

Abstract
Despite being one of the most promising amphiphilic block copolymers, use of Pluronic F68 in drug delivery is limited due to its high critical micelle concentration (CMC). In this study, we developed a novel F68 derivative, cholesterol-coupled F68 (F68-CHMC). This new derivative has a CMC of 10 μg/mL, which is 400-fold lower than that of F68. The drug-loading capacity of F68-CHMC was investigated by encapsulating cabazitaxel, a novel antitumor drug. Drug-loaded micelles were fabricated by a self-assembly method with simple dilution. The optimum particle size of the micelles was 17.5±2.1 nm, with an entrapment efficiency of 98.1% and a drug loading efficiency of 3.16%. In vitro release studies demonstrated that cabazitaxel-loaded F68-CHMC micelles had delayed and sustained-release properties. A cytotoxicity assay of S180 cells showed that blank F68-CHMC was noncytotoxic with a cell viability of nearly 100%, even at a concentration of 1,000 μg/mL. The IC50 revealed that cabazitaxel-loaded F68-CHMC micelles were more cytotoxic than Tween 80-based cabazitaxel solution and free cabazitaxel. In vivo antitumor activity against S180 cells also indicated better tumor inhibition by the micelles (79.2%) than by Tween 80 solution (56.2%, P<0.05). Based on these results, we conclude that the F68-CHMC copolymer may be a potential nanocarrier to improve the solubility and biological activity of cabazitaxel and other hydrophobic drugs.
Keywords: Pluronic F68, cholesterol, synthesis, cabazitaxel, micelles, cancer therapy
Introduction
Pluronic, which consists of hydrophilic ethylene oxide and hydrophobic propylene oxide units, is a well known amphiphilic block copolymer. Structural studies show that pluronic can self-assemble into micelles in aqueous solutions and has aroused wide concern in drug delivery and drug targeting.1–4 In these micelles, the hydrophobic propylene oxide core serves as a “pool” for incorporation of various insoluble compounds, whereas the hydrophilic ethylene oxide shell maintains the micelles in a dispersed state in aqueous solution and avoids renal clearance and nonspecific reticuloendothelial uptake in vivo.5 Several pluronic micelles have been studied, and found to be able to solubilize drugs and preserve their activity in a biological medium.3,6,7 In particular, a mixed micelle formulation of pluronic L61 and F127 loaded with doxorubicin showed a satisfactory antitumor effect in patients with metastatic adenocarcinoma of the esophagus, gastroesophageal junction, or stomach in a Phase II clinical trial.8,9 Further research showed that mixed micelles had the ability to deplete cancer stem cells and decrease the tumorigenicity of cancer cells in vivo.10
F68 is the only pluronic approved by the US Food and Drug Administration for intravenous injection. Michael et al reported that F68 was well tolerated clinically after continuous intravenous infusion over 48 hours at cumulative doses as high as 14,400 mg/kg (300 mg/kg/hour × 48 hours).11 However, the CMC of F68 is very high (4.8 × 10−4 M) due to the short propylene oxide segments, resulting in low drug loading and poor dilution stability.5 Importantly, this drawback of F68 may also lead to rapid drug release before it reaches the target site. To overcome the instability of F68 micelles, several stabilization approaches have been investigated, including mixed micelles and chemical modifications.12,13
Cholesterol, an essential membrane component in animal cells, is produced by the liver. Boyd et al found that cancer cells obtained a large amount of cholesterol from serum to support their rapid growth, which led to a metabolic disorder of cholesterol and hypocholesterolemia.14 These findings were confirmed in clinical trials, which showed a lower concentration of cholesterol in serum and a higher concentration in malignant cells from patients with cancer of the breast, prostate, liver, stomach, or esophagus.15–17 Cholesterol plays an important role in molecular events associated with the cell membrane, such as membrane fusion and endocytosis, and can also promote siRNA transfection.18 Studies have demonstrated that lipoplexes with a high cholesterol content show enhanced tumor distribution.19 Another study reported that lipoplexes containing folate-cholesterol showed a 50-fold increase in transfection compared with those with folate-DSPE (1,2-distearoyl-sn-glycero-3-phosphoethanolamine).20 In addition, cholesterol has excellent compatibility with the stereocenters in paclitaxel and may interact with those to form stereocomplexes. Lee et al showed that micelles fabricated from an amphiphilic cholesterol-bearing copolymer, ie, poly[N-methyldietheneamine sebacate-co(cholesteryl oxocarbonylamido ethyl) methyl bis (ethylene) ammonium bromide sebacate], achieved a high paclitaxel loading of about 14% with an encapsulation efficiency of about 92% using a simple self-assembly process without homogenization or sonication.21
Cabazitaxel (previously also known as taxoid, XRP6258, TXD258, or RPR 116258A) is a novel antineoplastic agent belonging to the taxane class. Due to its poor affinity for P-glycoprotein, cabazitaxel has demonstrated antitumor activity not only against docetaxel-sensitive tumor models but also against docetaxel-insensitive tumor models.22–24 Cabazitaxel was approved “for the treatment of patients with hormone-refractory metastatic prostate cancer previously treated with a docetaxel-containing regimen” by the US Food and Drug Administration in June 2010.25 Like other taxanes, cabazitaxel is poorly soluble in water because of its bulky polycyclic structure. A clinical formulation of cabazitaxel (Jevtana®, Sanofi, Bridgewater, NJ, USA) uses Tween 80 and ethanol to increase its solubility, which unfortunately may cause hypersensitivity, neurotoxicity, and other severe side effects.24,25 In this study, a novel copolymer cholesterol-coupled F68 (F68-CHMC) was synthesized and used to encapsulate cabazitaxel into micelles by a simple self-assembly method. To evaluate the potential use of these micelles as drug delivery vehicles, we investigated their physicochemical properties, cytotoxicity in vitro, and anti-tumor efficacy in vivo.
