

Abstract
Background
Recent work has shown that Fusarium species and genotypes most commonly associated with human infections, particularly of the cornea (mycotic keratitis), are the same as those most commonly isolated from plumbing systems. The species most dominant in plumbing biofilms is Fusarium keratoplasticum, a cosmopolitan fungus known almost exclusively from animal infections and biofilms. To better understand its diversity and population dynamics, we developed and utilized a nine-locus sequence-based typing system to make inferences about clonality, recombination, population structure, species boundaries and hybridization.
Results
High levels of genetic diversity and evidence for recombination and clonality were detected among 75 clinical and 156 environmental isolates of Fusarium keratoplasticum. The multilocus sequence typing system (MLST) resolved 111 unique nine-locus sequence types (STs). The single locus bifactorial determinants of mating compatibility (mating types MAT1-1 and MAT1-2), were found in a ratio of 70:30. All but one of the 49 isolates of the most common ST (FSSC 2d-2) came from human infections, mostly of the cornea, and from biofilms associated with contact lenses and plumbing surfaces. Significant levels of phylogenetic incongruence were found among loci. Putative clonal relationships among genotypes were estimated, showing a mixture of large clonal complexes and unrelated singletons. Discordance between the nuclear ribosomal rRNA and other gene genealogies is consistent with introgression of ribosomal RNA alleles of phylogenetic species FSSC 9 into F. keratoplasticum. No significant population subdivision based on clinical versus non-clinical sources was found.
Conclusions
Incongruent phylogenetic trees and the presence of both mating types within otherwise identical STs were observed, providing evidence for sexuality in F. keratoplasticum. Cryptic speciation suggested in a published three-locus MLST system was not supported with the addition of new loci, but evidence of introgression of ribosomal RNA genes from another strongly supported phylogenetic species (FSSC 9), also known from plumbing systems and human infections, was detected in two isolates. Overall, F. keratoplasticum is a diverse and geographically unstructured species with a mixed clonal and recombinant life history.
Keywords: Biofilm, Clonality, Fusarium, Hybridization, MLST, Mycotic pathogen, Population dynamics, Recombination
Background
Fusarium is a large cosmopolitan genus of filamentous fungi best-known for causing a plethora of economically important plant diseases and mycotoxicoses [1]. Fusaria also cause human infections ranging from sinusitis, pneumonia, and localized skin lesions to life-threatening disseminated mycoses in immune-compromised and immune-suppressed individuals [2]. Along with Aspergillus, it is a frequent cause of trauma-associated mycotic keratitis in the tropics [3] and it is a predominant cause of contact-lens associated mycotic keratitis worldwide [4-9]. Fusarium is noted for its resistance to the broad-spectrum of antifungal drugs currently available [10]. Multilocus sequence typing (MLST) is a common and powerful tool for elucidating the diversity and population biology of microbial pathogens [11-13]. Several MLST systems have been developed for studying the genetic diversity and population biology of fungal human pathogens including, among others, Aspergillus [14], Blastomyces [15], Candida [16], Coccidioides [17], Cryptococcus [18], Histoplasma [19], and Pneumocystis [20]. Several studies utilizing MLST and genealogical concordance phylogenetic species recognition in Fusarium have revealed strongly supported species boundaries and have elucidated the spectrum of fusaria associated with human pathogenicity [4,9,12,21-27]. The application of MLST to mycotic pathogens has identified epidemic clones [28], uncovered signatures of linkage equilibrium associated with sexual recombination [28-30], and revealed cryptic speciation [15,31,32]. Typing systems are also valuable for epidemiological inference and for providing a framework for understanding the distribution of important phenotypes within species such as antimicrobial drug resistance [24].
Knowledge of species boundaries is a prerequisite for studying intraspecific population dynamics [33], and through several molecular systematics studies, the phylogenetic diversity of clinical fusaria has been elucidated [22-24]. Molecular markers useful for phylogenetics tend to utilize coding and intron sequences of various genes and have proved useful for distinguishing not only species, but also common intraspecific sequence types (STs) [26,34-36]. A three-locus MLST system developed for clinically relevant members of the species-rich Fusarium solani species complex (FSSC), which are all nested with Clade 3, utilizes an intron-rich portion of the translation elongation factor 1-alpha gene (TEF), a portion of the nuclear ribosomal RNA gene repeat that includes the internal transcribed spacer (ITS) regions and D1-D2 region of the nuclear large ribosomal RNA subunit gene (rDNA), and the gene encoding RNA polymerase II second largest subunit (RPB2) [24]. This system resolves ~60 phylogenetic species within the FSSC and distinguishes over 300 genetically distinct STs within Clade 3 of the FSSC [24].
Fusarium continues to be a nosocomial problem and MLST studies have highlighted the prevalence of certain widespread STs in hospital environments [37]. Members of the F. oxysporum species complex (e.g., FOSC ST 33) [25,37] and the FSSC [38], specifically F. petroliphilum (e.g., FSSC 1-a) and F. keratoplasticum (e.g., FSSC 2-d) [39], are the fusaria most commonly identified in hospital water systems employing MLST. Clinically important STs within F. keratoplasticum and other fusaria appear to be ubiquitous in indoor plumbing [40,41]. Moreover, multiple STs that are associated with cases of contact lens-associated mycotic keratitis [4] may also be found in this environment [4,42]. In fact, based on current analyses [39,41], F. keratoplasticum is the single most common Fusarium species associated with human infections.
Although its monophyly is well-supported [9,21,24,43], F. keratoplasticum (FSSC 2) does not possess any distinctive morphological characters and is highly variable in culture [39], so morphological identification is impossible. Therefore, a three-locus MLST system was developed, which resolved 55 unique STs within F. keratoplasticum primarily from clinical sources [24]. In a subsequent expanded analysis, nearly half (107/231) of the isolates from clinical and environmental sources belonged to a single ST, FSSC 2-d [39]. In addition to supporting its monophyly, the combined three-locus phylogeny showed a moderately supported partition within F. keratoplasticum, suggesting that further genetic subdivision may exist within this species [24]. Also, evidence suggesting introgressive hybridization between F. keratoplasticum and a second FSSC species (FSSC 9) was reported [39].
To better understand the population biology of F. keratoplasticum, the three-locus MLST system was expanded to include six novel sequence-based markers, developed using the complete genome sequence of a related species, FSSC 11 (F. solani f. sp pisi) [44]. In addition, a PCR assay for mating-type determination [39] was utilized as a tenth locus to discriminate STs and investigate the potential for sexual reproduction. These markers were used to screen environmental and clinical isolates to infer the intraspecific diversity and relative roles of recombination, clonality, and hybridization in F. keratoplasticum.
Methods
Isolates of F. keratoplasticum
We analyzed 231 F. keratoplasticum isolates collected from a variety of sources and geographic origins in this study. Sources included human infections (N = 76), plumbing drains (N = 110), as well as veterinary infections, soil and various anthropogenic substrates (Additional file 1: Table S1). The human isolates presented in this paper have come from many sources over decades and were collected in a clinical setting. The isolates were sent from clinics for identification. We do not request or maintain personal information about the patients, and only keep data on the body part/disease associated with the isolate, and its general geographic source. Although isolations from six continents are represented, 84% of the isolates were from North America.
Locus development and primer design
Six new sequence markers (loci 3968, 3972, 4081, 6512, 5437, and 6549) were developed in this study (Tables 1 and 2), which were utilized in combination with the existing 3-locus system [21,24,45]. Based on the optical map of F. solani f. sp. pisi (FSSC 11; also known as ‘Nectria’ haematococca Mating Population VI (NhMPVI)), the six new markers are predicted to reside on four different chromosomes. Markers were chosen based on the presence of single-nucleotide polymorphisms (SNPs) as well as microsatellites and insertion/deletion polymorphisms (indels) in 20 isolates of F. keratoplasticum and other species in the FSSC (Table 1). The complete genome was downloaded [46] and searched for 500–800 bp regions containing perfect microsatellite repeats using Tandem Repeats Finder [47]. Flanking primers were designed using Primer3 [48].
Table 1.
