

Abstract
Elizabethkingia anophelis is a dominant bacterial species in the gut ecosystem of the malaria vector mosquito Anopheles gambiae. We recently sequenced the genomes of two strains of E. anophelis, R26T and Ag1, isolated from different strains of A. gambiae. The two bacterial strains are identical with a few exceptions. Phylogenetically, Elizabethkingia is closer to Chryseobacterium and Riemerella than to Flavobacterium. In line with other Bacteroidetes known to utilize various polymers in their ecological niches, the E. anophelis genome contains numerous TonB dependent transporters with various substrate specificities. In addition, several genes belonging to the polysaccharide utilization system and the glycoside hydrolase family were identified that could potentially be of benefit for the mosquito carbohydrate metabolism. In agreement with previous reports of broad antibiotic resistance in E. anophelis, a large number of genes encoding efflux pumps and β-lactamases are present in the genome. The component genes of resistance-nodulation-division type efflux pumps were found to be syntenic and conserved in different taxa of Bacteroidetes. The bacterium also displays hemolytic activity and encodes several hemolysins that may participate in the digestion of erythrocytes in the mosquito gut. At the same time, the OxyR regulon and antioxidant genes could provide defense against the oxidative stress that is associated with blood digestion. The genome annotation and comparative genomic analysis revealed functional characteristics associated with the symbiotic relationship with the mosquito host.
Introduction
The mosquito gut accommodates a diverse and dynamic microbiota [1]–[5], which has a profound impact on host metabolism, fecundity [6] and immunity [7], [8]. The gut microbiome is not a random assemblage; the common core taxa belong to Proteobacteria, Bacteroidetes and Actinobacteria [2]. To better understand its structure and function in the mosquito gut ecosystem, it is necessary to characterize abundant taxa. Elizabethkingia is a genus in the Flavobacteriaceae family of Bacteroidetes and represents a separate lineage from the Chryseobacterium-Bergeyella-Riemerella branch [9]. Elizabethkingia spp. has been found to be a predominant resident in the gut of Anopheles gambiae [1], [2], An. stephensi [4], [10] and Aedes aegypti [11]. Lately, the strain R26T of Elizabethkingia sp. was isolated from the midgut of An. gambiae, Ifakara strain [1]. The comparison of 16S ribosomal RNA (rRNA) gene sequences indicated closest similarity (98.6%) to that of Elizabethkingia meningoseptica. Subsequent hybridization experiments and fingerprint analyses together with biochemical tests, however, clearly separated R26T from E. meningoseptica and E. miricola. Hence, a novel species, Elizabethkingia anophelis, was proposed as a third member in the genus Elizabethkingia [12]. Similar to E. meningoseptica, E. anophelis is a non-motile, non-spore-forming Gram negative rod with natural resistance against several antibiotics [12]. Later, the strain Ag1 of E. anophelis was isolated from the G3 strain of An. gambiae. The genomes of both strains were sequenced and annotated [13]. Here we present the in silico annotation of the genome, particularly focusing on functional categories that provide insights regarding the ecological connection between the bacteria and the mosquito host.
Materials and Methods
Genome Project History
The strains of R26T and Ag1 of E. anophelis were isolated from the midgut of Anopheles gambiae maintained in the Faye laboratory in Stockholm University [1], [12] and the Xu laboratory in New Mexico State University, respectively. The genome sequencing, assembly and gene prediction were described in [13]. The draft genomes have been deposited at DDBJ/EMBL/GenBank. The R26T and Ag1 genomes are under the accession ANIW01000000 and AHHG00000000, respectively.
Functional Categorization
The predicted genes were functionally categorized using SEED subsystems [14] at the RAST server (http://rast.nmpdr.org) [15]. Conserved functional domains in protein sequences were identified using NCBI Conserved Domain Search Service (CD Search) [16]. Genes encoding candidate carbohydrate active enzymes, including enzymes of assembly (glycosyltransferases, GT) and deconstruction (glycoside hydrolases, GH, polysaccharide lyases, PL, carbohydrate esterases, CE) were detected with the CAZymes Analysis Toolkit (CAT) [17] using the Carbohydrate Active Enzyme (CAZy) database [18], [19].
Genomes used in the Analysis
The E. anophelis genome was compared to several available genomes in the Flavobacteriaceae family: E. meningoseptica ATCC 13253 (BioProject: PRJDB229), Chryseobacterium gleum ATCC 35910 (BioProject: PRJNA30953), Chryseobacterium sp. CF314 (BioProject: PRJNA83055) [20], Flavobacterium johnsoniae UW101 (BioProject: PRJNA58493) [21], Flavobacterium branchiophilum FL-15 (BioProject: PRJNA73421 [22] and Riemerella anatipestifer RA-CH-2 (BioProject: PRJNA183917). The bacterial 16S rDNA and rpoB, the gene that encodes the β subunit of bacterial RNA polymerase, were used to investigate phylogenetic relationships between the species in the Flavobacteriaceae family. The Multiple protein sequences were aligned using ClustalW and a neighbor joining phylogeny tree was constructed using MEGA 5.10 [23]. The Gamma distribution of substitution rate, Jones-Taylor-Thornton (JTT) model and pairwise deletion of gap/missing data were chosen for the tree construction. Bootstrap was conducted with 1000 replications. Synteny analysis was performed using Mauve Multiple Genome Alignment software [24]. A PCR assay was designed to verify an Ag1-present and R26T- absent segment. Primer sequences were: F1, CGGATCTTTTAATACCCAGCGT; R1, GGCATTTCCTGTCGTTACACC; F2, AACCTGCTGAACCTACAACGG; R2, GCCAATCTGTAAGTAGCGCC (Fig. S1).
Antimicrobial and Hemolysis Assays
Antibiotics and Cecropin A from Hyalophora cecropia were purchased from Sigma-Aldrich. The radial diffusion assay was performed as earlier described by Hultmark et al [25]. Blood agar plates (horse blood) and bacterial control strains were kindly provided by Ann-Beth Jonsson’s lab group. Hemolytic activity was evaluated following incubation at 37°C for up to 48 h.
Results and Discussion
Genome Properties
The draft genome of type strain R26T of Elizabethkingia anophelis is 4.03 Mbp in size with an average GC content of 35.4%. The genome was predicted to have 3687 protein coding sequences (CDS) and 44 RNA genes. A single copy of a 16 S rRNA gene is found in the genome. The genome statistics are presented in Table S1. Among the predicted protein coding genes, 2169 were assigned a putative function and 1518 were hypothetical proteins. To characterize the functions of the genome, the draft genome was annotated at RAST using the SEED subsystems [15]. Among the protein coding genes, 1174 were assigned into 308 subsystems (Fig. 1). The gene name, locus tag, protein ID and similarity comparison between the R26T and Ag1 genomes are presented in Table S2. The genomes of Ag1 and R26T are almost identical with a few exceptions. A small number of predicted ORFs (185 in R26T and 146 in Ag1) were not found in the other strain, likely because both genomes were incomplete. Strain specific genomic regions were, however, identified. Two R26T contigs, 104 (ANIW01000041; 28,343 bp) and 107 (ANIW01000035; 5,773 bp), are not found in the Ag1 genome. The R26T contig 104 contains six putative genes associated with conjugal elements, indicating the presence of a conjugative plasmid or transposon. In addition, a segment of 32,350 bp in the Ag1 contig 47 (AHHG01000028) is not present in the counterpart contig 16 (ANIW01000059) in the R26T genome. The presence of putative phage genes in the Ag1-segment indicates presence, or remnants, of a lysogenic phage. The insertion/deletion between Ag1 contig 47 and R26T contig 16 was verified by PCR and subsequent sequencing of the amplicons (Fig. S1). Further investigation is necessary to confirm the identity and nature of those strain-specific elements.
