

Abstract
The objective of this work was to investigate the role of acidic residues within the exposed middle segment of the central helix of cTnC in (1) cTnC-cTnI interactions, (2) Ca2+ binding and exchange with the regulatory N-domain of cTnC in increasingly complex biochemical systems, and (3) ability of the cTn complex to regulate actomyosin ATPase. In order to achieve this objective, we introduced the D87A/D88A and E94A/E95A/E96A mutations into the central helix of cTnC. The D87A/D88A and E94A/E95A/E96A mutations decreased affinity of cTnC for the regulatory region of cTnI. The Ca2+ sensitivity of the regulatory N-domain of isolated cTnC was decreased by the D87A/D88A, but not E94A/E95A/E96A mutation. However, both the D87A/D88A and E94A/E95A/E96A mutations desensitized the cTn complex and reconstituted thin filaments to Ca2+. Decreases in the Ca2+ sensitivity of the cTn complex and reconstituted thin filaments were, at least in part, due to faster rates of Ca2+ dissociation. In addition, the D87A/D88A and E94A/E95A/E96A mutations desensitized actomyosin ATPase to Ca2+, and decreased maximal actomyosin ATPase activity. Thus, our results indicate that conserved acidic residues within the exposed middle segment of the central helix of cTnC are important for the proper regulatory function of the cTn complex.
Keywords: Troponin C, Troponin I, Central helix, Fluorescence, Calcium binding
INTRODUCTION
Cardiac muscle utilizes troponin (cTn)1 complex to regulate contraction-relaxation cycles in response to changes in intracellular Ca2+. The hetero-trimeric cTn complex consists of cTnC (the Ca2+ binding subunit), cTnI (the inhibitory subunit), and cTnT (the tropomyosin (cTm)-binding subunit) (for review, see (1; 2)). At a resting level of intracellular Ca2+, the cTn complex keeps cTm in a position that prevents force-producing interactions between myosin heads and actin. Increase in intracellular Ca2+ results in a series of conformational rearrangements in the cTn-cTm complex, allowing myosin heads to strongly bind actin (for review, see (3–9)).
The Ca2+ sensor cTnC consists of the N-and C-terminal globular domains connected by a flexible central α-helix. Each domain contains a pair of EF-hand (helix-loop-helix) Ca2+ binding motifs, numbered I–IV. The α-helices flanking the Ca2+ binding loops are denoted A–H. An additional 14-residue α-helix, denoted the N-helix, is located at the amino terminus. The nine-turn central helix connecting the two globular domains includes the D-helix of the N-domain, D/E helical linker and the E-helix of the C-domain (10; 11). The helical segments at the ends of the central helix are at least partially buried within the globular domains, while the middle three-turn segment is exposed to solvent (10; 11). The critical role of the central helix appears to be in properly orienting and positioning the two globular domains of cTnC for interactions with its targets (12).
Ca2+ binding and exchange with the N-domain of cTnC play a direct role in regulating cardiac muscle contractility, while the C-domain is believed to play a structural role of anchoring cTnC into the cTn complex (13; 14). The response of the regulatory N-domain of cTnC to Ca2+ is modulated by cTnI and other regulatory muscle proteins (15–17). CTnI contains an N-terminal extension region, an IT-arm region, the inhibitory region (which binds actin), the switch region and the C-terminal mobile domain (which contains second actin-binding site) (for review, see (18; 19)). The inhibitory region was determined to be important for both activation and inhibition of actomyosin ATPase (20). In the absence of Ca2+, the inhibitory region binds to actin, preventing myosin heads from strongly binding actin. Binding of Ca2+ to the N-domain of cTnC allows the switch region of cTnI to bind to the hydrophobic patch on the N-domain of cTnC. This interaction leads to the removal of the inhibitory and C-domain actin-binding regions of cTnI from actin, resulting in shifting of the cTm position on the surface of actin and ultimately force generation (for review, see (6; 9)).
An overriding goal of our research is to elucidate the role of cTnC in the regulation of cardiac contractility. In this study, we have focused on the importance of acidic residues located within the exposed middle segment of the central helix of cTnC. Functionally important residues tend to be conserved within a protein family (21; 22). The exposed middle segment of the central helix of cTnC contains negatively charged residues Asp87, Asp88, Glu94, Glu95 and Glu96. Sequence analysis indicates that these negatively charged residues are highly conserved among TnCs from different species and muscle types (23). In the crystal structure of the Ca2+ saturated skeletal Tn (sTn) complex, acidic residues within the exposed segment of the central helix of sTnC are involved in electrostatic interactions with basic residues within the inhibitory region of sTnI (24). On the other hand, the inhibitory region of cTnI was not visible in the crystal structure of the cTn complex (25). Thus, the importance of electrostatic interactions between the central helix of cTnC and the inhibitory region of cTnI is under debate.
The objective of our current study was to investigate the role of acidic residues within the exposed middle segment of the central helix in the interactions of cTnC with the regulatory region of cTnI (which includes both the inhibitory and switch regions, cTnI128-180 (26; 27)). Furthermore, we wanted to determine whether these acidic residues influence Ca2+ binding and functional properties of cTnC. In order to achieve our objective, the Asp87/Asp88 or Glu94/Glu95/Glu96 residues within the central helix of cTnC were mutated to neutral Ala residues. Ala was selected as a replacement residue in order to eliminate the side-chain interactions while maintaining the backbone conformation. Based on the assumption that in solution the cTn complex adopts a structure similar to the crystal structure of the sTn complex (28; 29), we expected our results to demonstrate the functional importance of acidic residues within the exposed middle segment of the central helix of cTnC.
EXPERIMENTAL PROCEDURES
Materials
Phenyl-Sepharose CL-4B, CaCl2 and EGTA were purchased from Sigma-Aldrich (St. Louis, MO). IAANS and phalloidin were purchased from Invitrogen (Carlsbad, CA). Affi-Gel 15 affinity media were purchased from Bio-Rad (Hercules, CA). Malachite Green Oxalate and poly(vinyl alcohol) were purchased from Fisher Scientific (Pittsburgh, PA)
Protein Mutagenesis and Purification
The pET3a plasmid encoding human cTnC was a generous gift from Dr. Lawrence B. Smillie (University of Alberta, Edmonton, AB). The cTnC mutants were generated as previously described, and confirmed by DNA sequencing (15; 30). Expression and purification of cTnC and its mutants was carried out as previously described (15; 30; 31). The pET3d plasmids encoding human cTnI and human cTnT were generated by GenScript USA (Piscataway, NJ). The cTnI and cTnT subunits were bacterially expressed, purified and quantified as described (30). Rabbit fast skeletal actin and myosin S1, and bovine cTm were a general gift from Dr. Darl R. Swartz (Delaware Valley College (Doylestown, PA)).
Fluorescent Labeling of cTnC and its Mutants
cTnC proteins (with C35S/T53C/C84S substitution) were labeled with the environmentally sensitive thiol-reactive fluorescent probe IAANS on Cys53 as previously described (15; 30).
Determination of cTnI128-180 Peptide Affinities
Fluorescence of IAANS attached to Cys53 of cTnC was monitored at 450 nm with excitation at 330 nm. Microliter amounts of cTnI128-180 were added to 2 mL of each cTnC protein labeled with IAANS on Cys53 (0.15 μM) in buffer (10 mM MOPS, 150 mM KCl, 3 mM MgCl2, 1 mM CaCl2, 0.02 % Tween-20 and 1 mM DTT, at pH 7.0) at 15 °C with constant stirring. Each apparent peptide dissociation constant represents a means of at least three titrations ± SE fit to the root of quadratic equation for binary complex formation as previously described (30; 31).