Characteristics of loci employed
| Locus 1 | Genome position in MPVI 2 | P. I. chars. 3 | SNPs 4 | No. of indels/Msats: no. of alleles for each 5 | C. I. 6 |
|---|---|---|---|---|---|
|
TEF |
sca_2_chr3_3_0: 2429595-2430256 |
18 |
23 |
4 : 3,3,2,2s |
0.75 |
| rDNA |
Unknown |
9 |
6* |
None observed |
0.9 |
|
RPB2 |
sca_20_chr6_4_0: 1850977-1852794 |
14 |
21 |
None observed |
1 |
| 3968 |
sca_8_chr1_1_0: 1137337-1138068 |
11 |
17 |
2 : 2s, 2 |
0.95 |
| 3972 |
sca_82_chr10_2_0: 791230-792036 |
10 |
17 |
None observed |
1 |
| 4081 |
sca_8_chr1_1_0: 544046-544635 |
13 |
28 |
5 : 2,5,2s,2s,2s |
0.95 |
| 6512 |
sca_26_chr2_2_0: 74409-74990 |
9 |
12 |
2 : 2s, 2s |
0.93 |
| 5439 |
sca_37_chr_6_2_0: 374081-374828 |
6 |
8 |
4 : 2s,2,2s,2s |
1 |
| 5437 |
sca_37_chr_6_2_0: 78042-78546 |
25 |
27 |
2 : 11,2 |
0.95 |
| MAT | sca_2_chr3_3_0: N.A. | N.A. | N.A. | N.A. | N.A. |
1. Name of locus 2. Scaffold, chromosome and inclusive base pairs of targeted sequences in Fusarium solani f. sp. pisi. 3. Number of parsimony informative characters observed within F. keratoplasticum. 4. Number of single nucleotide polymorphisms observed within F. keratoplasticum. 5. Number of total insertion/deletion and microsatellite polymorphisms observed within F. keratoplasticum (underlined), followed by the number of alleles observed at each, i.e. locus 5437 contains 2 such polymorphisms, one of which has 11 alleles. Superscript s indicates that this polymorphism was found in only one isolate. 6. Consistency index of individual locus maximum parsimony trees. *Putative hybrids contained 4 more SNPs within their rDNA sequences.
Table 2.
Primer pairs used for PCR amplification and Sanger sequencing
| Locus | Fw primer | Rv primer | Annealing temp. (°C) | Size 1 (bp) | Seq. composition (bp) 2 | Alleles 3 | H 4 |
|---|---|---|---|---|---|---|---|
|
TEF |
EF1: ATGGGTAAGGARGACAAGAC |
EF2: GGARGTACCAGTSATCATGTT |
53 |
667 |
256 intronic; 411 exonic |
22 |
0.89 |
| rDNA |
ITS5: GGAAGTAAAAGTCGTAACAAGG |
NL4: GGTCCGTGTTTCAAGACGG |
53 |
1029 |
705 coding; 324 ITS |
6 |
0.35 |
|
RPB2 5-7 |
5f2: GGGGWGAYCAGAAGAAGGC |
7cr: CCCATRGCTTGYTTRCCCAT |
55 |
863 |
Exonic |
11 |
0.51 |
|
RPB2 7-11 |
7cf: ATGGGYAARCAAGCYATGGG |
11ar: GCRTGGATCTTRTCRTCSACC |
55 |
881 |
Exonic |
- |
- |
| 3968 |
3968fw: TGTTGGTTCGAGATGGTTGA |
3968rv: GAGAAGGGCAACTGGGAGA |
53 |
770 |
Intergenic |
12 |
0.68 |
| 3972 |
3972fw: TCTGATGCAGACTAGCACTCG |
3972rv: ATCGGACGAAACAGAGCAGG |
53 |
831 |
Intergenic |
13 |
0.45 |
| 4081 |
4081fw: TGACRAGGATGAATGAGCGA |
4081rv: TGACCAGCCTCCAAGSG |
56 |
642 |
Intergenic |
18 |
0.8 |
| 6512 |
6512fw: GGAGGACCAGGAGGAATAGG |
6512rv: CAAAGCAGATCGACTGAGGA |
53 |
644 |
Intergenic |
12 |
0.52 |
| 5439 |
5439fw: AATGGGAATACGAGCGTCAG |
5439rv: AGGGGCTGCTGTTAGTGAGA |
53 |
779 |
Intergenic |
10 |
0.48 |
| 5437 |
5437fw: AACAAGACAAGGCAGCAGGT |
5437rv: TCCAGAGGAACGACGAGGC |
56 |
544 |
213 exonic, 331 intergenic |
23 |
0.88 |
|
MAT1-1 |
MAT1-S-1 F: ATGGCTTTCCGCAGTAAGGA |
MAT1-S-1R: CATGATAGGGCAGCAAAGAG |
53 |
~200 |
N. A. |
N. A. |
N. A. |
| MAT1-2 | MAT2-S-1 F: GGGAATCTGAGAAAGATACGTAC | MAT2-S-1R: CGGTACTGGTAGTCGGGAT | 53 | ~800 | N. A. | N. A. | N. A. |
1. Number of characters in the aligned dataset. For different isolates, the exact length of any region may be variable due to indels and microsatellite repeats 2. ITS = internal transcribed spacers 1 and 2. Locus 5437 contains coding regions of a predicted hypothetical protein of unknown function. 3. Number of alleles observed in F. keratoplasticum 4. Average genetic diversity of each of the 9 loci calculated using LIAN accessed at http://pubmlst.org.
Nucleic acid manipulation
Isolates were grown in 5 mL of potato dextrose broth (PDB) in test tubes for 14 days. Mycelium was then collected, rinsed with sterile distilled water, dried with paper towels, and transferred to 2 mL tubes for lyophilization. Mycelium was ground to a fine powder using plastic pestles and total genomic DNA was isolated using a DNeasy Plant Minikit protocol (Qiagen, Valencia, CA, USA) and suspended in a 100 μl volume of elution buffer. PCR and sequencing was performed using GoTaq PCR Kits (Promega, Madison, WI, USA) in 50 μl volumes following the manufacturer’s instructions, with reactions subjected to 2 minutes at 94°C, 35 cycles of 1 minute at 94°C, 1 minute at the appropriate annealing temperature (Table 2), and 1 minute at 72°C, followed by 10 minutes at 72°C. PCR products were sequenced at the Penn State Nucleic Acid Facility, Huck Institute of Life Sciences, University Park, PA. A multiplex PCR for identifying MAT idiomorph was performed as previously described [39]. PCR products were visualized using a 1.5% w/v agarose gel and scored as MAT1-1 or MAT1-2 based on amplicon size (~200 and 800 bp, respectively). Portions of the nuclear ribosomal intergenic spacer region (IGS) were also amplified and sequenced in two putative hybrid isolates as previously described [22] using Invitrogen PlatinumTaq kits (Life Technologies, Carlsbad, CA USA) following the manufacturer’s instructions.
Sequence type (ST) diversity
Isolates were assigned to existing and novel STs as described previously [4,9,24]. Briefly, ST assignments were based on their position in terminal nodes in one of 200 maximum parsimony (MP) phylogenetic trees generated by implementation of the parsimony ratchet [49] on concatenated DNA sequence alignments in PAUP*4.0b. Chromatograms were then cross-checked for accuracy to confirm ST membership. Missing or ambiguous DNA positions were assigned Ns in the sequence alignments and treated as missing data. For the nine-locus MLST system, the previously assigned three-locus STs were amended with numerals indicating additional subdivision revealed by the six additional loci. For example, FSSC 2-d, the most common ST in the three-locus system, was subdivided into 18 nine-locus STs designated FSSC 2-d1 through FSSC 2-d18. SNP and insertion-deletion polymorphisms in the loci were used to distinguish STs. For certain MLST analyses, alleles at each locus were assigned simple numerical identifiers, which we refer to as digitized nine-locus STs; e.g., 16-1-1-4-6-2-3-4-13 represents a combination of unique alleles observed at the nine loci in ST FSSC 2-c1. 24/2079 (1.2%) of the nine loci sampled across all isolates produced partial or no sequence data. There were 21 and three isolates missing a significant portion of one or two loci, respectively, and 11 of these cases involved locus 5439. When determining the number of multilocus STs, none were scored as unique by virtue of missing or ambiguous nucleotide data. The number of nine-locus STs was calculated using the digitized nine-locus STs as input into the application DNAcollapser implemented in the website FaBox [50]. Mating type was then used as a tenth locus, with individuals assigned as 1 or 2 based on the detection of the MAT1-1 or MAT1-2 idiomorph and the number of ten-locus STs was calculated in the same way. Nine-locus ST diversity of the total sample was estimated using the formula [n/(n - 1)](1 - ∑x i 2), where x i is the frequency of the i th ST and n is the number of isolates [51]. Genetic diversity of the loci and mean genetic diversity were calculated from the digitized nine-locus STs using LIAN [52] accessed at (http://pubmlst.org). To test for patterns of population differentiation, the dataset of 231 isolates was divided into two groups comprising clinical (n = 76) and non-clinical (n = 155) isolates. These groups were clone corrected and an exact test of population differentiation was performed in Arlequin ver 3.11 [53] using the digitized nine-locus STs. Finally, a numerically coded, clone corrected nucleotide sequence dataset with constant characters removed was used for plotting the mean number of genotypes vs. the number of loci in MULTILOCUS, by randomly sampling 1–182 characters 100 times each [54]. Sequence data for the six new loci developed in this study have been deposited in NCBI GenBank (Table 3).