Figure 1. Subsystem category distribution statistics for the genome of E. Anophelis as annotated by RAST.

The pie chart represents relative abundance of each subsystem category and numbers depict subsystem feature counts.
Phylogenetic Relationship to Related Genomes
Upon comparison to available genomes in the family Flavobacteriaceae, E. anophelis is closer to E. meningoseptica, Riemerella anatipestifer RH-CH-2, Chryseobacterium gleum ATCC 35910 and Chryseobacterium sp. CF314 [20] and is divergent to taxa in the genus Flavobacterium. Table 1 shows the similarity of comparable CDS between E. anophelis R26T and six other genomes. The pattern was supported by the phylogenetic relationship inferred from the 16S rRNA (Fig. S2A) and the peptide sequences of RNA polymerase beta-subunit, rpoB (Fig. S2B). E. anophelis and E. meningoseptica are grouped as a clade sister to the genera Chryseobacterium and Riemerella, while the taxa of the genus Flavobacterium are clustered in a separate clade.
Table 1. The similarity of comparable CDS between R26T and related genomes in the family Flavobacteriaceae.
| Species | Identity | |||
| Comparable CDS | 80–100% | 50–79.9% | 20–49.9% | |
| Elizabethkingia meningoseptica ATCC 13253 | 3791 | 2481 (65.4) | 940 (24.8) | 370 (9.8) |
| Chryseobacterium gleum ATCC 35910 | 2921 | 578 (19.8) | 1537 (52.6) | 806 (27.6) |
| Chryseobacterium sp. CF314 | 2773 | 521 (18.8) | 1382 (49.8) | 870 (31.4) |
| Flavobacteriaceae bacterium 3519–10 | 2299 | 321 (14.0) | 1077 (46.8) | 901 (39.2) |
| Riemerella anatipestifer RA-CH-2 | 2021 | 306 (15.1) | 883 (28.6) | 832 (56.3) |
| Flavobacterium johnsoniae UW101 | 2569 | 34 (1.3) | 1001 (39.0) | 1534 (59.7) |
Numbers within parenthesis reflect the percentage of total comparable CDS.
TonB Dependent Transporters
The bacterial TonB-dependent transporters (TBDTs) are specialized elaborate machinery for active uptake of rare but essential nutrients and other substrates, such as iron complexes, vitamin B12, nickel, carbohydrates and colicin [26]–[30]. The total number of TBDTs is highly variable among bacterial genomes. Recently, Mirus et al. (2009) found putative TBDTs in 347 out of 686 sequenced bacterial genomes [31]. Among the investigated taxa of Bacteroidetes, all were found to endow over 50 TBDT genes. This is in line with our finding of 59 TBDT genes in E. anophelis. In the two other bacterial genomes that were isolated from the gut of An. gambiae in the Xu lab, Pseudomonas sp. Ag1 and Enterobacter sp. Ag1 possessed 55 and 20 TBDT genes, respectively [32], [33]. Different TBDTs are distinct in substrate specificity [31], [34]. The protein conserved domain analysis revealed various domains associated with different substrates. The conserved domains of 14 representative TBDTs are presented in Fig. S3. The architecture of mosaic conserved domains implies sophisticated interactions of TBDTs and biopolymers. The various TBDTs endow the bacterium with an uptake system for a variety of biopolymers, which have been demonstrated in many Gram negative bacteria, including Bacteroidetes in different metagenomic settings [35]–[37]. Further investigations are necessary to characterize the substrates of the TBDTs of E. anophelis and their contributions in the gut microbial community.
To energize the transport process, TBDTs interact with the TonB complex, a cytoplasmic transmembrane assembly of the proteins ExbB and ExbD, which couples with the TonB in periplasm. In the genome of E. anophelis, two ExbB, two ExbD genes and four TonB genes were found. A gene cluster was identified in the genome, where ExbB, ExbD and TonB as well as genes encoding an ABC transporter and a tetratricopeptide repeat containing protein are co-localized. The synteny of the gene cluster is conserved in taxa from all of the four existing classes of Bacteroidetes (Fig. 2). Such conservation indicates that it originated from a common ancestor and remained as an inheritable unit due to functional relationships among these syntenic genes.
Figure 2. Graphic view of a syntenic gene cluster that is conserved in five taxa of Bacteroidetes.
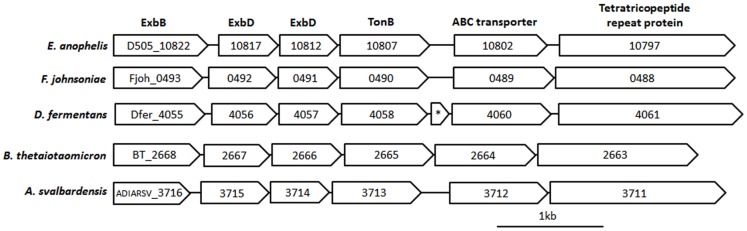
The genes encoding the components of TonB dependent transporters are ExbB, ExbD and TonB. Locus ID of each gene is given in the boxes, with taxon prefix (e.g. D505_ for E.a.) in the ExbB box for each species. The box with * in D. fermentans represents a predicted gene encoding a hypothetic protein. The scale bar represents 1 kb in length. Phylogenetically, E. anophelis and F. johnsoniae belong to the class Flavobacteria, Dyadobacter fermentans belongs to the class Cytophagia, Arcticibacter svalbardensis is in the class Sphingobacteriia, and Bacteroides thetaiotaomicron is located in the class Bacteroidia.
Polysaccharide Utilization Loci
Carbohydrates serve as a major carbon and energy source for bacteria. In Bacteroidetes, utilization of complex carbohydrates (glycans) involves polysaccharide sensing, degrading and import machinery. Such activities are mainly controlled by genes in various polysaccharide utilization loci (PULs) [29], [38]–[41]. The starch-utilization system (Sus) represents a typical example of glycan acquisition [29], [42]. The Sus like PUL consists of a cluster of genes encoding SusD (glycan-binding protein), SusC (Ton-B dependent transporter), SusE/SusF (carbohydrate-binding proteins without enzyme activity), SusA, SusB, SusG (enzymes for polysaccharide deconstruction) and SusR (an inner membrane-associated sensor-regulator system for transcriptional activation of Sus genes) [29], [42]. The genome of E. anophelis contains 28 pairs of SusD and SusC homologs. Fig. 3 shows four loci where putative Sus-like genes are located. Table S3 lists predicted CAZy proteins in the E. anophelis genome, including glycoside hydrolases (GH), such as α-glucosidase, β-glucosidases, α-galactosidase, β-galactosidase, α-mannosidase, α-amylase, β-mannosidase, β-glucanase, cellulase, maltodextrin glucosidase, xylosidase, and α-dextrin endo-1,6-α-glucosidase, which indicate a broad capability of degrading polysaccharides. Recently, Kolton et al. (2013) demonstrated that taxa in the genus Flavobacterium were separated into terrestrial and aquatic clades according to geographical distribution. The terrestrial taxa have greater capacity to degrade polysaccharides [43]. Interestingly, some CAZy enzymes of R26T have orthologs in F. johnsoniae, a terrestrial taxon, but not in the aquatic taxon F. branchiophilum. It is thus plausible that Elizabethkingia species have a similar ecological niche as the terrestrial taxa of flavobacteria.