Materials and methods
Materials
Pluronic F68 (molecular weight 8,400) was a gift from BASF Co, Ltd, (Mannheim, Germany). Cholesterol chloroformate was purchased from Tokyo Chemical Industry Co, Ltd, (Tokyo, Japan). Cabazitaxel was sourced from Taizhou Baolong Chemical Co, Ltd, (Zhejiang, People’s Republic of China). Tween 80 was obtained from Merck KGaA Co, Ltd, (Darmstadt, Germany). 4-Dimethylaminopyridine, triethylamine, and dichloromethane were purchased from Shandong Yuwang Co, Ltd, (Shandong, People’s Republic of China). 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), Roswell Park Memorial Institute 1640 medium (RPMI 1640), and fetal bovine serum were purchased from KeyGen Biotech Co, Ltd, (Nanjing, People’s Republic of China). S180 cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). All other reagents were of analytical grade and obtained from commercial sources.
Male Kunming mice weighing 18–22 g were purchased from the Central Animal Laboratory of Shenyang Pharmaceutical University (Shenyang, People’s Republic of China). All mice had free access to food and water. All experiments were carried out in accordance with the guidelines of the local animal welfare committee.
Synthesis and characterization of F68-CHMC
The F68-CHMC copolymer was synthesized using the method suggested in the literature.26 Briefly, a solution of cholesteryl chloroformate (0.24 mmol) in 16 mL of dichloromethane was added dropwise to F68 (0.04 mmol) at 0°C with gentle stirring, followed by addition of 4-dimethylaminopyridine (0.02 mmol) and triethylamine (20 μL). The mixture was reacted for 36 hours at 45°C under nitrogen gas. After the reaction was completed, the reaction mixture was dried by rotary evaporation. Five milliliters of cold ethyl ether was added to the residue and white crystals were precipitated subsequently. The crystals were washed three times with ethyl ether to eliminate excess cholesteryl chloroformate. The crude product was then redissolved with dichloromethane and washed with 100 mM HCl, saturated sodium chloride, and distilled water three times to remove the 4-dimethylaminopyridine and triethylamine. The organic layer was rotary evaporated and the product was again precipitated using cold ethyl ether. The purified copolymer was then dried under a vacuum and stored at −20°C. The accompanying reactions are outlined in Figure 1A.
Figure 1.

Synthesis and preparation of F68-CHMC micelles.
Notes: (A) Synthesis of F68-CHMC. (B) Preparation of cabazitaxel-loaded F68-CHMC micelles.
Abbreviations: CABA, cabazitaxel; CHM, cholesteryl chloroformate; F68-CHMC, cholesterol-coupled F68; DMAP, 4-dimethylaminopyridine; TEA, triethylamine; DCM, dichloromethane; Chol, cholesterol; PEO, poly(ethylene oxide); PPO, poly(propylene oxide).
Determination of critical micelle concentration
The CMC of F68-CHMC in deionized water was determined by pyrene as a fluorescence probe. Aliquots of pyrene in acetone solution (1×10−5 M, 100 μL) were added to volumetric flasks and nitrogen-dried to remove the solvent. Next, 10 mL aliquots of polymer solutions of varying concentrations were added to the volumetric flasks. The mixture obtained was left on the shaker to equilibrate for 24 hours in the dark at 25°C before fluorescence measurements were performed. The final concentration of pyrene in each volumetric flask was 1×10−7 M. The fluorescence spectra were recorded by a luminescence spectrometer (SpectraMax M3; Molecular Devices LLC, Sunnyvale, CA, USA) at room temperature. The excitation spectra were scanned at wavelengths from 300 nm to 350 nm with an emission wavelength of 397 nm. The fluorescence intensity ratio of I338/I334 from the excitation spectra was analyzed as a function of the micelle concentration.
Preparation of cabazitaxel-loaded F68-CHMC micelles
Cabazitaxel-loaded F68-CHMC micelles were prepared by a self-assembly method.27 In a typical experiment, 5 mg of cabazitaxel and 150 mg of F68-CHMC were dissolved in0.5 mL of dehydrated ethanol, and normal saline was rapidly injected into the homogeneous phase to obtain a clear micelle solution with a volume of 10 mL. The final preparation was filtered with a 0.22 μm pore size syringe filter.
