escherichia coli. Biochim. Biophys. Acta 904, 191–200 (1987). [DOI] [PubMed] [Google Scholar]
- Guan L., Jakkula S. V., Hodkoff A. A. & Su Y. Role of Gly117 in the cation/melibiose symport of MelB of Salmonella typhimurium. Biochemistry 51, 2950–2957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niiya S., Moriyama Y., Futai M. & Tsuchiya T. Cation coupling to melibiose transport in Salmonella typhimurium. J. Bacteriol. 144, 192–199 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassilana M., Pourcher T. & Leblanc G. Facilitated diffusion properties of melibiose permease in Escherichia coli membrane vesicles. Release of co-substrates is rate limiting for permease cycling. J. Biol. Chem. 262, 16865–16870 (1987). [PubMed] [Google Scholar]
- Bassilana M., Pourcher T. & Leblanc G. Melibiose permease of Escherichia coli. Characteristics of co-substrates release during facilitated diffusion reactions. J. Biol. Chem. 263, 9663–9667 (1988). [PubMed] [Google Scholar]
- Damiano-Forano E., Bassilana M. & Leblanc G. Sugar binding properties of the melibiose permease in Escherichia coli membrane vesicles. Effects of Na+ and H+ concentrations. J. Biol. Chem. 261, 6893–6899 (1986). [PubMed] [Google Scholar]
- Mus-Veteau I., Pourcher T. & Leblanc G. Melibiose permease of Escherichia coli: substrate-induced conformational changes monitored by tryptophan fluorescence spectroscopy. Biochemistry 34, 6775–6783 (1995). [DOI] [PubMed] [Google Scholar]
- Yousef M. S. & Guan L. A 3D structure model of the melibiose permease of Escherichia coli represents a distinctive fold for Na+ symporters. Proc. Natl Acad. Sci. USA 106, 15291–15296 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramson J. et al. Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615 (2003). [DOI] [PubMed] [Google Scholar]
- Guan L., Mirza O., Verner G., Iwata S. & Kaback H. R. Structural determination of wild-type lactose permease. Proc. Natl Acad. Sci. USA 104, 15294–15298 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Lemieux M. J., Song J., Auer M. & Wang D. N. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–620 (2003). [DOI] [PubMed] [Google Scholar]
- Yin Y., He X., Szewczyk P., Nguyen T. & Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 312, 741–744 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang S. et al. Structure of a fucose transporter in an outward-open conformation. Nature 467, 734–738 (2010). [DOI] [PubMed] [Google Scholar]
- Sun L. et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature 490, 361–366 (2012). [DOI] [PubMed] [Google Scholar]
- Pedersen B. P. et al. Crystal structure of a eukaryotic phosphate transporter. Nature 496, 533–536 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newstead S. et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 30, 417–426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purhonen P., Lundback A. K., Lemonnier R., Leblanc G. & Hebert H. Three-dimensional structure of the sugar symporter melibiose permease from cryo-electron microscopy. J. Struct. Biol. 152, 76–83 (2005). [DOI] [PubMed] [Google Scholar]
- Madej M. G., Dang S., Yan N. & Kaback H. R. Evolutionary mix-and-match with MFS transporters. Proc. Natl Acad. Sci. USA 110, 5870–5874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A., Singh S. K., Kawate T., Jin Y. & Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 (2005). [DOI] [PubMed] [Google Scholar]
- Kovalevsky A. Y. et al. Identification of the elusive hydronium ion exchanging roles with a proton in an enzyme at lower pH values. Angew. Chem. Int. Ed. Engl. 50, 7520–7523 (2011). [DOI] [PubMed] [Google Scholar]
- Zani M. L., Pourcher T. & Leblanc G. Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli. II. Effect on cationic selectivity and coupling properties. J. Biol. Chem. 268, 3216–3221 (1993). [PubMed] [Google Scholar]
- Pourcher T., Zani M. L. & Leblanc G. Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli. I. Effect on Na(+)-dependent transport and binding properties. J. Biol. Chem. 268, 3209–3215 (1993). [PubMed] [Google Scholar]
- Granell M., Leon X., Leblanc G., Padros E. & Lorenz-Fonfria V. A. Structural insights into the activation mechanism of melibiose permease by sodium binding. Proc. Natl Acad. Sci. USA 107, 22078–22083 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas D. C., Rogers D. M. & Jensen J. H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73, 765–783 (2008). [DOI] [PubMed] [Google Scholar]
- Pourcher T., Bassilana M., Sarkar H. K., Kaback H. R. & Leblanc G. The melibiose/Na+ symporter of Escherichia coli: kinetic and molecular properties. Phil. Trans. R. Soc. Lond. Ser. B Biol. Sci. 326, 411–423 (1990). [DOI] [PubMed] [Google Scholar]
- Faham S. et al. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V. & Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature 458, 47–52 (2009). [DOI] [PubMed] [Google Scholar]
- Guan L. & Kaback H. R. Lessons from lactose permease. Annu. Rev. Biophys. Biomol. Struct. 35, 67–91 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L., Sahin-Toth M. & Kaback H. R. Changing the lactose permease of Escherichia coli into a galactose-specific symporter. Proc. Natl Acad. Sci. USA 99, 6613–6618 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L., Sahin-Toth M., Kalai T., Hideg K. & Kaback H. R. Probing the mechanism of a membrane transport protein with affinity inactivators. J. Biol. Chem. 278, 10641–10648 (2003). [DOI] [PubMed] [Google Scholar]
- Cordat E., Leblanc G. & Mus-Veteau I. Evidence for a role of helix IV in connecting cation- and sugar-binding sites of Escherichia coli melibiose permease. Biochemistry 39, 4493–4499 (2000). [DOI] [PubMed] [Google Scholar]