Reconstitution of the cTn Complexes
The cTn complexes were prepared and reconstituted as previously described (15; 30).
Reconstitution of Thin Filaments
After exhaustive dialysis against reconstitution buffer (10 mM MOPS, 150 mM KCl, 3 mM MgCl2, and 1 mM DTT, at pH 7.0), actin was mixed with an equal molar ratio of phalloidin to stabilize actin filaments. Thin filaments were reconstituted as previously described (15; 30). Briefly, actin-phalloidin (4 μM) and cTm (0.57μM) were mixed in the reconstitution buffer and kept on ice for ~ 15 minutes. The cTn complexes (0.5 μM) were subsequently added, and reconstituted thin filaments were kept on ice for at least 15 minutes prior to the experiments. To examine the effect of the central helix mutations on the Ca2+ binding properties of reconstituted thin filaments in the presence of myosin S1, myosin S1 (1.14 μM) was subsequently added, and thin filaments were kept on ice for at least 3 minutes prior to the experiments. Therefore, the stoichiometry of reconstituted thin filaments was 7:1:0.88:2 (actin:cTm:cTn:myosin S1).
Determination of Ca2+ Binding Sensitivities
All steady-state fluorescence measurements were performed using a Perkin-Elmer LS55 fluorescence spectrometer at 15 °C. Trp fluorescence was excited at 285 nm and monitored at 345 nm as μL amounts of CaCl2 were added to 2 mL of each isolated cTnC protein (with F27W substitution (32)) in the titration buffer (200 mM MOPS (to prevent pH changes upon addition of Ca2+), 150 mM KCl, 2 mM EGTA, 3 mM MgCl2, and 1 mM DTT, at pH 7.0) with constant stirring. Fluorescence of IAANS attached to Cys53 of cTnC was excited at 330 nm and monitored at 450 nm or 490 nm as μL amounts of CaCl2 were added to 2 mL of each cTn complex in the titration buffer (with 0.02 % Tween-20), or to reconstituted thin filaments with constant stirring. Thin filaments were reconstituted as described above, and diluted in half with an appropriate solution to achieve the same titration buffer composition. The [Ca2+]free was calculated using the computer program EGCA02 developed by Robertson and Potter (33). The Ca2+ sensitivities of conformational changes were reported as a dissociation constant Kd, representing a mean of at least three titrations ± SE. The data were fit with a logistic sigmoid function (mathematically equivalent to the Hill equation), as previously described (34).
Determination of Ca2+ Dissociation Kinetics
All kinetic measurements were performed utilizing an Applied Photophysics Ltd. (Leatherhead, UK) model SX.18 MV stopped-flow instrument with a dead time of ~1.4 ms. The rates of conformational changes induced by EGTA (10 mM) removal of Ca2+ (500 μM) from the N-domain of isolated cTnC proteins (with F27W substitution) in the stopped-flow buffer (10 mM MOPS, 150 mM KCl, 3mM MgCl2, and 1 mM DTT, at pH 7.0) were measured following Trp fluorescence at 15°C and 5°C. The Trp fluorescence was excited at 285 nm, with emission monitored through a WG320 filter from Newport Corporation (Irvine, CA). The rates of conformational changes induced by EGTA (10 mM) removal of Ca2+ (200 μM) from the cTn complexes in the stopped-flow buffer (with 0.02 % Tween-20), or from reconstituted thin filaments were measured at 15°C following fluorescence of IAANS attached to Cys53 of cTnC. Thin filaments were reconstituted as described above, and diluted in half with the stopped-flow buffer prior to the experiments. The IAANS fluorescence was excited at 330 nm, with emission monitored through a 510 nm BrightLine Basic™ filter from Semrock (Rochester, NY) or 515 nm cut-on filter from Newport Corporation (Irvine, CA). The data were corrected for scattering artifacts as described previously (15; 30). The data were fit using a program (by P. J. King, Applied Photophysics Ltd) that utilizes the nonlinear Levenberg-Marquardt algorithm. Each koff represents an average of at least three separate experiments ± SE, each averaging at least five traces fit with a single exponential equation.
Actomyosin S1 ATPase Assay
Actomyosin ATPase assay was carried out as previously described (35). Briefly, reconstituted thin filaments (5μM actin, 1.0 μM cTm, 1.5 μM cTn, and 0.3 μM myosin S1) were formed at 25 °C in a buffer consisting of 50 mM MOPS, and 5 mM MgCl2, pH 7.0. EGTA (to a final concentration of 0.5 mM) and various amounts of CaCl2 were added to the 100 μL reaction mixture aliquots to achieve the desired pCa values. The ATPase reaction was initiated by addition of ATP (to a final concentration of 1 mM), and 10 μL aliquots were removed into 90 μL of 0.2M ice-cold perchloric acid in order to terminate the reaction. For determination of the Ca2+ dependence of actomyosin ATPase, the ATPase rates were measured at a single time point at which the reaction was still linear with time. In order to measure minimal and maximal specific actomyosin ATPase activities, 10 μL aliquots were terminated at 3 minute intervals (up to 12 minutes time course) by 90 μL of 0.2 M ice-cold perchloric acid. Actomyosin ATPase activity was determined by the amount of phosphate released. The amount of phosphate released was quantified using malachite green method, as previously described (30; 35).
Statistical Analysis
Statistical significance was determined by an unpaired two-sample t-test using the statistical analysis software Minitab (State College, PA). The two means were considered to be significantly different when the p value was < 0.05. All data is shown as a mean value ± SE.
RESULTS
Location of the Central Helix Residues within the Structure of cTnC Reconstituted into the cTn Complex
Figure 1 shows location of the Asp87/Asp88 and Glu94/Glu95/Glu96 residues within the structure of cTnC reconstituted into the cTn complex (25). Figure 1 also shows that the central helix of cTnC was disordered, while the inhibitory region of cTnI was not visualized in the crystal structure of the cTn complex.
Figure 1. Location of the central helix residues within the structure of cTnC reconstituted into the cTn complex.
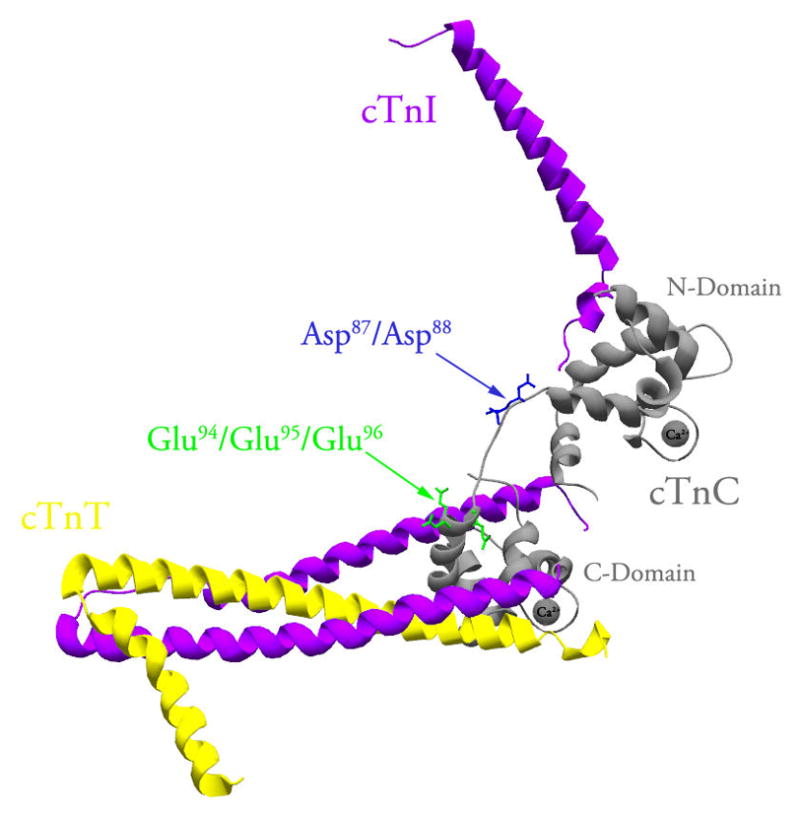
The figure shows a ribbon representation of the core domain of the cTn complex in the Ca2+ bound state (Protein Data Bank entry 1J1E (25)). CTnC, cTnI and cTnT are colored in grey, magenta and yellow, respectively. The Asp87/Asp88 residues (shown in blue) and Glu94/Glu95/Glu96 residues (shown in green) are located within the exposed middle segment of the central helix of cTnC. This figure was generated using Swiss-PdbViewer (56).