Characterization of cabazitaxel-loaded F68-CHMC micelles
Characterization of micelles
Cabazitaxel-loaded F68-CHMC micelles were identified by differential scanning calorimetry (DSC) and Fourier transform infrared (FTIR) spectroscopy. For the DSC experiments, samples were placed in open aluminum pans under a dry nitrogen atmosphere (50 mL per minute) and heated at 10°C per minute from 30°C to 200°C, using a DSC-SP Rheometric Scientific differential scanning calorimeter (Rheometric Scientific Inc., NJ, USA). For the FTIR experiments, the micelles were dispersed in KBr and then compressed to disks. FTIR spectra were scanned in the spectral range of 4,000–400 cm−1 using a spectrophotometer at 2 cm−1 resolution.
Drug-loading efficiency
The amount of cabazitaxel encapsulated in the micelles was measured by an ultraviolet detector at the λmax value of 230 nm. The micelle solution was suitably diluted with dimethylformamide and filtered with a 0.45 μm pore size filter before determination. Encapsulation efficiency (EE%) and drug loading (DL%) were calculated with the following equations: EE% = (actual amount of drug encapsulated in micelles)/(initial amount of drug used in micelles) × 100% and DL% = (weight of drug in micelles)/(weight of micelles) × 100%.
Particle size and morphology
The mean diameter and zeta potential of the micelles were estimated using dynamic laser light scattering (Nicomp™ 380 submicron particle sizer, Particle Sizing Systems, Goleta, CA, USA) operated at a wavelength of 632.8 nm at 25°C. Each sample was measured three times and the values were reported as the mean diameter ± standard deviation.
The surface morphology of the micelles was measured by transmission electron microscopy (TEM, JM-1200EX, JEOL Ltd, Japan). The micelle solutions were dropped onto carbon-coated copper and stained with osmium tetroxide (1%, w/v). The samples were then allowed to dry slowly in air before measurement.
In vitro release of micelles
The in vitro release behavior of cabazitaxel from F68-CHMC micelles was investigated using the dialysis method.28 Briefly, 2 mL of micelle solution was placed in a dialysis bag (molecular mass cutoff 5 kDa). The dialysis bag was then incubated in 200 mL of phosphate-buffered saline (pH 7.4) containing 0.5% (w/v) Tween 80 at 37°C with a stirring speed of 100 rpm. Aliquots of dissolution medium (1 mL) were withdrawn and replaced with 1 mL of fresh release medium at 2, 4, 6, 8, 12, 24, 48, 72, 96, 120, and 144 hours. The concentrations of the released cabazitaxel were determined by the same high-performance liquid chromatography procedure mentioned above.
Dilution stability of the micelles
The dilution stability of the drug-loaded F68-CHMC micelles was investigated by incubating them in phosphate-buffered saline (pH 7.4) in 5–100-fold dilutions at room temperature for 24 hours. The changes in particle size and entrapment efficiency were analyzed by the method mentioned above.
In vitro cytotoxicity assay
Cell culture
Mouse S180 ascites tumor cells were cultured in RPMI 1640 with 10% (v/v) heat-inactivated fetal bovine serum, penicillin G 100 U/mL, and streptomycin 100 μg/mL. Cells were cultivated in a humidified environment at 37°C with 5% CO2 (v/v). All experiments were performed on cells in the exponential growth phase.
In vitro cytotoxicity
The MTT method was used to evaluate the cytotoxic effects of blank F68-CHMC micelles and cabazitaxel-loaded F68-CHMC micelles against S180 cells. Cells were seeded at a density of 5×103 cells per well in 96-well plates and incubated for 24 hours. The cells were then exposed to various concentrations of the formulations at 37°C for 72 hours followed by addition of 20 μL of MTT solution and incubation for another 4 hours. The cell culture medium was removed after centrifugation (10,000 rpm, 10 minutes), and replaced with 200 μL of dimethylsulfoxide. Cells incubated with medium were used as the control. The absorbance readings of the formazan crystals were measured using a multiwall scanning spectrophotometer (Model 680, Bio-Rad, Hercules, CA, USA) at a wavelength of 570 nm. Based on these measurements, cell viability was calculated as a percentage by (absorbance of cells exposed to micelles or drug)/(absorbance of cells cultured without micelles or drug). All assays were performed in triplicate.
Antitumor activity test
The antitumor activity of the cabazitaxel preparations was investigated in a mouse S180 xenograft model. In brief, an S180 cell suspension (1 × 106 cells) was injected subcutaneously into the left axillary flank on a day designated as day 0. The animals were randomly assigned to three groups, with each group containing six mice. Mice in the treatment groups were injected intravenously with Tween 80-based cabazitaxel solution and cabazitaxel-loaded F68-CHMC micelles at a dose of 5 mg/kg through the tail vein. The control group received injections of normal saline (0.2 mL). Treatments were initiated as multiple doses on days 3, 6, 9, and 12, and the mice were euthanized on day 15. The tumor sizes were measured on alternate days using Vernier calipers and the tumor volumes (mm3) were calculated using the equation: V=0.5ab2, where length (a) was the longest diameter and width (b) was the shortest diameter perpendicular to length.
Statistical analysis
Statistical analysis of the samples was determined by one-way analysis of variance and P-values <0.05 were considered to be statistically significant. All data are presented as the mean ± standard deviation.