Figure 3. Graphic view of four Sus-like loci in the genome of E. anophelis R26T.
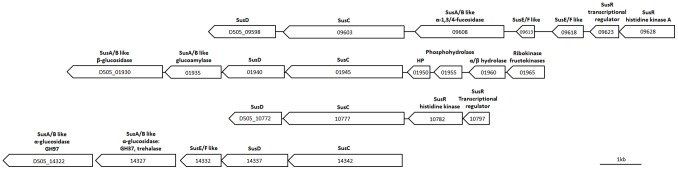
Locus ID of each gene is given in the boxes. HP: hypothetic protein. The scale bar represents 1 kb in length.
Sucrose and fructose are the most common sugars that mosquitoes ingest from floral nectar [44], [45]. Digestion of the sugar is carried out mainly by α-glucosidases that catalyze the hydrolysis of 1,4-α-glucosidic bonds to release α-glucose. The mosquito α-glucosidases have been characterized [46], [47]. In addition, mosquitoes can ingest plant tissue and cellulose particles from plants, especially in arid habitats where floral nectar is scarcely available [48]–[50]. Certain polysaccharides from plants (e.g. hemicelluloses, pectins, and starch) may contain several different monosaccharides and a variety of glycosidic linkages. Utilization of the polysaccharides with such a wide structural variety and fluctuating abundance requires sophisticated mechanisms to recognize and degrade them [41]. The predominance of E. anophelis in the sugar fed gut [2] and the possession of numerous Sus-like loci and GHs suggest that the bacterium may be capable of utilizing plant cellulose in the diet. Interestingly, no SusC and SusD were found in the genomes of Enterobacter and Pseudomonas strains that were isolated from the mosquito gut [32], [33]. The large capacity of polymer transport and utilization implies a potential ecological association between the mosquito and the gut microbiota in which certain microbial residents may support effective utilization of various types of polysaccharides that mosquitoes take in from nectar-rich or nectar-poor plants.
Antibiotic Resistance
E. anophelis is known for its intrinsic resistance to many antibiotics [12]. The resistance mechanisms employed by bacteria are manifold, such as the enzymatic degradation of the drug, the alteration of the target drug site and the direct extrusion of the drug from the cells through efflux pumps. The E. anophelis genome appears to contain a large number of resistance genes (Table 2, S4). This includes a large set of multidrug efflux pumps, predominantly members of the resistance-nodulation-division (RND) and major facilitator multidrug pumps (MFS) families. The RND transporters can mediate extrusion of a broad range of substrates, including heavy metals (heavy metal efflux, HME), multidrug hydrophobe/amphiphile efflux-1 (HAE1) and toxic chemical compounds to maintain homeostasis [51], [52]. The RND efflux pump is a tripartite assembly composed of an inner RND transport protein, a membrane fusion protein (MFP) and an outer membrane protein [53], [54]. There are 13 sets of genes encoding the components of the RND pumps in the genome of E. anophelis. In most cases, three genes were found adjacent to each other, likely in a single operon. The synteny is conserved in the flavobacteria compared in this study as well as in other taxa of Bacterioidetes (Fig. 4, Table S4). The role of RND-efflux pumps in intrinsic antibiotic resistance has been demonstrated in Chryseobacterium and F. johnsoniae [55], [56], as well as in Bacteroides fragilis [57]. The genome also encompasses genes encoding 44 MFS and two multidrug and toxic compound extrusion (MATE) transporters (Table S4). Generally, the MFS proteins mediate transport of a wide spectrum of substrates, including ions, carbohydrates, lipids, amino acids and peptides, nucleosides, and other molecules [58], [59].
Table 2. Summary of the functional subcategories of resistance genes.
| Number of Genes | Average identity (%) | ||||
| Functional classification | Ag1 | E. m. | C. g. | F. b. | |
| RND efflux pump | 37 | 100 | 83.5 | 66.5 | 27.7 |
| MFS transport | 44 | 100 | 73.7 | 55.5 | 25.1 |
| Resistance to β-lactams | 28 | 100 | 69.0 | 52.4 | 24.2 |
| Resistance to fluoroquinolones | 4 | 100 | 96.7 | 82.9 | 63.1 |
| Drug resistance: other | 19 | 100 | 76.8 | 52.1 | 17.8 |
| Heavy metal detoxification | 11 | 99.3 | 77.4 | 68.3 | 39.6 |
Average identity reflects the mean identity of genes within the subcategory compared to R26T. E. m., Elizabethkingia meningoseptica; C. g., Chryseobacterium gleum; F. b., Flavobacterium branchiophilum.
Figure 4. Graphic view of a locus where three component genes of an RND efflux pump are located syntenically in four taxa of Bacteroidetes.
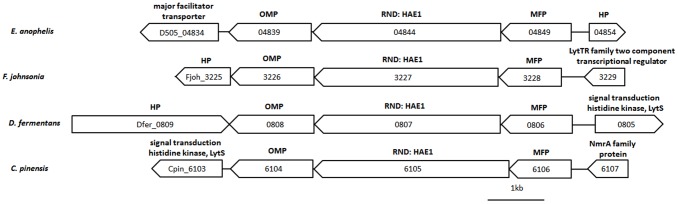
Gene name was given on the top of each box. Locus ID of each gene is given in the boxes. OMP, outer membrane protein; RND: HAE1, Hydrophobe/amphiphile efflux-1; MFP, membrane fusion protein; HP: hypothetic protein. The scale bar represents 1 kb in length.
The synthesis of β-lactamases is the most commonly employed strategy among Gram-negative bacteria to combat β-lactam antibiotics [60]. A large set of genes conferring resistance to β-lactams were annotated in the R26T genome, including 19 β-lactamases, four metallo-β-lactamases (MBLs) and four penicillin-binding proteins (Table S4). An overall lower degree of conservation between the compared species and R26T was observed in this subcategory. Phylogenetic analysis of selected lactam degrading enzymes suggests that in addition to vertical inheritance, certain β-lactamase genes may be acquired by lateral gene transfer. As an example of the former, the β-lactamase D505_10647 appears conserved and vertically transmitted in Flavobacteriaceae (Fig. 5A, Table S5). In contrast, D505_08675 is not found in other members of Flavobacteriaceae except E. meningoseptica. Orthologs are instead present in the taxa belonging to the class Sphingobacteria of Bacteroidetes (Fig. 5B). Moreover, the MBL encoding gene D505_08350 lacks orthologs in several closely related species including E. meningoseptica ATCC 13253 (Fig. 5C), and appears to share common ancestry with the homologues in some Proteobacteria, particularly of the genus Pseudomonas, which indicates a history of lateral transmission. It is noteworthy that Pseudomonas aeruginosa produces transferable MBLs that recently have been spread to Enterobacteriaceae, likely in a clinical environment [61]. MBLs are also present in environmental microbiota, suggesting that certain taxa, including E. anophelis, potentially could act as reservoirs for lateral gene transfers in nature [62]–[65]. These patterns suggest that the β-lactamase superfamily is adaptive in response to various antimicrobial compounds in different eco-contexts.
Figure 5. Phylogenetic relationship of homologues of three selected lactam degrading enzymes in E. anophelis and other taxa.