Table 3.
NCBI GenBank accessions for DNA sequence data for six loci developed for F. keratoplasticum
| Locus | GenBank accession numbers |
|---|---|
| 3968 |
JN585993-JN586184 |
| 3972 |
JN586185-JN586372 |
| 4081 |
JN586373-JN586556 |
| 5437 |
JN586557-JN586711 |
| 5439 |
JN586712-JN586899 |
| 6512 | JN586900-JN587087 |
Estimation of clonal relationships
To generate population snapshots and to estimate clonal relationships among genotypes, the set of 111 unique digitized nine-locus STs was used as input into an MLST analysis [55] implemented using the web application eBURST [56], at the single locus variant (SLV) level. The total set of 231 digitized nine-locus STs was also analyzed at the triple locus variant (TLV) level utilizing the goEBURST algorithm [57] implemented in PHYLOVIZ [58]. STs that were not part of any TLV group were added manually as standalone data points. Mating-type data and source information were manually superimposed upon the resulting display of the TLV analysis with high-level edges (connections between STs identical at 7/8 and 6/8 loci) displayed as dashed and dotted lines, respectively. STs of isolates previously reported as producing cyclosporins in vitro [39,59] were indicated on the population snapshots in order to visualize those associated with this phenotype.
Multilocus tests for linkage disequilibrium
To test for the non-random association of alleles, a clone corrected, digitized nine-locus ST dataset was used to calculate the Index of Association (I A ) [60] together with rBarD, an alternative measure of IA adjusted for the number of loci, using MULTILOCUS ver. 1.3b [54]. The observed I A and rBarD values were calculated for the nine-locus dataset and compared to 1000 randomized datasets. In addition, a standardized I A was calculated using LIAN [52]. Additionally, to account for potential effects due to physical linkage, I A and rBarD were recalculated on clone corrected datasets with each of the nine loci removed separately.
Detection of recombination
A clone corrected, concatenated NEXUS data file with constant characters removed was used to generate a NeighborNet in SPLITSTREE4 [61], and to calculate the dataset’s δ score (a measure of treelikeness, where higher δ values may reflect higher levels of recombination) [62] and phylogenetic diversity, a measure of diversity based on branch lengths [63]. A clone corrected, concatenated dataset including all nucleotide characters was used to perform the PHI test for recombination [64] implemented in SPLITSTREE4. Incompatibility among all 9 individual gene trees was assessed using COMPAT.PY, a program to detect topological conflict between supported clades in phylogenetic trees [65]. For each of the nine loci, one of 200 MP trees generated using PAUPRAT [49] with branch lengths included was used as input in a test for topological conflict.
Detection of interspecific introgression
Based on the discovery of two putative hybrids of F. keratoplasticum (FRC S-2406 and FRC S-2509) that possess identical rDNA sequences to those of FSSC 9 [39], sequence data from the nine-locus MLST system were generated for three members of FSSC 9 (FRC S-2485, FRC S-2530, FRC S-2540). For comparative purposes, portions of the nine loci were also sequenced for FSSC 5 (FRC S-2519), F. petroliphilum (FSSC 1, FRC S-2550), and F. solani f. sp pisi (FSSC 11) (http://genome.jgi.doe.gov/Necha2/Necha2.home.html). Individual unrooted MP gene trees were generated as described previously to assess whether other loci of the two putative hybrid isolates showed affiliation to FSSC 9. Finally, a portion of the intergenic spacer region of the nuclear ribosomal RNA gene repeat (IGS) was sequenced for the two putative hybrids, three FSSC 9 isolates, and one F. keratoplasticum isolate (NRRL 32710) and analyzed to determine whether this portion of the rRNA repeat was also introgressed.
Results
F. keratoplasticum shows high levels of ST diversity
The nine-locus genetic diversity ranged from 0.35 to 0.89 with an average of 0.62 (Table 3). One hundred and eleven nine-locus and 122 ten-locus STs (with mating-type as a tenth marker) reflected high levels of diversity in the total sample of 231 isolates. The number of MAT1-1 and MAT1-2 isolates observed was 162 (70%) and 69 (30%), respectively. Isolates of seven STs comprised both mating types (2d1, 2d2, 2d3, 2f2, 2f6, 2 k4, 2o2). ST diversity was 0.94 among the nine-locus STs, and phylogenetic diversity (PD) of the nine-locus dataset calculated in SPLITSTREE4 was 0.45 (Table 3). Even with 9 loci covering ~7650 bp, the plot of genotypes vs. number of loci sampled indicates a likelihood of more diversity present within this panel of isolates than was resolved (Figure 1). No strong evidence for cryptic species boundaries within F. keratoplasticum was observed, as comparisons between gene trees for each of the nine loci did not reveal any groups with bootstrap values >60% in all of the MP trees (data not shown). No population differentiation was detected using exact tests comparing ST diversity of isolates from clinical sources with those from all other sources.
Figure 1.
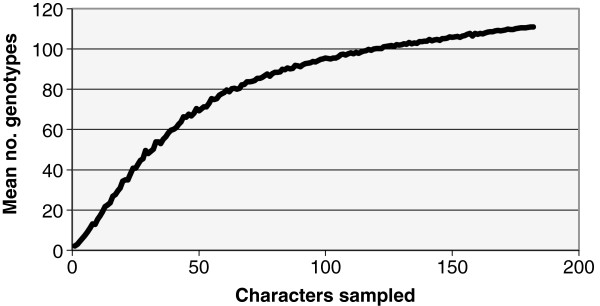
Plot of mean number of genotypes of F. keratoplasticum as a function of number of characters (SNPs and indels) sampled.
Inferred relationships among clones and clonal complexes
Evolutionary relationships among the 231 F. keratoplasticum isolates inferred using goeBURST resolved nine distinct clonal complexes (i.e. groups of hypothetically clonally related genotypes or clonets). These comprised 50 STs that were defined at the single locus variant (SLV) level (STs identical at 8/9 loci, identified by solid black lines) (Figure 2). Double and triple locus variants (DLV and TLV; STs identical at 7/9 and 6/9 loci, respectively) were also identified by dashed and dotted lines, (Figure 2). Lastly, goeBURST resolved 14 STs comprising 15 isolates that were different at four of more loci (displayed as unconnected STs in Figure 2). Interestingly, included among the singleton STs were the isolate that is the International Organization for Standardization (ISO), ATCC 36031 (ST 2-c1), as well as one of only two confirmed sexually fertile isolates of F. keratoplasticum (ST 2-ss1) [39]. Isolates previously known to produce cyclosporins in vitro were found to belong to F. keratoplasticum STs 2-d4, 2-d8, 2-d9, 2-d11, 2-ii2, 2-ll1, 2-k1, 2-pp3, 2-ss1, and 2-v1 (Figure 2).
Figure 2.

Population snapshots of F. keratoplasticum inferred using Phyloviz beta. Each node represents a unique ST (the prefix ″2-″ has been omitted for clarity); areas of nodes are proportional to the number of isolates. STs with 8/9 loci in common (SLVs) are connected with solid black lines, STs with 7/9 loci in common (DLVs) are connected with dashed lines, STs with 6/9 loci in common (TLVs) are connected with dotted lines. STs different at more than three loci not connected to a group. 9 SLV groups are shown. In A) light gray indicates proportion of STs that are MAT1-1; darker gray shading indicates proportion of STs that is MAT1-2. In B) light gray indicates isolates from clinical sources; darker gray indicates isolates from all other sources. Plus signs denote several STs containing isolates known to produce cyclosporin in vitro. Asterisks denote the ST of the ISO standard ATCC 36031 strain. White stars denote STs containing isolates successfully crossed in mating experiments.