Effect of the Central Helix Mutations the on the Ca2+ Binding Properties of Isolated cTnC
Due to substitutions of several loop residues, the first EF-hand of cTnC is unable to bind Ca2+ under physiological conditions (36). The F27W substitution (32) was used to follow Ca2+ binding and exchange with the second EF-hand of isolated cTnC. Use of the F27W substitution was necessitated by the fact that in isolated cTnC, IAANS probe attached to Cys53 reflects conformational changes occurring within the structural C-domain sites (data not shown). Results of NMR spectroscopy suggested that the F27W substitution increases the Ca2+ sensitivity of the N-domain of cTnC (37). However, neither the D87A/D88A nor E94A/E95A/E96A mutation is located in close proximity to the F27W substitution. Thus, any possible effects of the F27W substitution are likely to be similar between cTnC and central helix mutants, allowing us to accurately quantify differences in the Ca2+ sensitivity of the N-domain.
The Ca2+-induced increases in Trp fluorescence, which occur when Ca2+ binds to the regulatory N-domain of cTnC or its mutants, are shown in Figure 2A. Results are summarized in Table I. The cTnC exhibited a half-maximal Ca2+-dependent increase in Trp fluorescence at 28 ± 2 μM. The D87A/D88A mutation led to ~1.6-fold decrease in the Ca2+ sensitivity of cTnC, while the E94A/E95A/E96A mutation did not significantly affect the Ca2+ sensitivity of cTnC (Figure 2A and Table I). The Hill coefficients (as indicated by the nH values) for the Ca2+ titrations were between 0.88 and 1.03, consistent with binding of single Ca2+ ion to the second EF-hand of cTnC (Table I).
Figure 2. Effect of the central helix mutations on the Ca2+ binding properties of isolated cTnC.

Panel A shows increases in Trp fluorescence, which occur as Ca2+ binds to the N-domains of cTnC (■), cTnCD87A/D88A (●), or cTnCE94A/E95A/E96A (○) at 15°C. The cTnC proteins contained F27W substitution. The Trp fluorescence was excited at 285 nm and monitored at 345 nm. Panels B and C show the time course of decreases in Trp fluorescence as Ca2+ was removed by excess EGTA from the N-domains of isolated cTnC, cTnCD87A/D88A, or cTnCE94A/E95A/E96A. Panel B: measured at 15°C. Panel C: measured at 5°C. The data traces have been normalized and staggered for clarity. Each trace is an average of at least five traces fit with a single exponential equation. The Trp fluorescence was excited at 285 nm and monitored through a WG320 filter.
Table I.
Effect of the Central Helix Mutations on the Ca2+ Binding Properties of cTnC in Increasingly Complex Biochemical Systems
| Biochemical System | Ca2+ Kd (μM) | nH | Ca2+ koff (s−1) |
|---|---|---|---|
| cTnC | 28 ± 2 | 0.97 ± 0.01 | 1349 ± 56 |
| cTnCD87A/D88A | 46 ± 1a | 0.88 ± 0.01a | 1360 ± 120 |
| cTnCE94A/E95A/E96A | 24 ± 1 | 1.03 ± 0.01a | 1310 ± 77 |
| cTn | 0.83 ± 0.07 | 0.77 ± 0.02 | 42.3 ± 0.2 |
| cTnD87A/D88A | 2.5 ± 0.2b | 0.89 ± 0.07 | 82 ± 2b |
| cTnE94A/E95A/E96A | 1.8 ± 0.1b | 0.88 ± 0.04 | 63.7 ± 0.8b |
| cTn+actin+cTm | 2.15 ± 0.09 | 1.62 ± 0.04 | 93 ± 1 |
| cTnD87A/D88A + actin+cTm | 10 ± 1c | 1.16 ± 0.04c | 168 ± 2c |
| cTnE94A/E95A/E96A + actin+cTm | 12 ± 1c | 1.47 ± 0.09 | 164 ± 2c |
| cTn+actin+cTm+S1 | 0.29 ± 0.02 | 1.3 ± 0.1 | 11.7 ± 0.2 |
| cTnD87A/D88A + actin+cTm+S1 | 1.73 ± 0.05d | 0.93 ± 0.02 | 21.2 ± 0.3d |
| cTnE94A/E95A/E96A + actin+cTm+S1 | 3.7 ± 0.3d | 1.06 ± 0.07 | 20.7 ± 0.3d |
Significantly different from their respective cTnC values (p<0.05)
Significantly different from their respective cTn values (p<0.05)
Significantly different from their respective cTn+actin+cTm values (p<0.05)
Significantly different from their respective cTn+actin+cTm+S1 values (p<0.05)
Stopped-flow measurements, utilizing Trp fluorescence, were conducted to examine the effect of the central helix mutations on the rate of Ca2+ dissociation from the N-domain of isolated cTnC. Results are summarized in Table I. At 15°C, excess EGTA removed Ca2+ from the regulatory N-domain of cTnC at 1349 ± 56 s−1. At 15°C, the D87A/D88A and E94A/E95A/E96A mutations did not significantly affect rates of Ca2+ dissociation from the N-domain of isolated cTnC (Figure 2B and Table I). Since these koff values are close to the upper limit of rates that can be measured in the stopped-flow apparatus, rates of Ca2+ dissociation from the N-domain of isolated cTnC were also measured at 5°C. At 5°C, excess EGTA removed Ca2+ from the regulatory N-domain of cTnC, cTnCD87A/D88A, or cTnCE94A/E95A/E96A at 915 ± 9, 896 ± 8, or 886 ± 21 s−1, respectively (Figure 2C). Thus, our results demonstrate that the central helix mutations had very little effect on the rates of Ca2+ dissociation at either 15°C or 5°C.
Effect of the Central Helix Mutations on the Affinity of cTnC for the Regulatory Region of cTnI
The effect of the central helix mutations on the affinity of cTnC for the cTnI128-180 was measured following changes in fluorescence of IAANS attached to Cys53 of cTnC (30). Figure 3 shows that cTnI128-180 peptide binds to the Ca2+-saturated cTnC with a dissociation constant of 62 ± 5 nM. Figure 3 also shows that cTnI128-180 peptide binds to the Ca2+-saturated cTnCD87A/D88A or cTnCE94A/E95A/E96A with a dissociation constant of 174 ± 22 or 156 ± 6 nM, respectively. Thus, the D87A/D88A and E94A/E95A/E96A mutations significantly decreased the affinity of cTnC for cTnI128-180 by ~2.8- and 2.5-fold, respectively.
Figure 3. Effect of the central helix mutations on the affinity of cTnC for the regulatory region of cTnI.