Results and discussion
Characterization of F68-CHMC
In this study, hydrophobic cholesterol chloroformate was chosen to modify F68 via an esteratic reaction as described above (Figure 1A). The structure of F68-CHMC was characterized by FTIR, 1H-nuclear magnetic resonance (NMR), and 13C-NMR. Figure 2 presents the typical FTIR spectra of F68, cholesterol chloroformate, and F68-CHMC. In the spectrum of F68-CHMC, the band at approximately 1,742.3 cm−1 was assigned to the stretching vibration of the C=O bond of the carbonate ester, which was different from that of the acyl chloride, which was approximately 1,776.6 cm−1. The spectrum was consistent with the successful coupling of cholesterol with F68. Figure 3A shows the 1H-NMR spectrum of F68-CHMC. The major features of 1H-NMR reveal that the chemical shifts observed at 1.11, 3.38, and 3.50 ppm belonged to the protons of −CH3, −CH, and −CH2 in the poly(propylene oxide) (PPO) units of the F68 block, respectively, and the chemical shift at 3.62 ppm was attributed to methylene protons of −CH2CH2O in the poly(ethylene oxide) (PEO) units of the F68 block. Other chemical shifts were similar with the protons of cholesterol chloroformate. Figure 3B shows the 13C-NMR spectrum of F68-CHMC. The major features of 13C-NMR show that 167.75 ppm was the carbon of the ester bond formed by the acyl chloride of cholesterol chloroformate and the hydroxyl of F68. The peaks at 17.36, 70.50, and 73.37 ppm belonged to the carbons of −CH3, −CH, and −CH2 in the PPO units of the F68 block, respectively, and the peak at 75.33 ppm was attributed to the carbons of −CH2CH2O in the PEO units of the F68 block. Other chemical shifts were similar to the carbons of cholesterol chloroformate. All results from FTIR, 1H-NMR, and 13C-NMR demonstrate that F68-CHMC was synthesized successfully.
Figure 2.
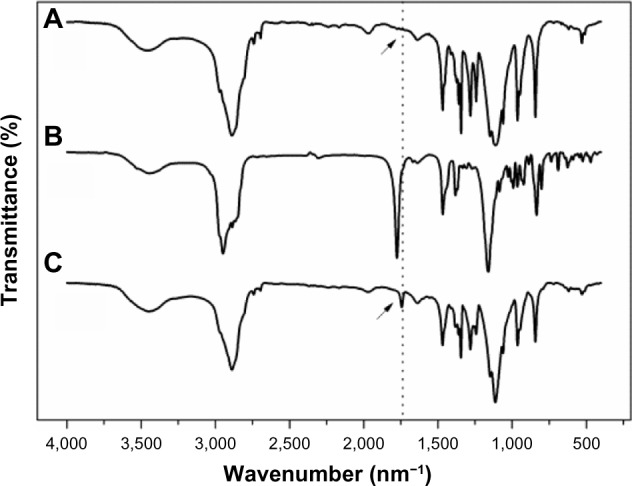
Fourier transform infrared spectra of (A) F68, (B) cholesteryl chloroformate, and (C) F68-CHMC.
Abbreviation: F68-CHMC, cholesterol-coupled F68.
Figure 3.
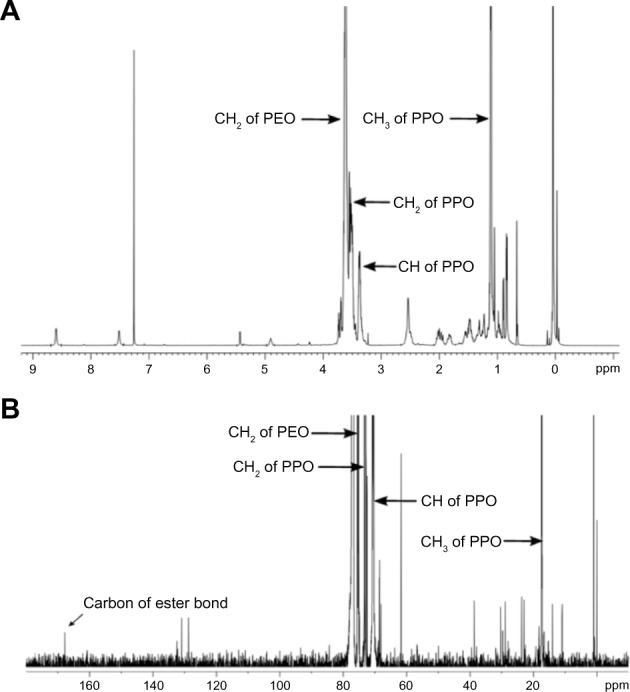
Typical 1H-NMR and 13C-NMR spectra of F68-CHMC.
Notes: (A)1H-NMR of F68-CHMC; (B)13C-NMR of F68-CHMC.
Abbreviations: F68-CHMC, cholesterol-coupled F68; PEO, poly(ethylene oxide); PPO, poly(propylene oxide); NMR, nuclear magnetic resonance.