Evidence for non-random association of alleles
Multiple analyses revealed a statistically significant association of alleles among loci, indicating that isolates may be related through asexual processes of population evolution. The index of association (I A ) and rBarD calculated from the nine-locus dataset of digitized STs were 0.47 and 0.06, respectively; both differed significantly from the ranges of these values observed in 1000 artificially recombined datasets (p < 0.01) (Table 4). A standardized (I A ) [52] of 0.059 was calculated using LIAN, and the null hypothesis of linkage equilibrium was rejected using 1000 resamplings in Monte Carlo and parametric tests (p < 0.001 and p < 3×10-21, respectively). To assess the potential contribution of linkage between loci, eight separate tests for linkage disequilibrium were also performed on clone corrected datasets with each locus removed individually. Each of these tests showed significant amounts of linkage disequilibrium (results not shown).
Table 4.
Summary statistics for the F. keratoplasticum data set
| No. isolates |
231 |
| No. nine-locus STs |
111 |
|
MAT1-1:MAT1-2 (total) |
162:69 |
|
MAT1-1:MAT1-2 (without 2-d types) |
64:60 |
| Allelic diversity1 |
0.94 |
| Average genetic diversity of loci2 |
0.62 |
| NeighborNet δ score3 |
0.28 |
| Phylogenetic Diversity4 |
0.45 |
| Index of Association5 |
0.47 |
| rBarD6 |
0.06 |
| Standardized I
A
7
|
0.05 |
| No. of eBURST SLV groups (clonal complexes) |
9 |
| No. STs connected at SLV level |
50 |
| No. isolates in SLV level STs |
162 |
| No. singletons at SLV level | 69 |
1. ST diversity calculated from Nei (43). 2. Average genetic diversity of the 9 loci calculated using LIAN (25). 3. Average δ score (26) of the clone corrected set of 111 unique STs calculated using SPLITSTREE. 4. Phylogenetic diversity (19) of the clone corrected set of 111 unique STs calculated using SPLITSTREE. 5. Index of Association (41) calculated using MULTILOCUS 1.3b (1) was found to be significantly different (p < 0.01) than 1000 randomizations, which showed an expected range of 2.86-4.09 (mean = 3.46). 6. rBarD calculated using MULTILOCUS 1.3b was found to be significantly different (p < 0.01) than 1000 randomizations, which showed an expected range of 0.0208-0.0283 (mean = 0.024). 7. Standardized I A calculated using LIAN (25); the null hypothesis of linkage equilibrium was rejected using 1000 resamplings in Monte Carlo tests and a parametric test, with p < 0.001 and p < 3×1021 respectively.
Evidence for recombination
A net-like and reticulated NeighborNet was observed (Figure 3) and the PHI test for recombination implemented in SPLITSTREE4 supported the presence of recombination (p < 0.0001). The average δ score for the 111 unique STs using the concatenated dataset showed a significant departure in treelikeness from zero (δ = 0.28) and could be due to the presence of historical recombination events (26). Individual F. keratoplasticum gene trees indicated various levels of intralocus homoplasy, with consistency indices (CI) ranging between 0.75 in TEF to 1.0 in RPB2, 3972, and 5439 (Table 1). COMPAT.PY detected multiple topological conflicts among the trees inferred from the nine loci (results not shown).
Figure 3.

Neighbor net of 111 unique nine-locus haplotypes inferred using SPLITSTREE4. A PHI test for recombination (7) implemented in SPLITSTREE4 suggests a statistically high likelihood of recombination in the full dataset (p < 0.0001).
Introgression between phylogenetic species
F. keratoplasticum isolates FRC S-2406 (from Honeymoon Island Park in Florida) and FRC S-2509 (from a shopping center in Georgia) possessed nuclear rDNA regions (ITS, the D1-D2 region of the large subunit, and IGS) that were perfect matches to those found in FSSC 9, suggesting some kind of genetic transfer between F. keratoplasticum and this species [39,41]. Phylogenetic analysis of the eight other loci grouped these isolates with F. keratoplasticum (Figure 4). Taken together, these analyses indicated that FRC S-2406 and FRC S-2509 possess ribosomal RNA repeats that are derived from FSSC 9, while the other eight loci have clear origins in F. keratoplasticum. Excluding the rDNA of these putative hybrids, the other loci supported reciprocal monophyly between F. keratoplasticum and FSSC 9.
Figure 4.

Nine individual locus unrooted maximum parsimony trees inferred using parsimony ratchet (72, 73) implemented in PAUP* 4.0. Isolates S-2406 and S-2510 are strongly supported members of F. keratoplasticum (FSSC 2) in eight of the trees. In the rDNA tree, however, these two isolates are strongly supported members of FSSC 9.
Discussion
The primary goal of this study was to apply novel sequence based markers to understand the diversity and population biology of F. keratoplasticum, the most common clinically relevant Fusarium [21,24]. In addition to their utility in addressing questions related to their basic ecology and epidemiology, MLST in F. keratoplasticum provides a framework for the study of biologically relevant phenotypes. For example, F. keratoplasticum STs vary in the types of secondary metabolites produced, including cyclosporins [39,59] (Figure 2). Sexual fertility also appears to be a variable phenotype within F. keratoplasticum. Perithecium formation was previously reported as very rare in crosses of opposite mating types, although a cross between STs 2-ss1 and 2-ll1 produced recombinant ascospores under laboratory conditions [39]. Finally, biofilm formation in F. keratoplasticum is another variable trait with practical significance. Although NRRL 22641 (=ATCC 36031) was recommended by the International Organization for Standardization guidelines for testing disinfectants, it does not form biofilms in in vitro models [66] and this singleton (ST 2-c1) appears to be quite divergent (Figure 2) and aberrant morphologically [39], casting further doubt on its choice for testing the efficacy of antimicrobials. It is unknown whether this isolate’s failure to form biofilms can be attributed to mutation, perhaps post-isolation, or phenotypes segregating in natural populations. In addition to those reported here, further tests of ATCC 36031 in comparison to other clinical isolates has led to the recommendation that multiple clinical isolates be utilized in disinfectant testing [67].
High levels of sequence type diversity in F. keratoplasticum
Polymorphisms within the nine loci employed in this study ranged from 6–33 alleles per locus. The six new markers developed in this study were originally targeted because they harbored microsatellite repeats in the F. solani f. sp pisi genome. Sequencing these regions in F. keratoplasticum revealed high levels of nucleotide polymorphisms in addition to polymorphic repeats, motivating us to utilize them more fully as sequence-based markers and also to avoid issues of convergence that can be problematic in interpreting alleles based on variable numbers of tandem repeats [68]. SNPs were the most common polymorphisms observed within F. keratoplasticum (163 out of ~7650 characters). Polymorphism per region sequenced was higher than observed in MLST systems of other fungi pathogenic to humans such as Aspergillus fumigatus [14], but lower than the dimorphic mycotic pathogens C. albicans, C. tropicalis and C. glabrata (6). Testing the primers designed in this study against other FSSC species indicated utility of these markers broadly in the complex (Figure 4), as they successfully amplified DNA from F. petroliphilum (FSSC 1), F. falciforme (FSSC 3 + 4), and FSSC phylogenetic species 5, 9, 18, 22 and 24. Many polymorphisms were found in other FSSC species, especially in the microsatellite repeats, which were extremely variable between species, though the same repeats motifs were often monomorphic within F. keratoplasticum. Although the six new loci developed in this study consisted almost entirely of non-coding sequence residing in intergenic regions, the level of genetic diversity observed at these loci within F. keratoplasticum were surprisingly similar to that of the protein-coding genes (Table 2).
Previous MLST studies indicated that environmental and human pathogenic fusaria are diverse, spanning multiple species complexes, species, and STs within species, but certain STs have been found to be dominant. For example, Fusarium oxysporum species complex ST 33 was identified as one such widespread clonal lineage based on analysis of TEF and the IGS region. Moreover, analysis of amplified fragment length polymorphisms (AFLPs) revealed seven distinct haplotypes within ST 33 [25]. We observed a similar trend within F. keratoplasticum with the addition of six markers to the 3-locus MLST scheme. The majority of previously defined 3-locus STs were further resolved into multiple nine-locus STs (Table 1), some of which appeared to be quite distinct (Figures 2 and 3). Furthermore, the plot of genotypic diversity vs. number of loci sampled indicated that additional diversity in the nine-locus STs remains to be resolved (Figure 1).