(A) D505_10647; (B) D505_08675; (C) D505_08350. Numbers above clades are bootstrap values (1,000 replicates). The trees were constructed by Neighbor Joining criterion implemented in MEGA 5.1. The GenBank accession numbers of the sequences were listed in Table S5.
E. meningoseptica endows two families of wide spectrum MBLs termed GOB [66], [67] and BlaB [68]. Of note, an exceptional genetic diversity of the family members has been reported between clinical isolates from Korea [69]. In line with this, we repeatedly observed a clearly stronger homology for the lactam degrading enzymes between R26T and E. meningoseptica strain 502 than the type strain ATCC 13253 (data not shown). This included the putative GOB/BlaB members, of which most are present E. meningoseptica 502 but only a few in ATCC 13253 [70]. This led us to compare the genome-wide CDS similarities between these two strains and R26T. In comparison to E. anophelis, 85.3% and 69.3% of CDS in E. meningoseptica 502 and ATCC 13253, respectively, display >80% identity. A similar comparison against CDS of E. meningoseptica 502 revealed 85.1% identity to R26T and only 58.7% to ATCC 13253. This unequivocally suggests that E. meningoseptica 502 is more related, albeit not identical to E. anophelis and raises concerns of potential misinterpretations when classifying strains of the genus. More thorough methods are hence needed in order to accurately determine whether isolates of Elizabethkingia sp. belong to either of the described species or represent a novel taxon.
The broad genetic capacity for antibiotic resistance is consistent with the observation that E. anophelis R26T has natural antibiotic resistance to ampicillin, chloramphenicol, kanamycin, streptomycin and tetracycline [12]. In addition, ciprofloxacin that previously has proven moderately potent against clinical isolates of E. meningoseptica [71]–[73] had limited effects on R26T growth (≥50 µg × ml−1), whereas rifampicin [73], [74] displayed cytotoxicity at moderate doses (≥3.125 µg × ml−1). A strong resistance against the insect antimicrobial peptide Cecropin A was also seen, as no bacterial clearance was observed at the highest dose (100 µM; Fig. S4).
The interactions among antibiotic-producing and resistant bacteria may be one of the determinants that shape and stabilize the community structure in the mosquito gut. Similar metagenomic contexts have been demonstrated in natural environments [75] and host associated microbiomes [76], [77]. The inhabitation of multidrug resistant E. anophelis and possibly other bacteria in the mosquito gut will affect the effectiveness of antibiotic treatment in mosquito microbiome research. Utilization of antibiotics may also perturb the community structure by only acting on sensitive bacteria. In summary, Table S4 presents the genes in the category of antibiotic resistance, efflux pumps and heavy metal detoxification, and the similarity to the orthologs in other taxa in the family Flavobacteriaceae.
The antibiotic resistance might have consequences for future work with the E. anophelis. Pathogenicity of the bacterium was recently demonstrated in a clinical case of meningitis in Africa [78] and a hospital outbreak in Singapore [79]. The isolates in both cases were resistant against a wide array of antibiotics. These case reports raised a concern regarding whether or not mosquitoes can pass E. anophelis and E. meningoseptica to humans in clinical situations and when handling mosquitoes in research. Further investigation is required to evaluate these potential risks.
Mevalonate Pathway Utilization for Isoprenoid Synthesis
Isoprenoids comprise the largest group of organic molecules in nature and are involved in a wide variety of biological functions. The universal isoprenoid precursor and building block isopentenyl pyrophosphate (IPP) and its isomer dimethylallyl pyrophosphate (DMAPP) can be synthesized by two independent pathways, the classical mevalonate (MVA) pathway and the alternative 2C-methyl-D-erythtritol 4-phosphate (MEP) pathway. While the MVA pathway is present in higher eukaryotes, plant cytoplasm and archaea, the MEP pathway is generally found in eubacteria and the plastids of plants and apicomplexan parasites. Remnants of enzyme sequences for either pathway are commonly found among bacterial genomes, and in a few bacteria both pathways are active (reviewed in [80]).
Interestingly, opposite to most eubacteria, E. anophelis and flavobacteria appear to utilize the MVA pathway for isoprenoid biosynthesis. Exemplified in Fig. 6, E. anophelis and F. branchiophilum possess all but one of the enzymes known in the MVA pathway and none in the MEP pathway, while Enterobacter sp. Ag1 and Pseudomonas sp. Ag1 have all enzymes in the MEP pathway. A comparison with six other related bacteria indicated this to be a general feature within the family Flavobacteriaceae (Table S6). Whereas the phosphomevalonate kinase (EC 2.7.4.2) seems to be missing, which is not uncommon in bacteria, the phosphorylation of phosphomevalonate is likely mediated via an alternate route in this family [81], [82]. A nonorthologous kinase in the galactokinase/homoserine kinase/mevalonate kinase/phosphomevalonate (GHMP) family could possibly play the role [82], [83]. In the E. anophelis genome, a homoserine kinase coding gene (D505_ 04189) and a galactokinase coding gene (D505_05014) were identified. Vinella et al. showed that the function of the MEP pathway is dependent on the ability to form Fe-S complexes in the IspG and IspH enzymes [84]. Fe-S complexes can be destroyed by nitric oxide (NO) [85], suggesting that possession of the MVA pathway would be conducive to living in a NO-rich environment. Because the production of NO is an essential part of both the mosquito anti-parasitic and anti-bacterial response [86]–[88], the MVA pathway utilizing E. anophelis and E. meningoseptica could have an advantage over other bacteria which have the MEP pathway in the mosquito midgut populations.
Figure 6. Isoprenoid synthesis pathway in four bacterial species.

The color code represents the enzymes that are present in the species.
Antioxidant Capacity
Hematophagous mosquitoes take a blood meal for egg production, the digestion of which changes the gut conditions drastically. Catabolism of hemoglobins results in the release of a large quantity of free heme in the gut lumen. The pro-oxidation of heme increases oxidative stress in the gut environment (reviewed in [89]). A gene encoding the heme-degrading protein HemS (D505_03742) was present in the genome with orthologs found in E. meningoseptica and C. gleum, but not in F. branchiophilum. The HemS protein has been shown to degrade heme in vitro and is required for the defense against oxidative stress upon hydrogen peroxide exposure in Bartonella henselae [90]. The genome also encompasses four hemolysins, one hemolysin D secretion protein and six hemolysin translocator HlyD proteins. These gene products may assemble hemolysin transporters, as demonstrated in E. coli [91], [92]. Hemolytic activity has been demonstrated in the fish pathogen F. psychrophilus [93] and E. meningoseptica [94]. In agreement with these findings, we found that E. anophelis also displays hemolytic activity and the ability to grow in the blood agar (Fig. S5). Following 24 h of incubation, E. anophelis caused a distinct brown discoloration of the blood agar (Fig. S5A). Despite a somewhat different appearance compared to Streptococcus pneumonia (Fig. S5B), the discoloration caused by either of these strains is a typical sign of α-hemolysis. Additional incubation for 24 h resulted in a clear, brown-colored zone adjacent to the growing E. anophelis bacteria. This effect was dependent on the bacterial density as single colonies caused discoloration but no clearance of the agar at this time point. Upon overall comparison, the peptide sequences of the orthologous genes were found with high similarity in E. meningoseptica, C. gleum, and to a less extent in the more divergent F. branchiophilum, indicating a broad conservation within the family Flavobacteriaceae. It is possible that the presence of hemolysins and heme degrading genes aids in the mosquito blood meal digestion. Similar synergism has been observed in Ae. aegypti [6].