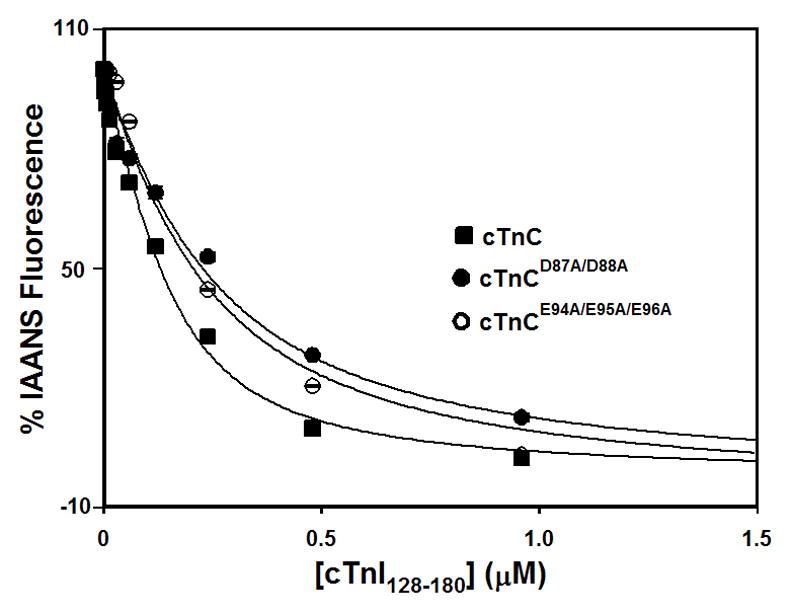
Figure shows the effect of central helix mutations on the cTnI128-180 binding properties of cTnC in the presence of 1 mM Ca2+ at 15°C. The cTnI128-180-dependent decreases in IAANS fluorescence are shown as a function of [cTnI128-180] for cTnC (■), cTnCD87A/D88A (●), or cTnCE94A/E95A/E96A (○). The cTnC proteins (with C35S/T53C/C84S substitution) were labeled with IAANS at Cys53. 100 % IAANS fluorescence corresponds to the Ca2+-bound state, whereas 0% fluorescence corresponds to the Ca2+-cTnI128-180 bound state for each individual cTnC protein.
Effect of the Central Helix Mutations on the Ca2+ Binding Properties of the cTn Complex
The Ca2+-induced decreases in IAANS fluorescence, which occur when Ca2+ binds to the regulatory N-domain of cTnC or its mutants reconstituted into the cTn complexes, are shown in Figure 4A. Results are summarized in Table I. The cTn complex exhibited a half-maximal Ca2+-dependent decrease in IAANS fluorescence at 0.83 ± 0.07 μM. The D87A/D88A and E94A/E95A/E96A mutations led to ~3.0- and 2.2-fold decreases, respectively, in the Ca2+ sensitivity of the cTn complex. Neither the D87A/D88A nor E94A/E95A/E96A mutation affected the cooperativity of Ca2+ binding to the cTn complex (as indicated by the nH values) (Table I).
Figure 4. Effect of the central helix mutations on the Ca2+ binding properties of the cTn complex.
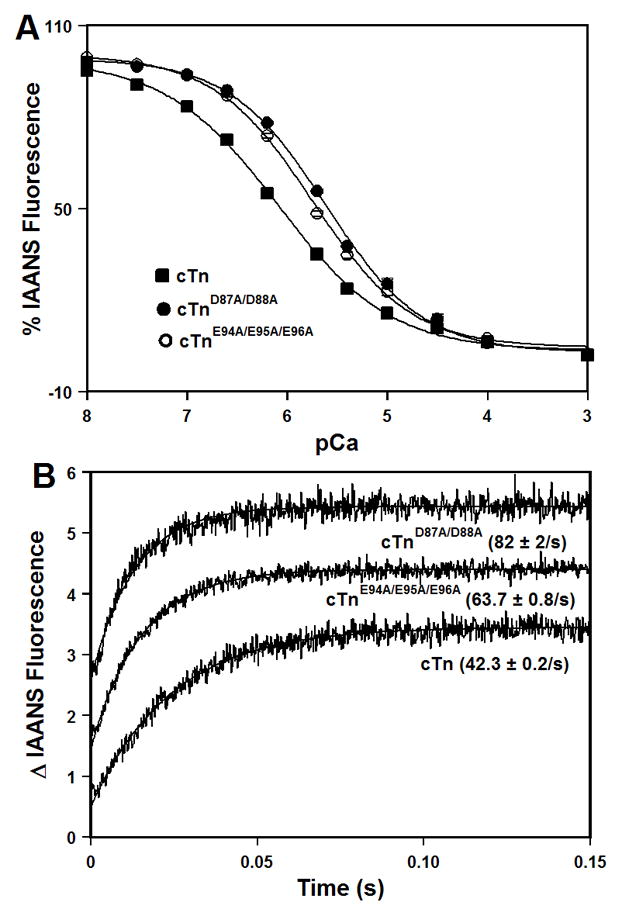
Panel A shows decreases in IAANS fluorescence, which occur as Ca2+ binds to the cTn (■), cTnD87A/D88A (●), or cTnE94A/E95A/E96A (○) complexes at 15°C. The cTnC proteins (with C35S/T53C/C84S substitution) were labeled with IAANS at Cys53. The IAANS fluorescence was excited at 330 nm and monitored at 450 nm. Panel B shows the time course of increases in IAANS fluorescence as Ca2+ was removed by excess EGTA from the cTn, cTnD87A/D88A, or cTnE94A/E95A/E96A complexes at 15°C. The data traces have been normalized and staggered for clarity. Each trace is an average of at least five traces fit with a single exponential equation. The IAANS fluorescence was excited at 330 nm and monitored through a 510 nm band-pass filter.
Stopped-flow measurements, utilizing IAANS fluorescence, were conducted to examine the effect of the central helix mutations on the rate of Ca2+ dissociation from the cTn complex. Results are summarized in Table I. Figure 4B shows that excess EGTA removed Ca2+ from the regulatory N-domain of cTnC reconstituted into the cTn complex at 42.3 ± 0.2 s−1. The D87A/D88A and E94A/E95A/E96A mutations led to ~1.9- and 1.5-fold faster, respectively, rates of Ca2+ dissociation from the cTn complex (Figure 4B and Table I).
Effect of the Central Helix Mutations on the Ca2+ Binding Properties of Reconstituted Thin Filaments
The Ca2+-induced changes in IAANS fluorescence, which occur when Ca2+ binds to the regulatory N-domain of the cTn, cTnD87A/D88A, or cTnE94A/E95A/E96A complexes reconstituted into thin filaments, are shown in Figure 5A. Results are summarized in Table I. After reconstitution into thin filaments, the cTn complex exhibited a half-maximal Ca2+- dependent increase in IAANS fluorescence at 2.15 ± 0.09 μM. The D87A/D88A and E94A/E95A/E96A mutations led to ~4.7- and 5.6- fold decreases, respectively, in the Ca2+ sensitivities of the cTn complex reconstituted into thin filaments (Figure 5A and Table I). Furthermore, the D87A/D88A mutation decreased the cooperativity of Ca2+ binding to reconstituted thin filaments (indicated by a lower nH), suggesting that this mutation affected near-neighbor regulatory unit interactions along the thin filament. The E94A/E95A/E96A mutation had no effect on cooperativity (Table I).
Figure 5. Effect of the central helix mutations on the Ca2+ binding properties of reconstituted thin filaments.