F. keratoplasticum has a mixed reproductive mode
Analyses of F. keratoplasticum isolates revealed evidence of clonality and recombination, mirroring reproductive modes observed in this species. FSSC species reproduce by clonally via asexual propagules (e.g., macroconidia, microconidia, chlamydospores) and several sexually through formation of perithecia and recombinant ascospores These potential modes of reproduction are known in F. keratoplasticum [39]. In contrast to the hypothesis of local sexual neighborhoods, where mating is thought to occur only between isolates of similar genotypes [69], isolates of F. keratoplasticum that produced recombinant ascospores shared only one identical sequence (locus 5439) among the nine loci. The observed frequencies of mating-types in F. keratoplasticum in the total sample deviated significantly from the expected 1:1 ratio (162:69 MAT1-1:MAT1-2; binomial cumulative probability p < 0.0001). This aberration may be due to sampling of the most common widespread clones, because when all 2-d types were removed, a ratio of 64:60 MAT1-1:MAT1-2 (binomial cumulative probability p > 0.39) was observed.
A robust estimate of the contribution of mutation relative to recombination may be calculated as the ratio of STs that form complexes of SLVs, which are assumed to have arisen via mutation, to those that do not. In F. keratoplasticum, 50 STs resolved into 9 clonal complexes of SLVs, comprising 2–22 unique STs each (Figure 2). The remaining number of STs that did not resolve into SLV clonal complexes was 61, giving an inferred 1:1.24 ratio of mutation:recombination. Without clone correction, however, 162 isolates are connected at the SLV level compared to 69 isolates that are identical at fewer than 8/9 loci, giving an inferred mutation:recombination ratio of 2.3:1. This estimate contrasts with the situation observed in the more clonal Talaromyces marneffei, where the mutation:recombination ratio was approximately 5:1 [69,70]. A mixed clonal/recombinant structure has also been reported in clinically relevant fungi including Aspergillus fumigatus[32], Cryptococcus gattii [29], Candida albicans [30,71], Candida glabrata [72], and Paracoccidioides brasiliensis [73].
F. keratoplasticum ST 2-d as a transcontinental epidemic clone
As previously mentioned, F. keratoplasticum 2-d is the three-locus ST most commonly associated with plumbing fixture biofilms, including hospital water systems, and human infections. It comprised 46% of the isolates in our dataset (107 isolates), and despite the incomplete resolution from nine loci, it is retained as a group of related, common and geographically widespread STs. The nine-locus MLST system divided 2-d into 18 unique types, all but one of which are connected through a series of SLVs, DLVs and TLVs (Figures 3 and 2). Based on sequence information, the dominant subtypes 2-d1 and 2-d2 appear to represent a widespread clonal lineage found in the United States, Puerto Rico, Germany, and Qatar (Table 1). Additional evidence of a strong clonal component was provided by the discovery that 94% of 2-d1 and 2-d2 isolates were MAT1-1 (Figure 2A). The remaining 6% of 2-d1 and 2-d2 isolates may represent one or multiple separate evolutionary lineages that have not been resolved, even with nine loci. The presence of haplotypes that are both extremely common and widely distributed has been observed in mycotic pathogens such as Cryptococcus gattii [28], in which the same common clone (C. gattii ST7) is present in Australia and the United States. In contrast, ST 2-k, previously shown to be among the six most common biofilm-associated human pathogenic fusaria [41], showed evidence for historical recombination (Figures 3 and 2), as 2-k2 and 2-k3 were members of an unconnected TLV group in the eBURST analysis (Figure 2). In support of this, 2-k isolates show a 9:10 MAT1-1:MAT1-2 mating-type ratio, which is similar to the 1:1 ratio expected among randomly mating populations. However, interfertility of the 2-k isolates has not been tested.
Introgression and hybridization
Introgression has been observed in other medically important fungi including Candida spp. [74], Cryptococcus [75], and Coccidioides [76], and is hypothesized to play an important role in producing diversity. Here, strong evidence of genetic exchange between two isolates of F. keratoplasticum and the unnamed species FSSC 9 is presented. FRC isolates S-2406 and S-2509 possessed RPB2 and TEF alleles that clearly placed them in F. keratoplasticum, but their rDNA appeared to have been derived from a FSSC 9 parent (100% sequence identity to all known FSSC 9 haplotypes). F. keratoplasticum and FSSC 9 are closely related, and both occur in plumbing biofilms, so the simplest explanation for this is introgression via an interspecific hybrid cross. Analyses of these putative hybrids showed that they possessed typical F. keratoplasticum alleles at all other loci, suggesting that if the introgression of rDNA was due to a hybridization event, the isolates were likely heavily backcrossed into the F. keratoplasticum background. Hybridization between closely related, phylogenetically defined species has been inferred in Fusarium in multiple instances [77-80]. The introgressed FSSC 9 rDNA included the ITS regions, the large subunit rRNA, and IGS region, and may encompass the entire rDNA gene repeat. Genome scale analyses are needed to fully assess what portion of the hybrid genomes was derived from a FSSC 9 parent. These analyses may help elucidate whether this introgression was mediated by a sexual or parasexual process. It is also possible that other unorthodox mechanisms may be at play in generating genetic diversity in F. keratoplasticum. Species that possess small conditionally dispensible (CD) chromosomes are known to be present within the FSSC [81] as well as in other fusaria [77]. The genome of F. solani f. sp pisi includes three CD chromosomes, which disproportionately harbor genes with no clear Fusarium orthologues, leading to the hypothesis that they were gained through unknown horizontal gene transfer (HGT) events [44]. While asexual transfer of CD chromosomes has not been demonstrated in the FSSC, it has been shown experimentally in F. oxysporum [82].
Conclusion
An extensive body of literature has connected many Fusarium infections, particularly ones suspected of being acquired nosocomially, to isolates resident in the patient indoor environment [21,38,41]. Similar to what has been observed in other common nosocomially acquired fungal pathogens, including A. fumigatus [83], we found no evidence differentiating clinical isolates from those collected from other sources, including biofilms in plumbing drains (Figure 2B). Although all known isolates of F. keratoplasticum are from highly anthropogenically influenced environments, nothing is known about a potential natural reservoir for this species [21,39,41]. While some STs show a broad geographic distribution, this MLST system may permit epidemiological inferences connecting patient and indoor environmental STs at a local scale. Diverse genotypes with patterns indicating historical recombination were observed, though there was also strong evidence for clonal expansion of epidemic STs. This mixed reproductive strategy, which is not unusual in fungal pathogens [28,73,84,85], enables the expansion of successful genotypes, at the same time allowing for adaptation to changing environments and antifungal treatment strategies. The detection of natural hybridization and introgression in this sample of isolates may be indicative of another prevalent mechanism permitting adaptation to the anthropogenic environments these fungi inhabit.
Availability of supporting data
Nucleotide sequence data for F. keratoplasticum from the six new sequence markers are available through NCBI GenBank with the following accession numbers: locus 3968: JN585993-JN586184; locus 3972: JN586185-JN586372; locus 4081: JN586373-JN586556; locus 5437: JN586557-JN586711; locus 5439: JN586712-JN586899; locus 6512: JN586900-JN587087.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DPGS performed all fungal culturing, DNA extraction, marker design and validation, PCR, sequencing, alignment, and analyses and drafted the manuscript. DG and KO conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
111 nine-locus sequence types and their frequencies based on the diversity of the 231 F. keratoplasticum isolates used in this study.
Contributor Information
Dylan PG Short, Email: DGeiser@PSU.edu.
Kerry O’Donnell, Email: kerry.odonnell@ars.usda.gov.
David M Geiser, Email: DGeiser@PSU.edu.
Acknowledgments
We thank Jean H. Juba for excellent technical assistance. This work was supported in part by a grant from the National Science Foundation, DEB 0089474 to DMG and Gary J. Samuels, National Institute of Food and Agriculture Project PEN 04527 at the Pennsylvania State Agricultural Experiment Station, and by Bausch and Lomb, Inc. DPGS was supported by NIFA grant 2010-65110-20488, Education in Genomics-Based Microbial Forensics. The mention of firm names or trade products does not imply that they are endorsed or recommended by the US Department of Agriculture over other firms or similar products not mentioned. The USDA is an equal opportunity provider and employer.