Prokaryotic cells employ two redox-sensing regulons, OxyR and SoxRS, to sense oxidative stress signals and subsequently activate defense mechanisms [95]. The SoxRS regulon has not been found in Bacteroidetes [96], [97]. The oxyR gene (D505_12281) is, however, present in the E. anophelis genome and is located in proximity to the antioxidant genes catalase (D505_12286) and manganese superoxide dismutase (MnSOD) (D505_12271). This genomic arrangement is unique to E. anophelis; no synteny was found in other flavobacteria in this study, although oxyR was found in other flavobacteria. In addition, other genes in the oxidative stress category are present, including those encoding alkyl hydroperoxide reductases (AhpC/F), DNA-binding protein from starved cells (Dps), thioredoxins (Trxs), glutaredoxins (Grxs) and glutathione (GSH) peroxidase. OxyR controls the expression of catalase, ahpC, ahpF and dps in response to H2O2 [97], [98]. The genetic antioxidant capacity may contribute to the persistence of E. anophelis in the gut [2].
Summary
The genome annotation provides insights into the capabilities of E. anophelis, and sheds light on its symbiotic relationships with the mosquito host and other members of the microbiome. The predominance of E. anophelis in the gut ecosystem of mosquitoes may represent an evolutionary fit. Firstly, the presence of a large number of TonB dependent receptors with numerous substrate specificities endows the bacteria with a sufficient capacity to acquire and utilize various biopolymers. Particularly, the polysaccharide utilization system supplies consumable carbohydrates for both microbial residents and host following the intake of nectar as well as cellulose. Secondly, the multidrug efflux pump genes of the RND and MATE superfamilies allow the bacteria to extrude heavy metals, microbicides and other toxic chemical compounds and maintain a homeostatic internal environment. Various antibiotic resistance mechanisms may serve as guardians for maintaining community structure in the gut microbial community. The interactions among antibiotic-producing and resistant bacteria may be one of the determinants that shape and stabilize the community structure. Similar metagenomic contexts have been demonstrated in natural environments [75] and host associated microbiomes [76], [77]. On the other hand, as an opportunistic pathogen, the multidrug resistance of E. anophelis renders infections very troublesome. Finally, the antioxidant capacity endows E. anophelis with defense against oxidative stress associated with blood digestion. The genome database provides a reference for further characterization of the mosquito gut microbiome and its impact on mosquito life traits.
Supporting Information
PCR verification of the putative phage insert in Ag1 not present in R26T. (A) Primers were designed to flank the insertion sites of a phage like segment in Ag1. Primer pairs F1–R1 and F2–R2 were expected to yield 650 and 500 bp amplicons, respectively, in Ag1. The F1–R2 pair was expected to yield a 566 bp amplicon in R26, whereas no amplification was expected in Ag1 due to the large size of the insert (>35 kb). (B) Expected PCR products and sizes were confirmed using agarose gel electrophoresis.
(TIF)
Phylogenetic relationship of E. anophelis relative to the taxa in the family Flavobacteriaceae inferred from 16 S ribosomal DNA (A) and rpoB gene (B). Numbers above clades are bootstrap values (1000 replicates). The trees were constructed by Neighbor Joining criterion implemented in MEGA 5.1.
(TIF)
Graphic view of conserved domains in TonB dependent transporters. The protein ID (GenBank accession #) was given for each protein. Detailed domain information can be found in NCBI Conserved Domains database.
(TIF)
Drug resistance and growth inhibitory capacity of E. anophelis . Representative figure of the drug resistance displayed by E. anophelis R26T. Plates were cast using a mixture of 5×104 bacteria in 6 ml Lysogeny broth and 1% SeaPlaque agarose (FMC BioProducts). Ciprofloxacin (100 µg/ml) and Cecropin A (100 µM) were added in a two-fold dilution series counter-clockwise in 2 mm wide holes with the lowest dose in the center and allowed to diffuse at ambient temperature for 30 min before incubation at 37°C for 24 h or until growth was apparent. E. coli strain D31 was used for comparison.
(TIF)
E. anophelis displays α-hemolytic activity. (A) Representative images taken from above (left plate) or underneath (right plate) at 48 h post inoculation. Black and white arrows depict the brown discoloration and clearance of the blood agar, respectively that were observed adjacent to the bacteria. The density dependence of the clearance zone was clear when observing the plate from underneath (right plate, white arrow). The right panel depicts the brown discoloration caused by individual colonies. (B) Control strains displaying the different types of hemolysis.
(TIF)
Draft genome statistics.
(XLSX)
Coding sequence comparison between R26T and Ag1 of E. anophelis .
(XLSX)
Carbohydrate-active-enzyme proteins in the E. anophelis genome.
(XLSX)
Drug resistance genes in E. anophelis and similarity to related genomes. Numbers reflect percent identity of the best hit in related genomes to each gene in R26T. E. m., Elizabethkingia meningoseptica; C. g., Chryseobacterium gleum; F. b., Flavobacterium branchiophilum.
(XLSX)
Sequence ID used in Figure 6 . Homologous sequences from related taxa and GenBank accession numbers were provided.
(XLSX)
Mevalonate pathway genes in different species. E. m., Elizabethkingia meningoseptica; C. g., Chryseobacterium gleum; F. b., Flavobacterium branchiophilum; R. a., Riemerella anatipestifer; F. j., Flavobacterium johnsoniae UW101; C. sp., Chryseobacterium sp. CF314; F. ba., Flavobacteriaceae bacterium 3519-10; F. sp., Flavobacterium sp. F52.