- Zani M. L., Pourcher T. & Leblanc G. Mutation of polar and charged residues in the hydrophobic NH2-terminal domains of the melibiose permease of Escherichia coli. J. Biol. Chem. 269, 24883–24889 (1994). [PubMed] [Google Scholar]
- Guan L., Hu Y. & Kaback H. R. Aromatic stacking in the sugar binding site of the lactose permease. Biochemistry 42, 1377–1382 (2003). [DOI] [PubMed] [Google Scholar]
- Vazquez-Ibar J. L., Guan L., Svrakic M. & Kaback H. R. Exploiting luminescence spectroscopy to elucidate the interaction between sugar and a tryptophan residue in the lactose permease of Escherichia coli. Proc. Natl Acad. Sci. USA 100, 12706–12711 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lipp K. et al. The inner interhelix loop 4-5 of the melibiose permease from Escherichia coli takes part in conformational changes after sugar binding. J. Biol. Chem. 281, 25882–25892 (2006). [DOI] [PubMed] [Google Scholar]
- Maehrel C., Cordat E., Mus-Veteau I. & Leblanc G. Structural studies of the melibiose permease of Escherichia coli by fluorescence resonance energy transfer. I. Evidence for ion-induced conformational change. J. Biol. Chem. 273, 33192–33197 (1998). [DOI] [PubMed] [Google Scholar]
- Quistgaard E. M., Low C., Moberg P., Tresaugues L. & Nordlund P. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat. Struct. Mol. Biol. 20, 766–768 (2013). [DOI] [PubMed] [Google Scholar]
- Yan H. et al. Structure and mechanism of a nitrate transporter. Cell Rep. 3, 716–723 (2013). [DOI] [PubMed] [Google Scholar]
- Zheng H., Wisedchaisri G. & Gonen T. Crystal structure of a nitrate/nitrite exchanger. Nature 497, 647–651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakkula S. V. & Guan L. Reduced Na+ affinity increases turnover of Salmonella enterica serovar Typhimurium MelB. J. Bacteriol. 194, 5538–5544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doki S. et al. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl Acad. Sci. USA 110, 11343–11348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej M. G., Soro S. N. & Kaback H. R. Apo-intermediate in the transport cycle of lactose permease (LacY). Proc. Natl Acad. Sci. USA 109, E2970–E2978 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski G. J. & Kaback H. R. Mechanism of lactose translocation in membrane vesicles from Escherichia coli. 1. Effect of pH on efflux, exchange, and counterflow. Biochemistry 18, 3691–3697 (1979). [DOI] [PubMed] [Google Scholar]
- Smirnova I., Kasho V., Sugihara J., Choe J. Y. & Kaback H. R. Residues in the H+ translocation site define the pKa for sugar binding to LacY. Biochemistry 48, 8852–8860 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova I. N., Kasho V. & Kaback H. R. Protonation and sugar binding to LacY. Proc. Natl Acad. Sci. USA 105, 8896–8901 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova I., Kasho V., Sugihara J., Vazquez-Ibar J. L. & Kaback H. R. Role of protons in sugar binding to LacY. Proc. Natl Acad. Sci. USA 109, 16835–16840 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco N. et al. Characterization of site-directed mutants in the lac permease of Escherichia coli. 2. Glutamate-325 replacements. Biochemistry 28, 2533–2539 (1989). [DOI] [PubMed] [Google Scholar]
- Pourcher T., Leclercq S., Brandolin G. & Leblanc G. Melibiose permease of Escherichia coli: large scale purification and evidence that H+, Na+, and Li+ sugar symport is catalyzed by a single polypeptide. Biochemistry 34, 4412–4420 (1995). [DOI] [PubMed] [Google Scholar]
- Chae P. S. et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback H. R. inMethods in Enzymology Vol. XXII, eds Kaplan Nathan P., Jakoby William B., Colowick Nathan P. 99–120Elsevier (1971). [Google Scholar]
- Short S. A., Kaback H. R. & Kohn L. D. D-lactate dehydrogenase binding in Escherichia coli dld- membrane vesicles reconstituted for active transport. Proc. Natl Acad. Sci. USA 71, 1461–1465 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordat E., Mus-Veteau I. & Leblanc G. Structural studies of the melibiose permease of Escherichia coli by fluorescence resonance energy transfer. II. Identification of the tryptophan residues acting as energy donors. J Biol Chem 273, 33198–33202 (1998). [DOI] [PubMed] [Google Scholar]
- Guan L., Smirnova I. N., Verner G., Nagamori S. & Kaback H. R. Manipulating phospholipids for crystallization of a membrane transport protein. Proc. Natl Acad. Sci. USA 103, 1723–1726 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. & Minor W. inMethods in Enzymology, Part A, Macromolecular Crystallography, Vol. 276, 307–326Academic Press (1997). [DOI] [PubMed] [Google Scholar]
- Winn M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla J. E. & Yeates T. O. A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta. Crystallogr. D. Biol. Crystallogr. 59, 1124–1130 (2003). [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Sawaya M. R. & Eisenberg D. Structure of superoxide dismutase from Pyrobaculum aerophilum presents a challenging case in molecular replacement with multiple molecules, pseudo-symmetry and twinning. Acta. Crystallogr. D. Biol. Crystallogr. 59, 2191–2199 (2003). [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W. G. & Cowtan K. Features and development of Coot. Acta. Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A., Sept D., Joseph S., Holst M. J. & McCammon J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger L. The PyMOL Molecular Graphics System Version 1.5 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1-S8