References
- Leslie JF, Summerell BA. The Fusarium Laboratory Manual. Ames, Iowa: Blackwell Publishing; 2006. [Google Scholar]
- Nucci M, Anaissie E. Fusarium infections in immunocompromised patients. Clin Microbiol Rev. 2007;20(4):695–704. doi: 10.1128/CMR.00014-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dóczi I, Gyetvai T, Kredics L, Nagy E. Involvement of Fusarium spp. in fungal keratitis. Clin Microbiol Infect. 2004;10(9):773–776. doi: 10.1111/j.1469-0691.2004.00909.x. [DOI] [PubMed] [Google Scholar]
- Chang DC, Grant GB, O’Donnell K, Wannemuehler KA, Noble-Wang J, Rao CY, Jacobson LM, Crowell CS, Sneed RS, Lewis FM, Schaffzin JK, Kainer MA, Genese CA, Alfonso EC, Jones DB, Srinivasan A, Fridkin SK, Park BJ. Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. J Am Med Assoc. 2006;296(8):953–963. doi: 10.1001/jama.296.8.953. [DOI] [PubMed] [Google Scholar]
- Daniel CS, Rajan MS, Saw VPJ, Claerhout I, Kestelyn P, Dart JKG. Contact lens-related Fusarium keratitis in London and Ghent. Eye. 2008;23(2):484–485. doi: 10.1038/eye.2008.188. [DOI] [PubMed] [Google Scholar]
- Gaujoux T, Chatel MA, Chaumeil C, Laroche L, Borderie VM. Outbreak of contact lens-related Fusarium keratitis in France. Cornea. 2008;27(9):1018–1021. doi: 10.1097/ICO.0b013e318173144d. [DOI] [PubMed] [Google Scholar]
- Khor WB, Aung T, Saw SM, Wong TY, Tambyah PA, Tan AL, Beuerman R, Lim L, Chan WK, Heng WJ, Lim J, Loh RSK, Lee SB, Tan DTH. An outbreak of Fusarium keratitis associated with contact lens wear in Singapore. J Am Med Assoc. 2006;295(24):2867–2873. doi: 10.1001/jama.295.24.2867. [DOI] [PubMed] [Google Scholar]
- Ma SE, So K, Chung P, Tsang HT, Chuang S. A multi-country outbreak of fungal keratitis associated with a brand of contact lens solution: the Hong Kong experience. Int J Infect Dis. 2009;13(4):443–448. doi: 10.1016/j.ijid.2007.12.018. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Sarver BA, Brandt M, Chang DC, Noble-Wang J, Park BJ, Sutton DA, Benjamin L, Lindsley M, Padhye A, Geiser DM, Ward TJ. Phylogenetic diversity and microsphere array-based genotyping of human pathogenic fusaria, including isolates from the multistate contact lens-associated US keratitis outbreaks of 2005 and 2006. J Clin Microbiol. 2007;45(7):2235–2248. doi: 10.1128/JCM.00533-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speeleveld E, Gordts B, Landuyt HWV, Vroey CD, Raes-Wuytack C. Susceptibility of clinical isolates of Fusarium to antifungal drugs. Mycoses. 1996;39(1–2):37–40. doi: 10.1111/j.1439-0507.1996.tb00081.x. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Fisher MC. Fungal multilocus sequence typing — it’s not just for bacteria. Curr Opin Microbiol. 2003;6(4):351–356. doi: 10.1016/S1369-5274(03)00088-2. [DOI] [PubMed] [Google Scholar]
- Balajee SA, Borman AM, Brandt ME, Cano J, Cuenca-Estrella M, Dannaoui E, Guarro J, Haase G, Kibbler CC, Meyer W, O’Donnell K, Petti CA, Rodriguez-Tudela JL, Sutton D, Velegraki A, Wickes BL. Sequence-based identification of Aspergillus, Fusarium, and Mucorales species in the clinical mycology laboratory: where are we and where should we go from here? J Clin Microbiol. 2009;47(4):877–884. doi: 10.1128/JCM.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LT, Carter DA. Looking for sex in the fungal pathogens Cryptococcus neoformans and Cryptococcus gattii. FEMS Yeast Res. 2006;6(4):588–598. doi: 10.1111/j.1567-1364.2006.00087.x. [DOI] [PubMed] [Google Scholar]
- Bain JM, Tavanti A, Davidson AD, Jacobsen MD, Shaw D, Gow NAR, Odds FC. Multilocus sequence typing of the pathogenic fungus Aspergillus fumigatus. J Clin Microbiol. 2007;45(5):1469–1477. doi: 10.1128/JCM.00064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, McTaggart LR, Zhang SX, Low DE, Stevens DA, Richardson SE. Phylogenetic analysis reveals a cryptic species Blastomyces gilchristii, sp. nov. within the human pathogenic fungus Blastomyces dermatitidis. PLoS One. 2013;8(3):e59237. doi: 10.1371/journal.pone.0059237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Chen YC, Lo HJ, Chen KW, Li SY. Assessment of Candida glabrata strain relatedness by pulsed-field gel electrophoresis and multilocus sequence typing. J Clin Microbiol. 2007;45(8):2452–2459. doi: 10.1128/JCM.00699-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt A, Dechairo BM, Koenig GL, Carter DA, White TJ, Taylor JW. Molecular markers reveal differentiation among isolates of Coccidioides immitis from California, Arizona and Texas. Mol Ecol. 1997;6(8):781–786. doi: 10.1046/j.1365-294X.1997.00245.x. [DOI] [PubMed] [Google Scholar]
- Meyer W, Aanensen DM, Boekhout T, Cogliati M, Diaz MR, Esposto MC, Fisher M, Gilgado F, Hagen F, Kaocharoen S. Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med Mycol. 2009;47(6):561–570. doi: 10.1080/13693780902953886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balajee SA, Hurst SF, Chang LS, Miles M, Beeler E, Hale C, Kasuga T, Benedict K, Chiller T, Lindsley MD. Multilocus sequence typing of Histoplasma capsulatum in formalin-fixed paraffin-embedded tissues from cats living in non-endemic regions reveals a new phylogenetic clade. Med Mycol. 2013;51(4):345–351. doi: 10.3109/13693786.2012.733430. [DOI] [PubMed] [Google Scholar]
- Maitte C, Leterrier M, Le Pape P, Miegeville M, Morio F. Multilocus sequence typing of Pneumocystis jirovecii from clinical samples: how many and which loci? J Clin Microbiol. 2013;59(9):2843–2849. doi: 10.1128/JCM.01073-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, O’Donnell K, Sutton DA, Nalim FA, Summerbell RC, Padhye AA, Geiser DM. Members of the Fusarium solani species complex that cause infections in both humans and plants are common in the environment. J Clin Microbiol. 2006;44(6):2186–2190. doi: 10.1128/JCM.00120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Gueidan C, Sink S, Johnston PR, Crous PW, Glenn A, Riley R, Zitomer NC, Colyer P, Waalwijk C, van der Lee T, Moretti A, Kang S, Kim HS, Geiser DM, Juba JH, Baayen RP, Cromey MG, Bithell S, Sutton DA, Skovgaard K, Ploetz R, Kistler HC, Elliott M, Davis M, Sarver BAJ. A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genet Biol. 2009;46(12):936–948. doi: 10.1016/j.fgb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Rinaldi MG, Gueidan C, Crous PW, Geiser DM. Novel multilocus sequence typing scheme reveals high genetic diversity of human pathogenic members of the Fusarium incarnatum-F. equiseti and F. chlamydosporum species complexes within the United States. J Clin Microbiol. 2009;47(12):3851–3861. doi: 10.1128/JCM.01616-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Fothergill A, McCarthy D, Rinaldi MG, Brandt ME, Zhang N, Geiser DM. Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J Clin Microbiol. 2008;46(8):2477–2490. doi: 10.1128/JCM.02371-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Rinaldi MG, Magnon KC, Cox PA, Revankar SG, Sanche S, Geiser DM, Juba JH, van Burik JH, Anaissie EJ, Francesconi A, Walsh TJ, Robinson JS. Genetic diversity of human pathogenic members of the Fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial origin. J Clin Microbiol. 