(XLSX)
Funding Statement
JX was supported by grants from the National Institute of General Medical Sciences, the National Institutes of Health (1SC2GM092789, 1SC1AI112786) and National Science Foundation (DMS-1222592). MS was an undergraduate research scholar supported by the NMSU Howard Hughes Medical Institute (HHMI) research scholars program. IF was supported by a grant from the Research Infrastructure - Integrating Activity project supported by the European Community (INFRAVEC) FP7 Infrastructures (grant agreement number: 228421). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lindh JM, Borg-Karlson AK, Faye I (2008) Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop 107: 242–250. [DOI] [PubMed] [Google Scholar]
- 2. Wang Y, Gilbreath TM 3rd, Kukutla P, Yan G, Xu J (2011) Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6: e24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gusmao DS, Santos AV, Marini DC, Bacci M Jr, Berbert-Molina MA, et al. (2010) Culture-dependent and culture-independent characterization of microorganisms associated with Aedes aegypti (Diptera: Culicidae) (L.) and dynamics of bacterial colonization in the midgut. Acta Trop 115: 275–281. [DOI] [PubMed] [Google Scholar]
- 4. Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK (2009) Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiology 9: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boissiere A, Tchioffo MT, Bachar D, Abate L, Marie A, et al. (2012) Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog 8: e1002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gaio Ade O, Gusmao DS, Santos AV, Berbert-Molina MA, Pimenta PF, et al. (2011) Contribution of midgut bacteria to blood digestion and egg production in aedes aegypti (diptera: culicidae) (L.). Parasit & vect 4: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dong Y, Dimopoulos G (2009) Anopheles fibrinogen-related proteins provide expanded pattern recognition capacity against bacteria and malaria parasites. J Biol Chem 284: 9835–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meister S, Agianian B, Turlure F, Relogio A, Morlais I, et al. (2009) Anopheles gambiae PGRPLC-mediated defense against bacteria modulates infections with malaria parasites. PLoS Pathog 5: e1000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim KK, Kim MK, Lim JH, Park HY, Lee ST (2005) Transfer of Chryseobacterium meningosepticum and Chryseobacterium miricola to Elizabethkingia gen. nov. as Elizabethkingia meningoseptica comb. nov. and Elizabethkingia miricola comb. nov. Int J Syst Evol Microbiol 55: 1287–1293. [DOI] [PubMed] [Google Scholar]
- 10. Ngwa CJ, Glockner V, Abdelmohsen UR, Scheuermayer M, Fischer R, et al. (2013) 16S rRNA gene-based identification of Elizabethkingia meningoseptica (Flavobacteriales: Flavobacteriaceae) as a dominant midgut bacterium of the Asian malaria vector Anopheles stephensi (Dipteria: Culicidae) with antimicrobial activities. J Med Entomol 50: 404–414. [DOI] [PubMed] [Google Scholar]
- 11. Terenius O, Lindh JM, Eriksson-Gonzales K, Bussiere L, Laugen AT, et al. (2012) Midgut bacterial dynamics in Aedes aegypti. FEMS Microbiol Ecol 80: 556–565. [DOI] [PubMed] [Google Scholar]
- 12. Kampfer P, Matthews H, Glaeser SP, Martin K, Lodders N, et al. (2011) Elizabethkingia anophelis sp. nov., isolated from the midgut of the mosquito Anopheles gambiae. Int J Syst Evol Microbiol 61: 2670–2675. [DOI] [PubMed] [Google Scholar]
- 13.Kukutla P, Lindberg BG, Pei D, Rayl M, Yu W, et al.. (2013) Draft Genome Sequences of Elizabethkingia anophelis Strains R26T and Ag1 from the Midgut of the Malaria Mosquito Anopheles gambiae. Genome Announc 1: pii: e01030–01013. [DOI] [PMC free article] [PubMed]
- 14. Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, et al. (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acid Research 33: 5691–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, et al. (2011) CDD: a Conserved Domain Database for the functional annotation of proteins. Nucl Acid Res 39: D225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC (2010) CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiol 20: 1574–1584. [DOI] [PubMed] [Google Scholar]
- 18. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, et al. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucl Acid Res 37: D233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acid Res 42: D490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown SD, Utturkar SM, Klingeman DM, Johnson CM, Martin SL, et al. (2012) Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoides. J Bacteriol 194: 5991–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McBride MJ, Xie G, Martens EC, Lapidus A, Henrissat B, et al. (2009) Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl Environ Microbiol 75: 6864–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Touchon M, Barbier P, Bernardet JF, Loux V, Vacherie B, et al. (2011) Complete genome sequence of the fish pathogen Flavobacterium branchiophilum. Appl Environ Microbiol 77: 7656–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS One 5: e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hultmark D, Engstrom A, Bennich H, Kapur R, Boman HG (1982) Insect immunity: isolation and structure of cecropin D and four minor antibacterial components from Cecropia pupae. Eur J Biochem 127: 207–217. [DOI] [PubMed] [Google Scholar]
- 26. Noinaj N, Guillier M, Barnard TJ, Buchanan SK (2010) TonB-dependent transporters: regulation, structure, and function. Ann Rev Microbiol 64: 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schauer K, Rodionov DA, de Reuse H (2008) New substrates for TonB-dependent transport: do we only see the ‘tip of the iceberg’? Trends Biochem Sci 33: 330–338. [DOI] [PubMed] [Google Scholar]
- 28. Krewulak KD, Vogel HJ (2011) TonB or not TonB: is that the question? Biochem Cell Biol 89: 87–97. [DOI] [PubMed] [Google Scholar]
- 29. Martens EC, Koropatkin NM, Smith TJ, Gordon JI (2009) Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem 284: 24673–24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jakes KS, Cramer WA (2012) Border crossings: colicins and transporters. Ann Rev Genet 46: 209–231. [DOI] [PubMed] [Google Scholar]
- 31. Mirus O, Strauss S, Nicolaisen K, von Haeseler A, Schleiff E (2009) TonB-dependent transporters and their occurrence in cyanobacteria. BMC Biol 7: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alvarez C, Kukutla P, Jiang J, Yu W, Xu J (2012) Draft genome sequence of Pseudomonas sp. strain Ag1, isolated from the midgut of the malaria mosquito Anopheles gambiae. J Bacteriol 194: 5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang J, Alvarez C, Kukutla P, Yu W, Xu J (2012) Draft genome sequences of Enterobacter sp. isolate Ag1 from the midgut of the malaria mosquito Anopheles gambiae. J Bacteriol 194: 5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schauer K, Rodionov DA, de Reuse H (2008) New substrates for TonB-dependent transport: do we only see the ‘tip of the iceberg’? Trends Biochem Sci 33: 330–338. [DOI] [PubMed] [Google Scholar]
- 35. Fernandez-Gomez B, Richter M, Schuler M, Pinhassi J, Acinas SG, et al. (2013) Ecology of marine Bacteroidetes: a comparative genomics approach. The ISME journal 7: 1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang K, Jiao N, Liu K, Zhang Y, Li S (2012) Distribution and functions of TonB-dependent transporters in marine bacteria and environments: implications for dissolved organic matter utilization. PloS One 7: e41204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, et al. (2007) Plant carbohydrate scavenging through tonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One 2: e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, et al. (2003) A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299: 2074–2076. [DOI] [PubMed] [Google Scholar]