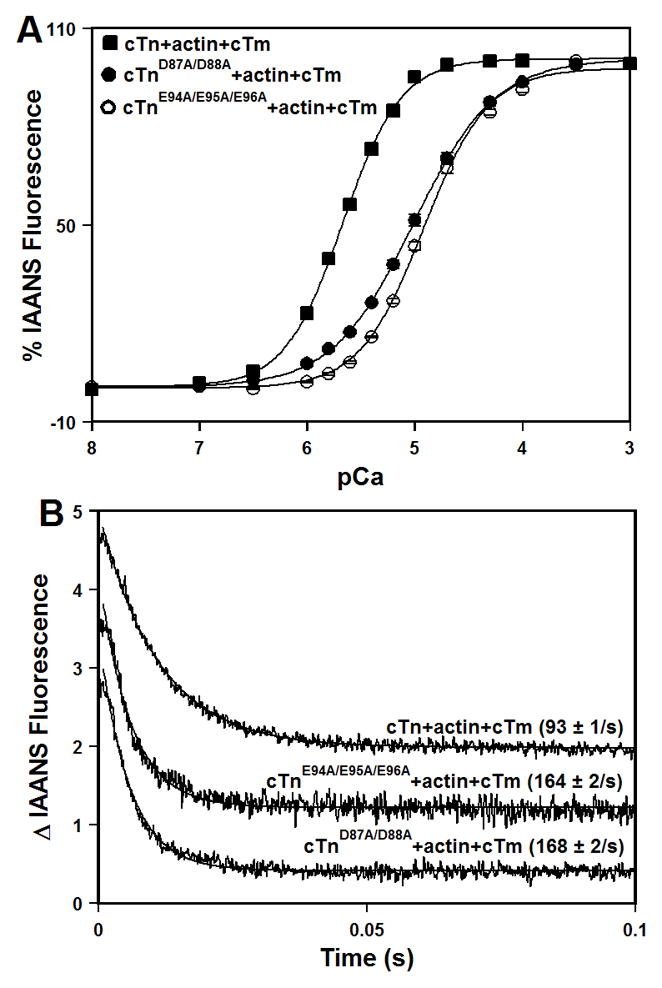
Panel A shows increases in IAANS fluorescence, which occur as Ca2+ binds to the cTn (■), cTnD87A/D88A (●), or cTnE94A/E95A/E96A (○) complexes reconstituted into thin filaments at 15°C. The cTnC proteins (with C35S/T53C/C84S substitution) were labeled with IAANS at Cys53. The IAANS fluorescence was excited at 330 nm and monitored at 450 nm. Panel B shows the time course of decreases in IAANS fluorescence as Ca2+ was removed by excess EGTA from the cTn, cTnD87A/D88A, or cTnE94A/E95A/E96A complexes reconstituted into thin filaments at 15°C. The data traces have been normalized and staggered for clarity. Each trace is an average of at least five traces fit with a single exponential equation. The IAANS fluorescence was excited at 330 nm and monitored through a 510 nm band-pass filter.
Stopped-flow measurements, utilizing IAANS fluorescence, were conducted to determine the effect of the central helix mutations on the kinetics of Ca2+ dissociation from the regulatory N-domain of the cTn complex reconstituted into thin filaments. The results are summarized in Table I. Figure 5B shows that excess EGTA removed Ca2+ from the cTn complex reconstituted into thin filaments at 93 ± 1 s−1. The D87A/D88A and E94A/E95A/E96A mutations led to ~1.8-fold acceleration in the rate of Ca2+ dissociation from the cTn complex reconstituted into thin filaments (Figure 5B and Table I).
Effect of the Central Helix Mutations on the Ca2+ Binding Properties of Reconstituted Thin Filaments in the Presence of Myosin S1
The Ca2+-induced increases in IAANS fluorescence, occurring when Ca2+ binds to the regulatory N-domain of the cTn, cTnD87A/D88A, or cTnE94A/E95A/E96A complexes, reconstituted into thin filaments in the presence of myosin S1, are shown in Figure 6A and summarized in Table I. In the presence of two myosin S1 molecules per stoichiometric unit, the cTn complex reconstituted into thin filaments exhibited a half-maximal Ca2+ dependent increase in IAANS fluorescence at 0.29 ± 0.02 μM. Our results show that in the presence of myosin S1, the D87A/D88A mutation led to ~6.0-fold decrease in the Ca2+ sensitivity of reconstituted thin filaments, while the E94A/E95A/E96A mutation led to ~12.8-fold decrease in the Ca2+ sensitivity of reconstituted thin filaments (Figure 6A and Table I). Furthermore, our results show that neither the D87A/D88A nor E94A/E95A/E96A mutation significantly affected the cooperativity of Ca2+ binding to reconstituted thin filaments in the presence of myosin S1 (Table I).
Figure 6. Effect of the central helix mutations on the Ca2+ binding properties of reconstituted thin filaments in the presence of myosin S1.
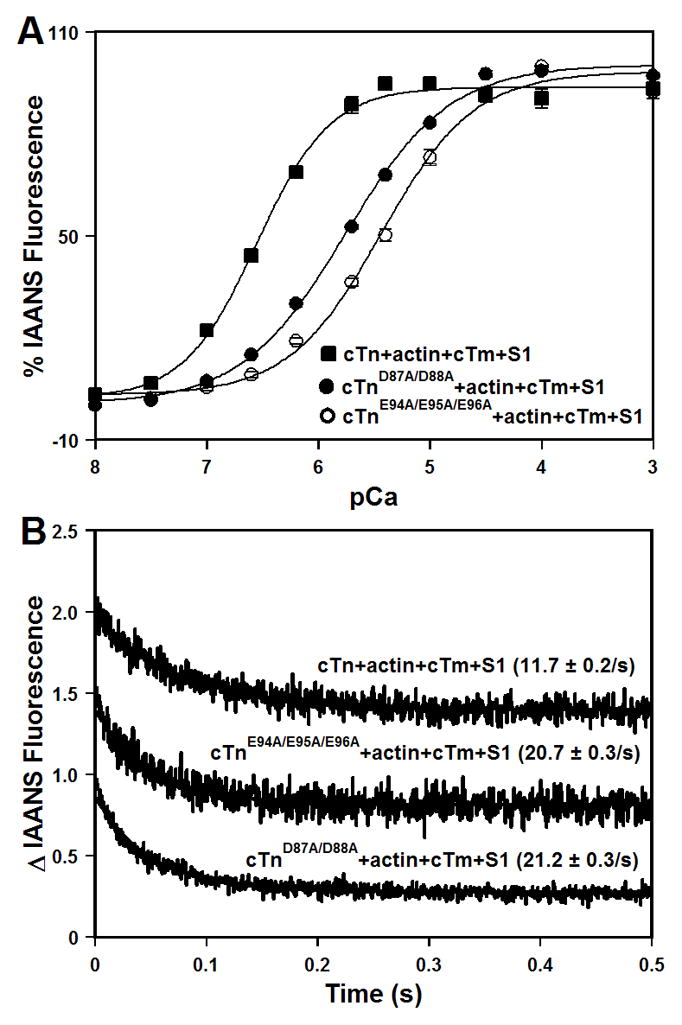
Panel A shows increases in IAANS fluorescence, which occur as Ca2+ binds to the cTn (■), cTnD87A/D88A (●), or cTnE94A/E95A/E96A (○) complexes reconstituted into thin filaments in the presence of myosin S1 at 15°C. The cTnC proteins (with C35S/T53C/C84S substitution) were labeled with IAANS at Cys53. The IAANS fluorescence was excited at 330 nm and monitored at 490 nm. Panel B shows the time course of decreases in IAANS fluorescence as Ca2+ was removed by excess EGTA from the cTn, cTnD87A/D88A, or cTnE94A/E95A/E96A complexes reconstituted into thin filaments in the presence of myosin S1 at 15°C. The data traces have been normalized and staggered for clarity. Each trace is an average of at least five traces fit with a single exponential equation. The IAANS fluorescence was excited at 330 nm and monitored through a 515 nm cut-on filter.