2004;42(11):5109–5120. doi: 10.1128/JCM.42.11.5109-5120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Sutton DA, Rinaldi MG, Sarver BA, Balajee SA, Schroers HJ, Summerbell RC, Robert VA, Crous PW, Zhang N, Aoki T, Jung K, Park J, Lee YH, Kang S, Park B, Geiser DM. Internet-accessible DNA sequence database for identifying fusaria from human and animal infections. J Clin Microbiol. 2010;48(10):3708–3718. doi: 10.1128/JCM.00989-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xiao M, Kong F, Chen S, Dou HT, Sorrell T, Li RY, Xu YC. Accurate and practical identification of 20 Fusarium species by seven-locus sequence analysis and reverse line blot hybridization, and an in vitro antifungal susceptibility study. J Clin Microbiol. 2011;49(5):1890–1898. doi: 10.1128/JCM.02415-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriconde F, Gilgado F, Arthur I, Ellis D, Malik R, van de Wiele N, Robert V, Currie BJ, Meyer W. Clonality and α-a recombination in the Australian Cryptococcus gattii VGII population-an emerging outbreak in Australia. PLoS One. 2011;6(2):e16936. doi: 10.1371/journal.pone.0016936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LT, Currie BJ, Krockenberger M, Malik R, Meyer W, Heitman J, Carter D. Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii. Eukaryot Cell. 2005;4(8):1403–1409. doi: 10.1128/EC.4.8.1403-1409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavanti A, Gow NAR, Maiden MCJ, Odds FC, Shaw DJ. Genetic evidence for recombination in Candida albicans based on haplotype analysis. Fungal Genet Biol. 2004;41(5):553–562. doi: 10.1016/j.fgb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Koufopanou V, Burt A, Szaro T, Taylor JW. Gene genealogies, cryptic species, and molecular evolution in the human pathogen Coccidioides immitis and relatives (Ascomycota, Onygenales) Mol Biol Evol. 2001;18(7):1246–1258. doi: 10.1093/oxfordjournals.molbev.a003910. [DOI] [PubMed] [Google Scholar]
- Pringle A, Baker DM, Platt JL, Wares JP, Latge JP, Taylor JW. Cryptic speciation in the cosmopolitan and clonal human pathogenic fungus Aspergillus fumigatus. Evolution. 2005;59(9):1886–1899. doi: 10.1111/j.0014-3820.2005.tb01059.x. [DOI] [PubMed] [Google Scholar]
- Taylor JW, Geiser DM, Burt A, Koufopanou V. The evolutionary biology and population genetics underlying fungal strain typing. Clin Microbiol Rev. 1999;12(1):126–146. doi: 10.1128/cmr.12.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debourgogne A, Gueidan C, de Hoog S, Lozniewski A, Machouart M. Comparison of two DNA sequence-based typing schemes for the Fusarium solani species complex and proposal of a new consensus method. J Microbiol Methods. 2012;9(1):65–72. doi: 10.1016/j.mimet.2012.07.012. [DOI] [PubMed] [Google Scholar]
- Debourgogne A, Gueidan C, Hennequin C, Contet-Audonneau N, de Hoog S, Machouart M. Development of a new MLST scheme for differentiation of Fusarium solani Species Complex (FSSC) isolates. J Microbiol Methods. 2010;82(3):319–323. doi: 10.1016/j.mimet.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Park B, Park J, Cheong KC, Choi J, Jung K, Kim D, Lee YH, Ward TJ, O’Donnell K, Geiser DM. Cyber infrastructure for Fusarium: three integrated platforms supporting strain identification, phylogenetics, comparative genomics and knowledge sharing. Nucleic Acids Res. 2011;39(suppl 1):D640–D646. doi: 10.1093/nar/gkq1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel CM, Hurst SF, Barreiros G, Akiti T, Nucci M, Balajee SA. Molecular analyses of Fusarium isolates recovered from a cluster of invasive mold infections in a Brazilian hospital. BMC Infect Dis. 2013;13(1):49. doi: 10.1186/1471-2334-13-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaissie EJ, Kuchar RT, Rex JH, Francesconi A, Kasai M, Müller FC, Mario L, Summerbell RC, Dignani MC, Chanock SJ, Walsh TJ. Fusariosis associated with pathogenic Fusarium species colonization of a hospital water system: a new paradigm for the epidemiology of opportunistic mold infections. Clin Infect Dis. 2001;33(11):1871–1878. doi: 10.1086/324501. [DOI] [PubMed] [Google Scholar]
- Short DPG, O’Donnell K, Thrane U, Nielsen KF, Zhang N, Juba JH, Geiser DM. Phylogenetic relationships among members of the Fusarium solani species complex in human infections and the descriptions of F. keratoplasticum sp. nov. and F. petroliphilum stat. nov. Fungal Genet Biol. 2013;53:59–70. doi: 10.1016/j.fgb.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Mehl HL, Epstein L. Sewage and community shower drains are environmental reservoirs of Fusarium solani species complex group 1, a human and plant pathogen. Environ Microbiol. 2008;10(1):219–227. doi: 10.1111/j.1462-2920.2007.01446.x. [DOI] [PubMed] [Google Scholar]
- Short DPG, O’Donnell K, Zhang N, Juba JH, Geiser DM. Widespread occurence of human pathogenic types of the fungus Fusarium detected in plumbing drains. J Clin Microbiol. 2011;49:4262–4272. doi: 10.1128/JCM.05468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso EC, Rosa RH, Miller D. Fungal keratitis. Cornea. 1997;2:1253–1265. [Google Scholar]
- O’Donnell K, Ward TJ, Aberra D, Kistler HC, Aoki T, Orwig N, Kimura M, Bjørnstad Å, Klemsdal SS. Multilocus genotyping and molecular phylogenetics resolve a novel head blight pathogen within the Fusarium graminearum species complex from Ethiopia. Fungal Genet Biol. 2008;45(11):1514–1522. doi: 10.1016/j.fgb.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S, Schwartz DC, Freitag M, Ma LJ, Danchin EGJ, Henrissat B, Coutinho PM, Nelson DR, Straney D, Napoli CA, Barker BM, Gribskov M, Rep M, Kroken S, Molnár I, Rensing C, Kennell JC, Zamora J, Farman ML, Selker EU, Salamov A. et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5(8):e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell K, Cigelnik E, Nirenberg HI. Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia. 1998;90(3):465–493. doi: 10.2307/3761407. [DOI] [Google Scholar]
- Nectria haematococca v2.0. http://genome.jgi.doe.gov/Necha2/Necha2.home.html.
- Tandem Repeats Finder. http://tandem.bu.edu/trf/trf.html.
- Primer3web version 4.0.0. http://frodo.wi.mit.edu/primer3.
- Sikes DS, Lewis PO. Beta software, version 1. PAUPRat: PAUP* implementation of the parsimony ratchet. Distributed by the authors. Department of Ecology and Evolutionary Biology. Storrs, CT, USA: University of Connecticut; 2001. [Google Scholar]
- Villesen P. FaBox: an online toolbox for fasta sequences. Mol Ecol Notes. 2007;7(6):965–968. doi: 10.1111/j.1471-8286.2007.01821.x. [DOI] [Google Scholar]
- Nei M, Tajima F. DNA polymorphism detectable by restriction endonucleases. Genetics. 1981;97(1):145–163. doi: 10.1093/genetics/97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubold B, Hudson RR. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics. 2000;16(9):847–849. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47. [PMC free article] [PubMed] [Google Scholar]
- Agapow PM, Burt A. Indices of multilocus linkage disequilibrium. Mol Ecol Notes. 2001;1(1–2):101–102. [Google Scholar]
- Spratt BG, Hanage WP, Li B, Aanensen DM, Feil EJ. Displaying the relatedness among isolates of bacterial species – the eBURST approach. FEMS Microbiol Lett. 2004;241(2):129–134. doi: 10.1016/j.femsle.2004.11.015. [DOI] [PubMed] [Google Scholar]
- eBURSTv3. http://eburst.mlst.net.