The CMC indicates the micellar stability in vitro and in vivo. A low critical micelle concentration confers great resistance to dilution. In this study, we used pyrene as a fluorescence probe to determine the CMC of F68-CHMC.29 Using this method, the CMC of the copolymer was 10 μg/mL (Figure 4), which was 400 times lower than that of F68 (4.8 × 10−4 M). This result indicates that the micelle structure formed by F68-CHMC would confer great resistance to dissociation, even upon extreme dilution in the body, and could keep the micelles stable in the bloodstream following intravenous injection. The obvious decrease in the CMC of F68-CHMC was thought to be due to the extreme hydrophobicity of the cholesterol.
Figure 4.
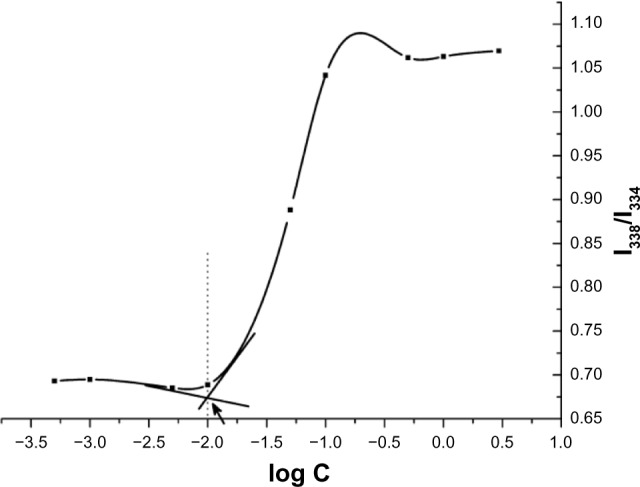
CMC determination of F68-CHMC.
Abbreviation: CMC, critical micelle concentration; F68-CHMC, cholesterol-coupled F68.
Preparation and characterization of the micelles
There are several methods that can be used for preparation of micelles, including the self-assembly method, the film-casting method, the dialysis method, and the oil in water emulsion method.27,30–32 Because of its simplicity, the self-assembly method is recognized as being outstanding, and has the potential for clinical application.27 In this study, we used the self-assembly method to prepare drug-loaded micelles. The micelles were clear and colorless with a high EE of 98.1%.
DSC and FTIR measurements were done to determine if cabazitaxel, a highly hydrophobic drug, was present in the core area of the micelles. The drug content of the physical mixtures was exactly the same as that of the drug-loaded micelles. The DSC data are shown in Figure 5A. A broad melting peak was observed at approximately 167.2°C for free cabazitaxel and for the mixture of cabazitaxel and empty micelles. However, this characteristic peak disappeared in the cabazitaxel-loaded micelles, indicating that cabazitaxel was in an amorphous state in the micelles. The FTIR spectra for the above four samples are shown in Figure 5B. In the FTIR spectrum for free cabazitaxel, the sharp peaks at 3,541.1 cm−1 and 1,519.2 cm−1 were assigned to the absorption band of the amide linkage of cabazitaxel. The peak at 1,721.0 cm−1 was the stretching vibration of C=O bonds in the carboxylic ester bond. These peaks disappeared in both the empty and cabazitaxel-loaded micelles, but still existed in the physical mixture. The FTIR results suggest that cabazitaxel was molecularly dispersed in the micelles and no chemical reaction occurred. From the results of DSC and FTIR, we conclude that cabazitaxel was stabilized in the micelles.
Figure 5.
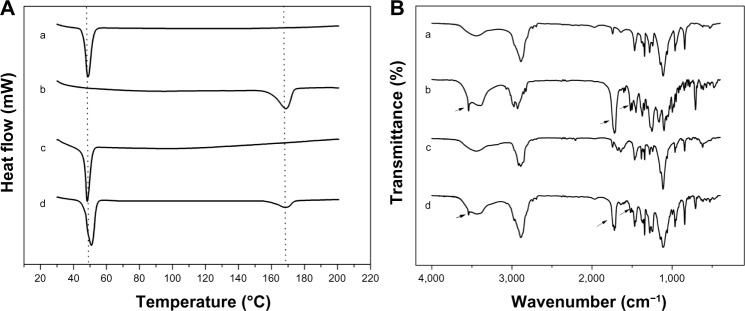
DSC theremograms and FTIR spectrograms of (a) empty F68-CHMC micelles, (b) free cabazitaxel, (c) cabazitaxel-loaded micelles, and (d) physical mixture of cabazitaxel and empty F68-CHMC micelles.
Notes: (A) DSC thermograms; (B) FTIR spectrograms.
Abbreviation: DSC, differential scanning calorimetry; F68-CHMC, cholesterol-coupled F68.
Particle size and morphology
Particle size has a direct effect on circulation time and biodistribution of carriers in vivo. Particles with a mean diameter below 200 nm can reduce uptake by the reticuloendothelial system and provide an efficiently passive tumor-targeting ability via the enhanced permeability and retention effect.33,34 In this study, the mean diameter of the empty F68-CHMC micelles was close to 14.1±1.3 nm with a narrow size distribution (approximately 0.1 of the coefficient of variation). When the empty micelles were used to load cabazitaxel, the average size was increased to 17.5±2.1 nm. The surface morphology of the drug-loaded micelles was identified by TEM. The TEM image in Figure 6 indicates that the micelles existed as homogeneous spheres with a mean diameter of approximately 20 nm, and no drug crystal was visible.