Stopped-flow measurements, utilizing IAANS fluorescence, were conducted to determine the effect of the central helix mutations on the kinetics of Ca2+ dissociation from the regulatory N-domain of the cTn complex reconstituted into thin filaments in the presence of myosin S1. The results are summarized in Table I. Figure 6B shows that excess EGTA removed Ca2+ from the cTn complex reconstituted into thin filaments in the presence of myosin S1 at 11.7 ± 0.2 s−1. Our results show that both the D87A/D88A and E94A/E95A/E96A mutations led to ~1.8-fold faster rate of Ca2+ dissociation from the cTn complex reconstituted into thin filaments in the presence of myosin S1 (Figure 6B and Table I).
Effect of the Central Helix Mutations on the Specific Activities and Ca2+ Sensitivities of Actomyosin ATPase
First, we evaluated the effect of the central helix mutations on the specific activities of actomyosin ATPase at pCa 9.0 and 4.5. The results are summarized in Table II and are shown in Figure 7A. Our results demonstrate that the central helix mutations enhanced the ability of the cTn complex to inhibit actomyosin ATPase in the absence of Ca2+. Our results also show that in the presence of saturating Ca2+, the central helix mutations substantially impaired the ability of the cTn complex to activate actomyosin ATPase.
Table II.
Effect of the Central Helix Mutations on the Properties of Actomyosin ATPase.
| Biochemical System | Activity at pCa 9.0 (mol Pi/s)/mol S1 | Activity at pCa 4.0 (mol Pi/s)/mol S1 | Ca2+ Kd (μM) | nH |
|---|---|---|---|---|
| cTn+actin+cTm+S1 | 0.0133 ± 0.0004 | 0.046 ± 0.001 | 1.49 ± 0.03 | 1.44 ± 0.06 |
| cTnD87A/D88A+actin+cTm+S1 | 0.0094 ± 0.0001a | 0.0284 ± 0.0004a | 2.9 ± 0.2a | 1.4 ± 0.1 |
| cTnE94A/E95A/E96A+actin+cTm+S1 | 0.0090 ± 0.0002a | 0.0173 ± 0.0004a | 2.84 ± 0.05a | 1.4 ± 0.1 |
Significantly different from their respective control values (p<0.05)
Figure 7. Effect of the central helix mutations on the specific activities and Ca2+ sensitivities of actomyosin ATPase.
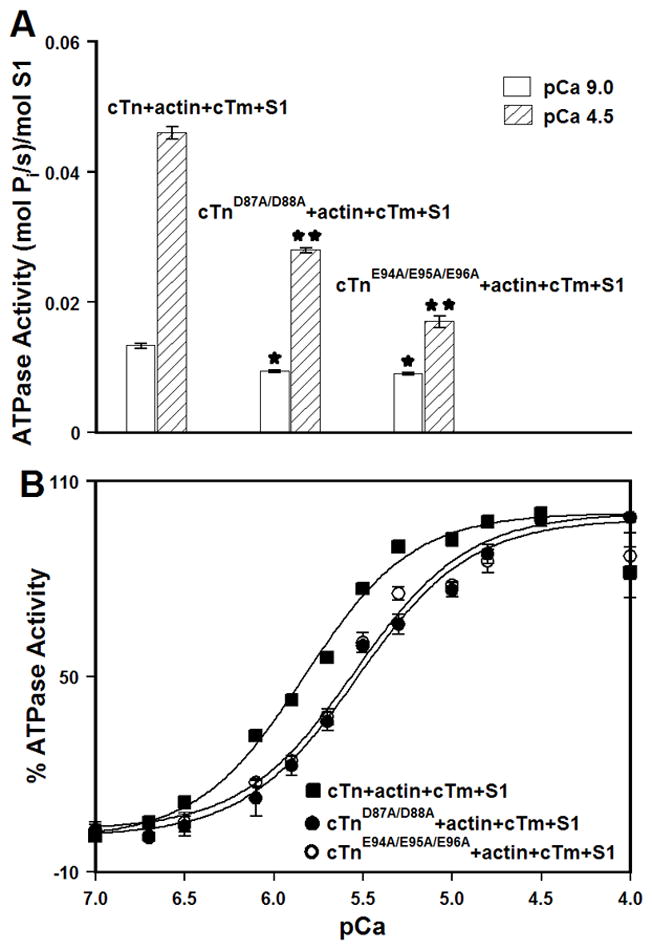
Panel A shows the specific actomyosin ATPase activity of the cTn, cTnD87A/D88A, or cTnE94A/E95A/E96A complexes reconstituted into thin filaments (in the presence of myosin S1) at pCa 9.0 (open bars) or pCa 4.5 (bars filled with slanted lines). The cTnC proteins (with C35S/T53C/C84S substitution) were labeled with IAANS at Cys53. Each data point represents a mean ± SE of three separate experiments (each carried out in triplicate). Values marked with * are significantly different from the control values at pCa 9.0 (p< 0.05). Values marked with ** are significantly different from the control values at pCa 4.5 (p<0.05). Panel B shows the Ca2+ dependent activity of actomyosin ATPase for the cTn (■), cTnD87A/D88A (●), or cTnE94A/E95A/E96A (○) complexes reconstituted into thin filaments (in the presence of myosin S1) as a function of pCa. Each data point represents a mean ± SE of at least three separate experiments (each carried out in triplicate). Data sets were individually normalized for each mutant cTn complex reconstituted into thin filaments (in the presence of myosin S1), and fit with logistic sigmoid.
Next, we examined the effect of the central helix mutations on the Ca2+ sensitivity of actomyosin ATPase. For thin filaments reconstituted with the cTn complex, half-maximal Ca2+ activation occurred at 1.49 ± 0.03 μM (Figure 7B and Table II). Our results indicate that both the D87A/D88A and E94A/E95A/E96A mutations led to a ~1.9-fold decrease in the Ca2+ sensitivity of actomyosin ATPase (Figure 7B and Table II). The nH values for actomyosin ATPase curves were not significantly affected by either the D87A/D88A or E94A/E95A/E96A mutation (Table II).
DISCUSSION
In this study, we investigated the molecular and functional consequences of the D87A/D88A and E94A/E95A/E96A mutations. We determined that the D87A/D88A but not E94A/E95A/E96A mutation resulted in the lower Ca2+ sensitivity of the N-domain of isolated cTnC. However, both the D87A/D88A and E94A/E95A/E96A mutations resulted in the substantially lower affinity of Ca2+ saturated cTnC for the regulatory region of cTnI (cTnI128-180), potentially due to the loss of electrostatic interactions between acidic residues within the central helix of cTnC and basic residues within the inhibitory region of cTnI.