- 39. Flint HJ, Scott KP, Duncan SH, Louis P, Forano E (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3: 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, et al. (2011) Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol 9: e1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bolam DN, Koropatkin NM (2012) Glycan recognition by the Bacteroidetes Sus-like systems. Current Opinion Structural Biol 22: 563–569. [DOI] [PubMed] [Google Scholar]
- 42. Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, et al. (2010) Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell 141: 1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kolton M, Sela N, Elad Y, Cytryn E (2013) Comparative Genomic Analysis Indicates that Niche Adaptation of Terrestrial Flavobacteria Is Strongly Linked to Plant Glycan Metabolism. PloS One 8: e76704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gary RE Jr, Foster WA (2004) Anopheles gambiae feeding and survival on honeydew and extra-floral nectar of peridomestic plants. Med Vet Entomol 18: 102–107. [DOI] [PubMed] [Google Scholar]
- 45. Impoinvil DE, Kongere JO, Foster WA, Njiru BN, Killeen GF, et al. (2004) Feeding and survival of the malaria vector Anopheles gambiae on plants growing in Kenya. Med Vet Entomol 18: 108–115. [DOI] [PubMed] [Google Scholar]
- 46. Souza-Neto JA, Machado FP, Lima JB, Valle D, Ribolla PE (2007) Sugar digestion in mosquitoes: identification and characterization of three midgut alpha-glucosidases of the neo-tropical malaria vector Anopheles aquasalis (Diptera: Culicidae). Comp Biochem Physiol A Mol Integr Physiol 147: 993–1000. [DOI] [PubMed] [Google Scholar]
- 47. Billingsley PF, Hecker H (1991) Blood digestion in the mosquito, Anopheles stephensi Liston (Diptera: Culicidae): activity and distribution of trypsin, aminopeptidase, and alpha-glucosidase in the midgut. J Med Entomol 28: 865–871. [DOI] [PubMed] [Google Scholar]
- 48. Schlein Y, Muller G (1995) Assessment of Plant Tissue Feeding by Sand Flies (Diptera: Psychodidae) and Mosquitoes (Diptera: Culicidae). J Med Entomol 32: 882–887. [DOI] [PubMed] [Google Scholar]
- 49. Junnila A, Muller GC, Schlein Y (2010) Species identification of plant tissues from the gut of An. sergentii by DNA analysis. Acta Trop 115: 227–233. [DOI] [PubMed] [Google Scholar]
- 50. Muller G, Schlein Y (2005) Plant tissues: the frugal diet of mosquitoes in adverse conditions. Medical and veterinary entomology 19: 413–422. [DOI] [PubMed] [Google Scholar]
- 51. Saier MH Jr, Paulsen IT (2001) Phylogeny of multidrug transporters. Semin Cell Dev Biol 12: 205–213. [DOI] [PubMed] [Google Scholar]
- 52. Wong K, Ma J, Rothnie A, Biggin PC, Kerr ID (2014) Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem Sci 39: 8–16. [DOI] [PubMed] [Google Scholar]
- 53. Alvarez-Ortega C, Olivares J, Martinez JL (2013) RND multidrug efflux pumps: what are they good for? Front Microbiol 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blair JM, Piddock LJ (2009) Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr Opin Microbiol 12: 512–519. [DOI] [PubMed] [Google Scholar]
- 55. Michel C, Matte-Tailliez O, Kerouault B, Bernardet JF (2005) Resistance pattern and assessment of phenicol agents’ minimum inhibitory concentration in multiple drug resistant Chryseobacterium isolates from fish and aquatic habitats. J Appl Microbiol 99: 323–332. [DOI] [PubMed] [Google Scholar]
- 56. Clark SE, Jude BA, Danner GR, Fekete FA (2009) Identification of a multidrug efflux pump in Flavobacterium johnsoniae. Vet Res 40: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wexler HM (2012) Pump it up: occurrence and regulation of multi-drug efflux pumps in Bacteroides fragilis. Anaerobe 18: 200–208. [DOI] [PubMed] [Google Scholar]
- 58. Saidijam M, Benedetti G, Ren Q, Xu Z, Hoyle CJ, et al. (2006) Microbial drug efflux proteins of the major facilitator superfamily. Curr Drug Targets 7: 793–811. [DOI] [PubMed] [Google Scholar]
- 59. Law CJ, Maloney PC, Wang DN (2008) Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62: 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fisher JF, Meroueh SO, Mobashery S (2005) Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem Rev 105: 395–424. [DOI] [PubMed] [Google Scholar]
- 61. Walsh TR, Toleman MA, Poirel L, Nordmann P (2005) Metallo-beta-lactamases: the quiet before the storm? Clin Microbiol Rev 18: 306–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Saavedra MJ, Peixe L, Sousa JC, Henriques I, Alves A, et al. (2003) Sfh-I, a subclass B2 metallo-beta-lactamase from a Serratia fonticola environmental isolate. Antimicrob Agents Chemother 47: 2330–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rossolini GM, Condemi MA, Pantanella F, Docquier JD, Amicosante G, et al. (2001) Metallo-beta-lactamase producers in environmental microbiota: new molecular class B enzyme in Janthinobacterium lividum. Antimicrob Agents Chemother 45: 837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Simm AM, Higgins CS, Pullan ST, Avison MB, Niumsup P, et al. (2001) A novel metallo-beta-lactamase, Mbl1b, produced by the environmental bacterium Caulobacter crescentus. FEBS Lett 509: 350–354. [DOI] [PubMed] [Google Scholar]
- 65. Stoczko M, Frere JM, Rossolini GM, Docquier JD (2006) Postgenomic scan of metallo-beta-lactamase homologues in rhizobacteria: identification and characterization of BJP-1, a subclass B3 ortholog from Bradyrhizobium japonicum. Antimicrob Agents Chemother 50: 1973–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moran-Barrio J, Gonzalez JM, Lisa MN, Costello AL, Peraro MD, et al. (2007) The metallo-beta-lactamase GOB is a mono-Zn(II) enzyme with a novel active site. J Biol Chem 282: 18286–18293. [DOI] [PubMed] [Google Scholar]
- 67. Horsfall LE, Izougarhane Y, Lassaux P, Selevsek N, Lienard BM, et al. (2011) Broad antibiotic resistance profile of the subclass B3 metallo-beta-lactamase GOB-1, a di-zinc enzyme. FEBS J 278: 1252–1263. [DOI] [PubMed] [Google Scholar]
- 68. Gonzalez LJ, Vila AJ (2012) Carbapenem resistance in Elizabethkingia meningoseptica is mediated by metallo-beta-lactamase BlaB. Antimicrob Agents Chemother 56: 1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yum JH, Lee EY, Hur SH, Jeong SH, Lee H, et al. (2010) Genetic diversity of chromosomal metallo-beta-lactamase genes in clinical isolates of Elizabethkingia meningoseptica from Korea. J Microbiol 48: 358–364. [DOI] [PubMed] [Google Scholar]
- 70.Matyi SA, Hoyt PR, Hosoyama A, Yamazoe A, Fujita N, et al.. (2013) Draft Genome Sequences of Elizabethkingia meningoseptica. Genome Announc 1. [DOI] [PMC free article] [PubMed]
- 71. Amer MZ, Bandey M, Bukhari A, Nemenqani D (2011) Neonatal meningitis caused by Elizabethkingia meningoseptica in Saudi Arabia. J Infect Dev Ctries 5: 745–747. [DOI] [PubMed] [Google Scholar]
- 72. Neuner EA, Ahrens CL, Groszek JJ, Isada C, Vogelbaum MA, et al. (2012) Use of therapeutic drug monitoring to treat Elizabethkingia meningoseptica meningitis and bacteraemia in an adult. J Antimicrob Chemother 67: 1558–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lin XH, Xu YH, Sun XH, Huang Y, Li JB (2012) Genetic diversity analyses of antimicrobial resistance genes in clinical Chryseobacterium meningosepticum isolated from Hefei, China. Int J Antimicrob Agents 40: 186–188. [DOI] [PubMed] [Google Scholar]
- 74. Di Pentima MC, Mason EO Jr, Kaplan SL (1998) In vitro antibiotic synergy against Flavobacterium meningosepticum: implications for therapeutic options. Clin Infect Dis 26: 1169–1176. [DOI] [PubMed] [Google Scholar]
- 75. Monier JM, Demaneche S, Delmont TO, Mathieu A, Vogel TM, et al. (2011) Metagenomic exploration of antibiotic resistance in soil. Curr Opin Microbiol 14: 229–235. [DOI] [PubMed] [Google Scholar]