Figure 6.

Transmission electron micrograph of cabazitaxel-loaded F68-CHMC micelles.
Abbreviation: F68-CHMC, cholesterol-coupled F68.
The zeta potential also has an important effect on the stability of a colloid dispersion system. As shown in Table 1, both empty and drug-loaded micelles were negatively charged, with zeta potentials of about −2.48 mV and −3.46 mV, respectively. The low zeta potential value was due to the uncharged structure of F68-CHMC. Although there was no electrostatic repulsion between the micelles, the hydration shell formed by PEO could also keep them sterically stable. The stability of micelles can be demonstrated indirectly via the data on drug release and dilution stability.
Table 1.
Physical characteristics of empty and cabazitaxel-loaded F68-CHMC micelles
| Mean size (nm) | Coefficient of variation | Zeta potential (mV) | EE (%) | DL (%) | |
|---|---|---|---|---|---|
| Empty F68-CHMC micelles | 14.1±1.3 | 0.11±0.01 | −2.48±0.21 | – | – |
| Cabazitaxel-loaded F68-CHMC micelles | 17.5±2.1 | 0.12±0.01 | −3.46±0.35 | 98.1 | 3.16 |
Abbreviations: DL, drug loading; EE, entrapment efficiency; F68-CHMC, cholesterol-coupled F68.
In vitro drug release
In vitro release of cabazitaxel from F68-CHMC micelles was investigated under sink conditions by the dialysis method with phosphate-buffered saline (pH 7.4) containing 0.5% (w/v) Tween 80. The cumulative release of cabazitaxel from the different formulations was plotted against time, as shown in Figure 7. Free cabazitaxel could completely permeate through the dialysis tubing within 24 hours, while approximately 50% was released from Tween 80 solution and 32% from the micelles during the same period. After 144 hours, the cumulative drug release from Tween 80 solution and F68-CHMC micelles was 88% and 59%, respectively. This means that almost all of the drug was released from the Tween 80 solution and approximately 40% of the drug was retained in the micelles. The slow release of cabazitaxel-loaded F68-CHMC micelles demonstrated that the drug stayed firmly in the core of the micelles because disintegration of the micelles after dilution was a relatively slow process, mainly involving drug diffusion and micelle deaggregation.35 The release behavior of cabazitaxel-loaded F68-CHMC micelles ensures that the micelles keep in effective concentration for a long time and have the ability to sustain the treatment to the cancer in vivo.
Figure 7.
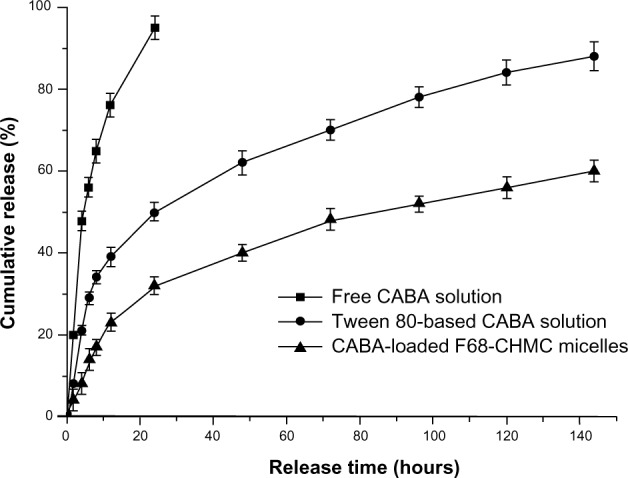
In vitro cabazitaxel release profile from dimethylsulfoxide solution, Tween 80 solution, and F68-CHMC micelles.
Notes: Phosphate-buffered saline (pH 7.4) with 0.5% (w/v) Tween 80 was used as the release medium.
Abbreviations: CABA, cabazitaxel; F68-CHMC, cholesterol-coupled F68.
Dilution stability of F68-CHMC micelles
The dilution stability of cabazitaxel-loaded F68-CHMC micelles was studied and no significant change in particle size or entrapment efficiency was found, even when diluted to 100-fold (Figure 8). One reason for this is that the concentration of F68-CHMC was 0.15 mg/mL when diluted to 100-fold, which is still above the CMC of the copolymer. Another reason may lie in the structure of F68-CHMC, which has two hydrophobic groups coexisting in the core of the micelles (see Figure 1B). The literature reports that mixed micelles could improve the micellar stability and drug loading efficiency more than their individual components.2,36 To some extent, F68-CHMC also corresponds to mixed micelles and has synergistic properties to stabilize the preparation. Further, research by Lee et al shows that paclitaxel has an affinity for cholesterol. As a structural analog of paclitaxel, cabazitaxel probably has the same affinity for cholesterol, which may be another reason for the stability of cabazitaxel in the core of the micelles.37
Figure 8.

Particle size and entrapment efficiency of F68-CHMC micelles diluted 5–100-fold with phosphate-buffered saline (pH 7.4).