- Francisco AP, Bugalho M, Ramirez M, Carriço JA. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinforma. 2009;10(1):152. doi: 10.1186/1471-2105-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carriço J. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinforma. 2012;13(1):87. doi: 10.1186/1471-2105-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Barr JR, Barr DB, Brock JW, Elie CM, Ueno Y, Patterson DG Jr, Potter ME, Reiss E. Physiological characteristics and mycotoxins of human clinical isolates of Fusarium species. Mycol Res. 1999;103(11):1462–1468. doi: 10.1017/S095375629900862X. [DOI] [Google Scholar]
- Smith JM, Smith NH, O’Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci U S A. 1993;90(10):4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14(1):68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- Holland BR, Huber KT, Dress A, Moulton V. δ plots: a tool for analyzing phylogenetic distance data. Mol Biol Evol. 2002;19(12):2051–2059. doi: 10.1093/oxfordjournals.molbev.a004030. [DOI] [PubMed] [Google Scholar]
- Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61(1):1–10. doi: 10.1016/0006-3207(92)91201-3. [DOI] [Google Scholar]
- Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172(4):2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauff F, Lutzoni F. Phylogeny of the Gyalectales and Ostropales (Ascomycota, Fungi): among and within order relationships based on nuclear ribosomal RNA small and large subunits. Mol Phylogenet Evol. 2002;25(1):138–156. doi: 10.1016/S1055-7903(02)00214-2. [DOI] [PubMed] [Google Scholar]
- Imamura Y, Chandra J, Mukherjee PK, Lattif AA, Szczotka-Flynn LB, Pearlman E, Lass JH, O’Donnell K, Ghannoum MA. Fusarium and Candida albicans biofilms on soft contact lenses: model development, influence of lens type, and susceptibility to lens care solutions. Antimicrob Agents Chemother. 2008;52(1):171–182. doi: 10.1128/AAC.00387-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume EBH, Flanagan J, Masoudi S, Zhu H, Cole N, Willcox MD. Soft contact lens disinfection solution efficacy: clinical Fusarium isolates vs. ATCC 36031. Optom Vis Sci. 2009;86(5):415–419. doi: 10.1097/OPX.0b013e31819fa239. [DOI] [PubMed] [Google Scholar]
- Avise JC. Molecular Markers, Natural History, and Evolution Second Edition. Sunderland MA: Sinauer Associates; 2004. [Google Scholar]
- Henk DA, Shahar-Golan R, Devi KR, Boyce KJ, Zhan N, Fedorova ND, Nierman WC, Hsueh PR, Yuen KY, Sieu TPM, Van Kinh N, Wertheim H, Baker SG, Day JN, Vanittanakom N, Bignell EM, Andrianopoulos A, Fisher MC. Clonality despite sex: the evolution of host-associated sexual neighborhoods in the pathogenic fungus Penicillium marneffei. PLoS Pathog. 2012;8(10):e1002851. doi: 10.1371/journal.ppat.1002851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M. In: Sex in Fungi: Molecular Determination and Evolutionary Implications. Heitman J, Kronstad JW, Taylor JW, Casselton LA, editor. Washington, DC: ASM Press; 2005. The evolutionary implications of an asexual lifestyle manifested by Penicillium marneffei. [Google Scholar]
- Gräser Y, Volovsek M, Arrington J, Schönian G, Presber W, Mitchell TG, Vilgalys R. Molecular markers reveal that population structure of the human pathogen Candida albicans exhibits both clonality and recombination. Proc Natl Acad Sci U S A. 1996;93(22):12473–12477. doi: 10.1073/pnas.93.22.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson AR, Pujol C, Pfaller MA, Denning DW, Soll DR. Evidence for recombination in Candida glabrata. Fungal Genet Biol. 2005;42(3):233–243. doi: 10.1016/j.fgb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Matute DR, McEwen JG, Puccia R, Montes BA, San-Blas G, Bagagli E, Rauscher JT, Restrepo A, Morais F, Niño-Vega G, Taylor JW. Cryptic speciation and recombination in the fungus Paracoccidioides brasiliensis as revealed by gene genealogies. Mol Biol Evol. 2006;23(1):65–73. doi: 10.1093/molbev/msj008. [DOI] [PubMed] [Google Scholar]
- Pujol C, Daniels KJ, Lockhart SR, Srikantha T, Radke JB, Geiger J, Soll DR. The closely related species Candida albicans and Candida dubliniensis can mate. Eukaryot Cell. 2004;3(4):1015–1027. doi: 10.1128/EC.3.4.1015-1027.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh LA, Fraser JA, Dietrich FS. Recent evolution of the human pathogen Cryptococcus neoformans by intervarietal transfer of a 14-gene fragment. Mol Biol Evol. 2006;23(10):1879–1890. doi: 10.1093/molbev/msl070. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, Barker BM, Sharpton TJ, Stajich JE, Park DJ, Whiston E, Hung CY, McMahan C, White J, Sykes S, Heiman D, Young S, Zeng Q, Abouelleil A, Aftuck L, Bessette D, Brown A, FitzGerald M, Lui A, Macdonald JP, Priest M, Orbach MJ, Galgiani JN, Kirkland TN, Cole GT, Birren BW, Henn MR, Taylor JW, Rounsley SD. Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res. 2010;20(7):938–946. doi: 10.1101/gr.103911.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgenson JE, Bowden RL, Zeller KA, Leslie JF, Alexander NJ, Plattner RD. A genetic map of Gibberella zeae (Fusarium graminearum) Genetics. 2002;160(4):1451–1460. doi: 10.1093/genetics/160.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasson MT, O’Donnell K, Rooney AP, Sink S, Ploetz RC, Ploetz JN, Konkol JL, Carrillo D, Freeman S, Mendel Z, Smith JA, Black AW, Hulcr J, Bateman C, Stefkova K, Campbell PR, Geering ADW, Dann EK, Eskalen A, Mohotti K, Short DPG, Aoki T, Fenstermacher KA, Davis DD, Geiser DM. An inordinate fondness for Fusarium: phylogenetic diversity of fusaria cultivated by ambrosia beetles in the genus Euwallacea on avocado and other plant hosts. Fungal Genet Biol. 2013;56:147–157. doi: 10.1016/j.fgb.2013.04.004. [DOI] [PubMed] [Google Scholar]
- O’Donnell K, Kistler HC, Tacke BK, Casper HH. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc Natl Acad Sci U S A. 2000;97(14):7905–7910. doi: 10.1073/pnas.130193297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller KA, Bowden RL, Leslie JF. Population differentiation and recombination in wheat scab populations of Gibberella zeae from the United States. Mol Ecol. 2004;13(3):563–571. doi: 10.1046/j.1365-294X.2004.02098.x. [DOI] [PubMed] [Google Scholar]
- Miao VP, Covert SF, Van Etten HD. A fungal gene for antibiotic resistance on a dispensable (“ B”) chromosome. Science. 1991;254(5039):1773–1776. doi: 10.1126/science.1763326. [DOI] [PubMed] [Google Scholar]
- Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman PM, Kang S, Shim WB, Woloshuk C, Xie X, Xu JR, Antoniw J, Baker SE, Bluhm BH, Breakspear A, Brown DW, Butchko RAE, Chapman S, Coulson R, Coutinho PM, Danchin EGJ, Diener A, Gale LR, Gardiner DM, Goff S. et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464(7287):367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti M, Rydholm C, Schwier EU, Anderson MJ, Szakacs G, Lutzoni F, Debeaupuis JP, Latgé JP, Denning DW, Dyer PS. Evidence for sexuality in the opportunistic fungal pathogen Aspergillus fumigatus. Curr Biol. 2005;15(13):1242–1248. doi: 10.1016/j.cub.2005.05.045. [DOI] [PubMed] [Google Scholar]
- Odds FC, Bougnoux ME, Shaw DJ, Bain JM, Davidson AD, Diogo D, Jacobsen MD, Lecomte M, Li SY, Tavanti A, Maiden MCJ, Gow NAR, d'Enfert C. Molecular phylogenetics of Candida albicans. Eukaryot Cell. 2007;6(6):1041–1052. doi: 10.1128/EC.00041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter DA, Burt A, Taylor JW, Koenig GL, White TJ. Clinical isolates of Histoplasma capsulatum from Indianapolis, Indiana, have a recombining population structure. J Clin Microbiol. 1996;34(10):2577–2584. doi: 10.1128/jcm.34.10.2577-2584.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
111 nine-locus sequence types and their frequencies based on the diversity of the 231 F. keratoplasticum isolates used in this study.