- 76. Schmieder R, Edwards R (2012) Insights into antibiotic resistance through metagenomic approaches. Future Microbiol 7: 73–89. [DOI] [PubMed] [Google Scholar]
- 77. Sommer MO, Church GM, Dantas G (2010) The human microbiome harbors a diverse reservoir of antibiotic resistance genes. Virulence 1: 299–303. [DOI] [PubMed] [Google Scholar]
- 78. Frank T, Gody JC, Nguyen LB, Berthet N, Le Fleche-Mateos A, et al. (2013) First case of Elizabethkingia anophelis meningitis in the Central African Republic. Lancet 381: 1876. [DOI] [PubMed] [Google Scholar]
- 79. Teo J, Tan SY, Tay M, Ding Y, Kjelleberg S, et al. (2013) First case of E anophelis outbreak in an intensive-care unit. Lancet 382: 855–856. [DOI] [PubMed] [Google Scholar]
- 80. Heuston S, Begley M, Gahan CG, Hill C (2012) Isoprenoid biosynthesis in bacterial pathogens. Microbiol 158: 1389–1401. [DOI] [PubMed] [Google Scholar]
- 81. Miziorko HM (2011) Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys 505: 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lombard J, Moreira D (2011) Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol Biol Evol 28: 87–99. [DOI] [PubMed] [Google Scholar]
- 83. Andreassi JL 2nd, Leyh TS (2004) Molecular functions of conserved aspects of the GHMP kinase family. Biochem 43: 14594–14601. [DOI] [PubMed] [Google Scholar]
- 84. Vinella D, Brochier-Armanet C, Loiseau L, Talla E, Barras F (2009) Iron-sulfur (Fe/S) protein biogenesis: phylogenomic and genetic studies of A-type carriers. PLoS Genetics 5: e1000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Reddy D, Lancaster JR Jr, Cornforth DP (1983) Nitrite inhibition of Clostridium botulinum: electron spin resonance detection of iron-nitric oxide complexes. Science 221: 769–770. [DOI] [PubMed] [Google Scholar]
- 86. Luckhart S, Vodovotz Y, Cui LW, Rosenberg R (1998) The mosquito Anopheles stephensi limits malaria parasite development with inducible synthesis of nitric oxide. Proc Natl Acad Sci U S A 95: 5700–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hillyer JF, Estevez-Lao TY (2010) Nitric oxide is an essential component of the hemocyte-mediated mosquito immune response against bacteria. Dev Comp Immunol 34: 141–149. [DOI] [PubMed] [Google Scholar]
- 88. Kumar S, Molina-Cruz A, Gupta L, Rodrigues J, Barillas-Mury C (2010) A peroxidase/dual oxidase system modulates midgut epithelial immunity in Anopheles gambiae. Science 327: 1644–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Graca-Souza AV, Maya-Monteiro C, Paiva-Silva GO, Braz GR, Paes MC, et al. (2006) Adaptations against heme toxicity in blood-feeding arthropods. Insect Biochem Mol Biol 36: 322–335. [DOI] [PubMed] [Google Scholar]
- 90. Liu M, Boulouis HJ, Biville F (2012) Heme degrading protein HemS is involved in oxidative stress response of Bartonella henselae. PloS One 7: e37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lee M, Jun SY, Yoon BY, Song S, Lee K, et al. (2012) Membrane fusion proteins of type I secretion system and tripartite efflux pumps share a binding motif for TolC in gram-negative bacteria. PloS one 7: e40460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pimenta AL, Racher K, Jamieson L, Blight MA, Holland IB (2005) Mutations in HlyD, part of the type 1 translocator for hemolysin secretion, affect the folding of the secreted toxin. J Bacteriol 187: 7471–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hogfors-Ronnholm E, Wiklund T (2010) Hemolytic activity in Flavobacterium psychrophilum is a contact-dependent, two-step mechanism and differently expressed in smooth and rough phenotypes. Microbial Pathogenesis 49: 369–375. [DOI] [PubMed] [Google Scholar]
- 94. Kawai Y, Yano I, Kaneda K (1988) Various kinds of lipoamino acids including a novel serine-containing lipid in an opportunistic pathogen Flavobacterium. Their structures and biological activities on erythrocytes. Eur J Biochem 171: 73–80. [DOI] [PubMed] [Google Scholar]
- 95. Zheng M, Storz G (2000) Redox sensing by prokaryotic transcription factors. Biochem Pharmacol 59: 1–6. [DOI] [PubMed] [Google Scholar]
- 96. Dietrich LE, Teal TK, Price-Whelan A, Newman DK (2008) Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321: 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rocha ER, Herren CD, Smalley DJ, Smith CJ (2003) The complex oxidative stress response of Bacteroides fragilis: the role of OxyR in control of gene expression. Anaerobe 9: 165–173. [DOI] [PubMed] [Google Scholar]
- 98. Sund CJ, Rocha ER, Tzianabos AO, Wells WG, Gee JM, et al. (2008) The Bacteroides fragilis transcriptome response to oxygen and H2O2: the role of OxyR and its effect on survival and virulence. Mol Microbiol 67: 129–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PCR verification of the putative phage insert in Ag1 not present in R26T. (A) Primers were designed to flank the insertion sites of a phage like segment in Ag1. Primer pairs F1–R1 and F2–R2 were expected to yield 650 and 500 bp amplicons, respectively, in Ag1. The F1–R2 pair was expected to yield a 566 bp amplicon in R26, whereas no amplification was expected in Ag1 due to the large size of the insert (>35 kb). (B) Expected PCR products and sizes were confirmed using agarose gel electrophoresis.
(TIF)
Phylogenetic relationship of E. anophelis relative to the taxa in the family Flavobacteriaceae inferred from 16 S ribosomal DNA (A) and rpoB gene (B). Numbers above clades are bootstrap values (1000 replicates). The trees were constructed by Neighbor Joining criterion implemented in MEGA 5.1.
(TIF)
Graphic view of conserved domains in TonB dependent transporters. The protein ID (GenBank accession #) was given for each protein. Detailed domain information can be found in NCBI Conserved Domains database.
(TIF)
Drug resistance and growth inhibitory capacity of E. anophelis . Representative figure of the drug resistance displayed by E. anophelis R26T. Plates were cast using a mixture of 5×104 bacteria in 6 ml Lysogeny broth and 1% SeaPlaque agarose (FMC BioProducts). Ciprofloxacin (100 µg/ml) and Cecropin A (100 µM) were added in a two-fold dilution series counter-clockwise in 2 mm wide holes with the lowest dose in the center and allowed to diffuse at ambient temperature for 30 min before incubation at 37°C for 24 h or until growth was apparent. E. coli strain D31 was used for comparison.
(TIF)
E. anophelis displays α-hemolytic activity. (A) Representative images taken from above (left plate) or underneath (right plate) at 48 h post inoculation. Black and white arrows depict the brown discoloration and clearance of the blood agar, respectively that were observed adjacent to the bacteria. The density dependence of the clearance zone was clear when observing the plate from underneath (right plate, white arrow). The right panel depicts the brown discoloration caused by individual colonies. (B) Control strains displaying the different types of hemolysis.
(TIF)
Draft genome statistics.
(XLSX)
Coding sequence comparison between R26T and Ag1 of E. anophelis .
(XLSX)
Carbohydrate-active-enzyme proteins in the E. anophelis genome.
(XLSX)
Drug resistance genes in E. anophelis and similarity to related genomes. Numbers reflect percent identity of the best hit in related genomes to each gene in R26T. E. m., Elizabethkingia meningoseptica; C. g., Chryseobacterium gleum; F. b., Flavobacterium branchiophilum.
(XLSX)
Sequence ID used in Figure 6 . Homologous sequences from related taxa and GenBank accession numbers were provided.
(XLSX)
Mevalonate pathway genes in different species. E. m., Elizabethkingia meningoseptica; C. g., Chryseobacterium gleum; F. b., Flavobacterium branchiophilum; R. a., Riemerella anatipestifer; F. j., Flavobacterium johnsoniae UW101; C. sp., Chryseobacterium sp. CF314; F. ba., Flavobacteriaceae bacterium 3519-10; F. sp., Flavobacterium sp. F52.
(XLSX)