Abbreviation: F68-CHMC, cholesterol-coupled F68.
Cytotoxicity of cabazitaxel-loaded F68-CHMC micelles
The cytotoxicity of empty and cabazitaxel-loaded F68-CHMC micelles was investigated in S180 cancer cells. Cell cultures were used as a blank control. Cell inhibition was analyzed using the MTT assay after 72 hours of incubation. In the testing period, empty F68-CHMC micelles showed negligible toxicity to the cancer cells, and cell viability was nearly 100%, even at the concentration of 1,000 μg/mL, whereas the viability of cells exposed to empty Tween 80 solution decreased to 70% (shown in Figure 9A). In the case of drug-containing formulations, all of the samples displayed cytotoxicity, which was enhanced as the drug concentration increased (shown in Figure 9B). The IC50 value was 27.51±2.37 ng/mL for micelles, 35.81±2.58 ng/mL for Tween 80 solution, and 39.73±2.16 ng/mL for free cabazitaxel. The micelles showed the lowest IC50 as compared with the Tween 80 solution (P<0.05) and free caba-zitaxel (P<0.05). The superior cytotoxicity of the micelles might be due to better uptake by tumor cells via an endocytic mechanism, and cholesterol may also have a synergistic effect in this process, given that it is essential for proliferation of tumor cells. Further, Tween 80-based cabazitaxel solution also showed higher cytotoxicity than the free drug. We presume that this is due to the high permeability of the cell membrane caused by Tween 80. Simoes et al reported that Tween 80 can solubilize fatty molecules and bilayer membranes, and it can deliver more drug into the cells to exert its antitumor effect.38
Figure 9.
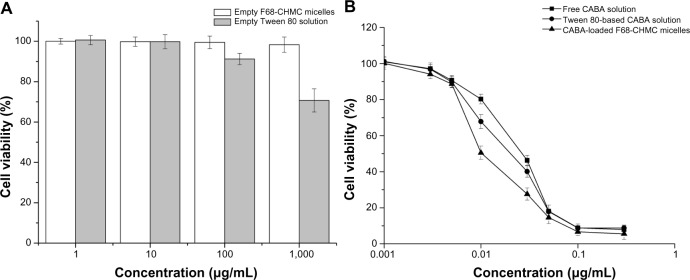
In vitro cytotoxicity of varying concentration of excipients and cabazitaxel formulations in an S180 cell line after 72 hours of incubation.
Notes: (A) excipients; (B) cabazitaxel formulations.
Abbreviations: CABA, cabazitaxel; F68-CHMC, cholesterol-coupled F68.
Antitumor activity
In recent years, xenografts of human tumors in immunodeficient mice, such as the nude mouse and SCID mouse models, were considered to be a valuable method for predicting clinical results of experimental treatment.39 To some extent, these models do have some correlations with human tumors, but they are inconsistent with the objective reality of patients with normal immune systems seen in the clinical setting. The literature shows that a healthy immune system is necessary for the control of malignant disease, and immune suppression associated with cancer contributes to its progression.40 Therefore, in our study, murine S180 cells, which have the characteristics of rapid growth, strong angiogenesis, and a good enhanced permeability and retention effect, were chosen for transplantation in Kunming mice.
Eighteen S180-bearing mice were randomly divided into three groups. Mice in the treatment groups received injections of cabazitaxel-loaded F68-CHMC micelles and Tween 80-based cabazitaxel solution at a dosage of 5.0 mg/kg. Meanwhile, control animals were injected with 0.2 mL of normal saline. No toxicity-induced deaths were observed in any of the groups during the study period, and the relative tumor volumes for the various formulations are shown in Figure 10A. Tumor sizes were similar in the treatment groups on day 6 when the animals received their second injection. By day 9, tumors in the micelle group increased less in size compared with tumors in the Tween 80 group. On the day of euthanasia, the antitumor efficacy of the micelles was 79.2%, which was significantly higher than that of the Tween 80 solution (56.2%, P<0.05). After the mice were euthanized, the xenografted tumors were excised and photographed (see Figure 10B). Typical photographs of the excised sarcomas clearly demonstrate the superior tumor suppression effect of the F68-CHMC micelles, and this result is consistent with the in vitro cytotoxicity assay. One reason for the significant tumor inhibition achieved by the F68-CHMC micelles may be passive targeting. The suitable size and unique core-shell architecture of the micelles can extend the circulation time and increase the drug concentration in tumor tissue via the enhanced permeability and retention effect.41 Another reason may be the cholesterol resident in F68-CHMC micelles, which is needed by the tumor cells and may help the cells absorb more of the drug.
Figure 10.
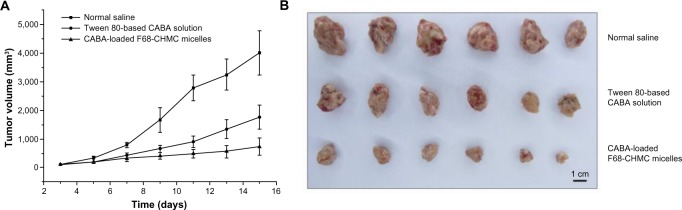
Antitumor activity of cabazitaxel-loaded F68-CHMC micelles in mice bearing S180 tumor xenografts.