In muscle, cTnC does not function in isolation but as a part of the cTn complex. Binding of cTnI substantially increases the Ca2+ sensitivity of cTnC (17), by stabilizing the “open” state of the regulatory N-domain (38; 39). Our results indicate that desensitization of the cTn complex to Ca2+ by the D87A/D88A and E94A/E95A/E96A mutations was largely due to the faster rates of Ca2+ dissociation. The D87A/D88A and E94A/E95A/E96A mutations had an even larger impact on the Ca2+ desensitization of the cTn complex after reconstitution into thin filaments (in the absence or presence of myosin S1). Decreased affinity of cTnC for the regulatory region of cTnI could lead to the increased probability of interactions between the inhibitory region of cTnI and actin, thus enhancing ability of the central helix mutations to desensitize thin filaments to Ca2+. The Ca2+ desensitization was not solely due to the faster rates of Ca2+ dissociation, indicating that the central helix mutations decreased the rates of Ca2+ association to the cTn complex reconstituted into thin filaments (in the absence or presence of myosin S1). Thus, central helix mutations appear to stabilize the “closed” state of the regulatory N-domain of cTnC within the cTn complex reconstituted into thin filaments (in the absence or presence of myosin S1), in addition to destabilizing the “open” state. Our findings are consistent with those from earlier studies, which showed that naturally occurring and engineered cTnC mutations can alter the rates of Ca2+ association to the cTn complex reconstituted into increasingly complex biochemical systems (40).
TnC exists in two isoforms, one found in cardiac and slow skeletal muscle (cTnC), the other found in fast skeletal muscle (sTnC). A number of earlier studies focused on elucidating the role of central helix in the biophysical properties and biological function of sTnC (41–48). Flexibility and length of the central helix were shown to play a role in the proper regulatory function of sTnC (42; 48). Much less is known about the significance of central helix to the function of cTnC. For instance, an earlier study determined that the D88A mutation did not affect the Ca2+ sensitivity of isolated cTnC (49). However, that study (49) did not examine the effect of the D88A mutation on the Ca2+ binding properties of cTnC reconstituted into more complex biochemical systems. Crystal structures of Ca2+ bound cTn (25) and sTn (24) complexes show similar overall organization of the subunits. However, there are a number of differences, including the orientation of the regulatory N-domain and conformation of the central helix of TnC, and the structure of the inhibitory region of TnI. In the crystal structure of the sTn complex, well ordered inhibitory region of sTnI was shown to interact with the rigid central helix of sTnC. On the other hand, in the crystal structure of the cTn complex, the central helix of cTnC was disordered, and the inhibitory region of cTnI was not visualized. Hydrogen-deuterium exchange mass spectroscopy and 15N NMR relaxation studies suggest that in solution the cTn complex adopts a structure more similar to that of the crystal structure of the sTn complex (28; 29). Our results are consistent with the idea that, in the presence of Ca2+, acidic residues within the exposed segment of the central helix of cTnC come in close contact with basic residues in the inhibitory region of cTnI, such as Lys139, Arg140, Arg144 and Arg145.
Both the D87A/D88A and E94A/E95A/E96A mutations desensitized actomyosin ATPase to Ca2+ and decreased maximal ATPase activity. Regarding the functional significance of the Asp87/Asp88 cluster, our results are in agreement with those observed for sTnC, where the D89A mutation in chicken sTnC (which corresponds to the D88A mutation in cTnC) led to diminished ability of the sTn complex to activate actomyosin ATPase in the presence of saturating Ca2+ (41). Similarly, the E88K mutation in chicken sTnC was shown to decrease the Ca2+ sensitivity of force development and reduce tension recovery in skinned muscle fibers (44). In this work, we show for the first time that the Glu94/Glu95/Glu96 cluster is as important to the proper regulatory function of the cTn complex as the Asp87/Asp88 cluster.
It is important to note that we used skeletal actin and myosin S1 in this study because of our extensive prior work using these proteins (15; 30; 35; 40; 50). However, skeletal and cardiac actins are 99 % identical, differing by only four amino acids. In addition, results of a recent study (51) examining effects of human recombinant cardiac myosin S1 (with both rigor and cycling myosin S1) on the Ca2+ sensitivity of thin filaments agreed with the results from our earlier study which used skeletal myosin S1(15). Thus, it is highly unlikely that use of skeletal actin and myosin S1 had a major effect on the outcomes of our current study.
It is well accepted that altered Ca2+ binding and exchange with thin filaments can lead to adverse physiological consequences. Mutations of sarcomeric proteins associated with dilated cardiomyopathy (DCM) tend to reduce Ca2+ sensitivity of actomyosin ATPase and lower maximal ATPase activity (52–55). Functional consequences of the D87A/D88A and E94A/E95A/E96A mutations are consistent with those of known mutations associated with the DCM phenotype. In vivo studies are needed to determine whether central helix mutations examined in this work can recapitulate the pathogenesis of DCM.
In conclusion, our results demonstrate the importance of acidic residues within the exposed middle segment of the central helix of cTnC in controlling: 1) affinity of cTnC for the regulatory fragment of cTnI, 2) Ca2+ binding and exchange with the cTn complex in increasingly complex biochemical systems, and 3) ability of the cTn complex to regulate actomyosin ATPase.
D87A/D88A and E94A/E95A/E96A mutations were introduced into the central helix of cTnC
Central helix mutations decreased affinity of Ca2+ saturated cTnC for cTnI128-180
Central helix mutations desensitized the cTn complex and thin filaments to Ca2+
Central helix mutations decreased the Ca2+ sensitivity of actomyosin ATPase
Central helix mutations decreased maximal activity of actomyosin ATPase
Acknowledgments
We thank Dr. Lawrence Smillie (University of Alberta) for the generous gift of the human cTnC plasmid. We also thank Dr. Darl Swartz (Delaware Valley College) for the generous gift of actin, cTm and myosin S1. Research reported in this publication was supported by the NHLBI institute of NIH under Award Number R15HL117034 (to S.B.T). The content is solely the responsibility of the authors, and does not necessarily represent the official views of the NIH.
Footnotes
Abbreviations: TnC, troponin C; TnI, troponin I; cTnC, cardiac troponin C; cTnCD87A/D88A, cTnC with the D87A/D88A mutation; cTnCE94A/E95A/E96A, cTnC with the E94A/E95A/E96A mutation; cTnI, cardiac troponin I; cTnI128-180, peptide corresponding to residues 128-180 of human cTnI; cTnT, cardiac troponin T; cTn, cardiac troponin complex: (cTnC-cTnI-cTnT); cTnD87A/D88A, the cTn complex containing cTnCD87A/D88A: (cTnCD87A/D88A-cTnI-cTnT); cTnE94A/E95A/E96A, the cTn complex containing cTnCE94A/E95A/E96A: (cTnCE94A/E95A/E96A-cTnI-cTnT); sTnC, skeletal troponin C; sTnI, skeletal troponin I; sTn, skeletal troponin complex; IAANS, 2-(4′-(iodoacetamido)anilino)naphthalene-6-sulfonic acid; EGTA, ethylene glycol-bis(2-aminoethyl)-N,N,N′,N′-tetraacetic acid; DTT, dithiothreitol; MOPS, 3-(N-morpholino)propanesulfonic acid; Kd, dissociation constant; koff, dissociation rate.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Filatov VL, Katrukha AG, Bulargina TV, Gusev NB. Biochemistry (Mosc) 1999;64:969–985. [PubMed] [Google Scholar]
- 2.Farah CS, Reinach FC. Faseb J. 1995;9:755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- 3.Davis JP, Tikunova SB. Cardiovasc Res. 2008;77:619–626. doi: 10.1093/cvr/cvm098. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi T, Jin L, de Tombe PP. Pflugers Arch. 2008;457:37–46. doi: 10.1007/s00424-008-0511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi T, Solaro RJ. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 6.Gordon AM, Homsher E, Regnier M. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 7.Li MX, Wang X, Sykes BD. J Muscle Res Cell Motil. 2004;25:559–579. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- 8.Tobacman LS. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 9.Gordon AM, Regnier M, Homsher E. News Physiol Sci. 2001;16:49–55. [PubMed] [Google Scholar]
- 10.Sundaralingam M, Bergstrom R, Strasburg G, Rao ST, Roychowdhury P, Greaser M, Wang BC. Science. 1985;227:945–948. doi: 10.1126/science.3969570. [DOI] [PubMed] [Google Scholar]
- 11.Herzberg O, James MN. Nature. 1985;313:653–659. doi: 10.1038/313653a0. [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto F, Makino K, Maeda K, Patzelt H, Maeda Y, Fujiwara S. J Mol Biol. 2004;342:1209–1221. doi: 10.1016/j.jmb.2004.07.086. [DOI] [PubMed] [Google Scholar]
- 13.Reinach FC, Farah CS, Monteiro PB, Malnic B. Cell Struct Funct. 1997;22:219–223. doi: 10.1247/csf.22.219. [DOI] [PubMed] [Google Scholar]
- 14.Potter JD, Gergely J. J Biol Chem. 1975;250:4628–4633. [PubMed] [Google Scholar]
- 15.Davis JP, Norman C, Kobayashi T, Solaro RJ, Swartz DR, Tikunova SB. Biophys J. 2007;92:3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi T, Solaro RJ. J Biol Chem. 2006;281:13471–13477. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 17.Johnson JD, Collins JH, Robertson SP, Potter JD. J Biol Chem. 1980;255:9635–9640. [PubMed] [Google Scholar]
- 18.Solaro RJ, Rosevear P, Kobayashi T. Biochem Biophys Res Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tardiff JC. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi T, Patrick SE, Kobayashi M. J Biol Chem. 2009;284:20052–20060. doi: 10.1074/jbc.M109.001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guharoy M, Chakrabarti P. BMC Bioinformatics. 2010;11:286. doi: 10.1186/1471-2105-11-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godzik A, Sander C. Protein Eng. 1989;2:589–596. doi: 10.1093/protein/2.8.589. [DOI] [PubMed] [Google Scholar]
- 23.Collins JH. J Muscle Res Cell Motil. 1991;12:3–25. doi: 10.1007/BF01781170. [DOI] [PubMed] [Google Scholar]
- 24.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Proc Natl Acad Sci U S A. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda S, Yamashita A, Maeda K, Maeda Y. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 26.Sorsa T, Pollesello P, Permi P, Drakenberg T, Kilpelainen I. J Mol Cell Cardiol. 2003;35:1055–1061. doi: 10.1016/s0022-2828(03)00178-0. [DOI] [PubMed] [Google Scholar]
- 27.Sorsa T, Pollesello P, Rosevear PR, Drakenberg T, Kilpelainen I. Eur J Pharmacol. 2004;486:1–8. doi: 10.1016/j.ejphar.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 28.Lindhout DA, Boyko RF, Corson DC, Li MX, Sykes BD. Biochemistry. 2005;44:14750–14759. doi: 10.1021/bi051580l. [DOI] [PubMed] [Google Scholar]
- 29.Kowlessur D, Tobacman LS. J Biol Chem. 2010;285:2686–2694. doi: 10.1074/jbc.M109.062349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, Davis JP. Biochemistry. 2010;49:1975–1984. doi: 10.1021/bi901867s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swindle N, Tikunova SB. Biochemistry. 2010;49:4813–4820. doi: 10.1021/bi100400h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tikunova SB, Davis JP. J Biol Chem. 2004;279:35341–35352. doi: 10.1074/jbc.M405413200. [DOI] [PubMed] [Google Scholar]
- 33.Robertson S, Potter JD. Methods in Pharmacology. 1984;5:63–75. [Google Scholar]
- 34.Tikunova SB, Rall JA, Davis JP. Biochemistry. 2002;41:6697–6705. doi: 10.1021/bi011763h. [DOI] [PubMed] [Google Scholar]
- 35.Albury AN, Swindle N, Swartz DR, Tikunova SB. Biochemistry. 2012;51:3614–3621. doi: 10.1021/bi300187k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Eerd JP, Takahashi K. Biochem Biophys Res Commun. 1975;64:122–127. doi: 10.1016/0006-291x(75)90227-2. [DOI] [PubMed] [Google Scholar]
- 37.Gillis TE, Blumenschein TM, Sykes BD, Tibbits GF. Biochemistry. 2003;42:6418–6426. doi: 10.1021/bi0340494. [DOI] [PubMed] [Google Scholar]
- 38.Li MX, Spyracopoulos L, Sykes BD. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 39.Dong WJ, Xing J, Villain M, Hellinger M, Robinson JM, Chandra M, Solaro RJ, Umeda PK, Cheung HC. J Biol Chem. 1999;274:31382–31390. doi: 10.1074/jbc.274.44.31382. [DOI] [PubMed] [Google Scholar]
- 40.Liu B, Tikunova SB, Kline KP, Siddiqui JK, Davis JP. PLoS One. 2012;7:e38259. doi: 10.1371/journal.pone.0038259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramakrishnan S, Hitchcock-DeGregori SE. Biochemistry. 1996;35:15515–15521. doi: 10.1021/bi961788u. [DOI] [PubMed] [Google Scholar]
- 42.Babu A, Rao VG, Su H, Gulati J. J Biol Chem. 1993;268:19232–19238. [PubMed] [Google Scholar]
- 43.Dobrowolski Z, Xu GQ, Hitchcock-DeGregori SE. J Biol Chem. 1991;266:5703–5710. [PubMed] [Google Scholar]
- 44.Fujimori K, Sorenson M, Herzberg O, Moult J, Reinach FC. Nature. 1990;345:182–184. doi: 10.1038/345182a0. [DOI] [PubMed] [Google Scholar]
- 45.Ding XL, Akella AB, Su H, Gulati J. Protein Sci. 1994;3:2089–2096. doi: 10.1002/pro.5560031122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sundaralingam M, Drendel W, Greaser M. Proc Natl Acad Sci U S A. 1985;82:7944–7947. doi: 10.1073/pnas.82.23.7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobayashi T, Zhao X, Wade R, Collins JH. Biochim Biophys Acta. 1999;1430:214–221. doi: 10.1016/s0167-4838(99)00002-3. [DOI] [PubMed] [Google Scholar]
- 48.Ramakrishnan S, Hitchcock-DeGregori SE. Biochemistry. 1995;34:16789–16796. doi: 10.1021/bi00051a029. [DOI] [PubMed] [Google Scholar]
- 49.Pollesello P, Ovaska M, Kaivola J, Tilgmann C, Lundstrom K, Kalkkinen N, Ulmanen I, Nissinen E, Taskinen J. J Biol Chem. 1994;269:28584–28590. [PubMed] [Google Scholar]
- 50.Liu B, Lee RS, Biesiadecki BJ, Tikunova SB, Davis JP. J Biol Chem. 2012;287:20027–20036. doi: 10.1074/jbc.M111.334953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sommese RF, Nag S, Sutton S, Miller SM, Spudich JA, Ruppel KM. PLoS One. 2013;8:e83403. doi: 10.1371/journal.pone.0083403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M, Sasaguri T, Ohtsuki I. Proc Natl Acad Sci U S A. 2002;99:913–918. doi: 10.1073/pnas.022628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinto JR, Siegfried JD, Parvatiyar MS, Li D, Norton N, Jones MA, Liang J, Potter JD, Hershberger RE. J Biol Chem. 2011;286:34404–34412. doi: 10.1074/jbc.M111.267211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mirza M, Marston S, Willott R, Ashley C, Mogensen J, McKenna W, Robinson P, Redwood C, Watkins H. J Biol Chem. 2005;280:28498–28506. doi: 10.1074/jbc.M412281200. [DOI] [PubMed] [Google Scholar]
- 55.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Circ Res. 2007;101:1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 56.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]