Notes: (A) The tumor volume changes of S180 tumor-bearing mice post IV injection of normal saline, Tween 80-based cabazitaxel solution and cabazitaxel loaded F68-CHMC micelles. (B) The images of excised tumor tissue from S180 tumor-bearing mice at the time of euthanized day.
Abbreviations: F68-CHMC, cholesterol-coupled F68; CABA, cabazitaxel.
Conclusion
In this study, a novel F68 derivative, ie, F68-CHMC, was synthesized successfully. The CMC of this novel copolymer was 10 μg/mL, which is 400-fold lower than that of F68. An antitumor drug, cabazitaxel, was homogenously incorporated into F68-CHMC micelles via a simple self-assembly method with a high entrapment efficiency and good dilution stability. The cabazitaxel-loaded F68-CHMC micelles showed higher cytotoxicity against S180 cells than the conventional formulation, while blank micelles showed no cytotoxicity. The drug-loaded F68-CHMC micelles had enhanced antitumor activity in vivo compared with a Tween 80-based drug solution. Our findings suggest that the F68-CHMC copolymer would be an ideal carrier for the delivery of cabazitaxel, and potentially other hydrophobic antitumor drugs as well, and therefore deserves more intensive investigation.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (81373334). We are grateful to Meixia Zhang for her helpful advice and Yingchun Qu for assistance with the in vitro cytotoxicity experiment.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343–1355. doi: 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- 2.Wei Z, Hao J, Yuan S, et al. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376(1):176–185. doi: 10.1016/j.ijpharm.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 3.Sahu A, Kasoju N, Goswami P, Bora U. Encapsulation of curcumin in pluronic block copolymer micelles for drug delivery applications. J Biomater Appl. 2011;25(6):619–639. doi: 10.1177/0885328209357110. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Sha X, Jiang X, Chen Y, Ren Q, Fang X. Pluronic P105/F127 mixed micelles for the delivery of docetaxel against Taxol-resistant non-small cell lung cancer: optimization and in vitro, in vivo evaluation. Int J Nanomedicine. 2013;8:73–84. doi: 10.2147/IJN.S38221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82(2):189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 6.Oh KT, Bronich TK, Kabanov AV. Micellar formulations for drug delivery based on mixtures of hydrophobic and hydrophilic Pluronic® block copolymers. J Control Release. 2004;94(2):411–422. doi: 10.1016/j.jconrel.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Zhang Z, Chen T, Guo X, Zhou S. Preparation and characterization of thermosensitive pluronic F127-b-poly (ε-caprolactone) mixed micelles. Colloids Surf B Biointerfaces. 2011;86(1):45–57. doi: 10.1016/j.colsurfb.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong A, Brewer J, Newman C, et al. SP1049C as first-line therapy in advanced (inoperable or metastatic) adenocarcinoma of the oesophagus: a phase II window study. J Clin Oncol. 2006;24(Suppl 18):4080. [Google Scholar]
- 9.Valle JW, Armstrong A, Newman C, et al. A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Investig New Drugs. 2011;29(5):1029–1037. doi: 10.1007/s10637-010-9399-1. [DOI] [PubMed] [Google Scholar]
- 10.Alakhova DY, Zhao Y, Li S, Kabanov AV. Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS One. 2013;8(8):e72238. doi: 10.1371/journal.pone.0072238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grindel JM, Jaworski T, Piraner O, Emanuele RM, Balasubramanian M. Distribution, metabolism, and excretion of a novel surface-active agent, purified poloxamer 188, in rats, dogs, and humans. J Pharm Sci. 2002;91(9):1936–1947. doi: 10.1002/jps.10190. [DOI] [PubMed] [Google Scholar]
- 12.Zhao L, Du J, Duan Y, et al. Curcumin loaded mixed micelles composed of Pluronic P123 and F68: preparation, optimization and in vitro characterization. Colloids Surf B Biointerfaces. 2012;97:101–108. doi: 10.1016/j.colsurfb.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Cha M-H, Choi J, Choi BG, et al. Synthesis and characterization of novel thermo-responsive F68 block copolymers with cell-adhesive RGD peptide. J Colloid Interface Sci. 2011;360(1):78–85. doi: 10.1016/j.jcis.2011.04.041. [DOI] [PubMed] [Google Scholar]
- 14.Boyd N, Connelly P, Lynch H, et al. Plasma lipids, lipoproteins, and familial breast cancer. Cancer Epidemiol Biomarkers Prev. 1995;4(2):117–122. [PubMed] [Google Scholar]
- 15.Danilo C, Frank PG. Cholesterol and breast cancer development. Curr Opin Pharmacol. 2012;12(6):677–682. doi: 10.1016/j.coph.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 16.Freeman MR, Solomon KR. Cholesterol and prostate cancer. J Cell Biochem. 2004;91(1):54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 17.Dessì S, Batetta B, Pulisci D, et al. Cholesterol content in tumor tissues is inversely associated with high-density lipoprotein cholesterol in serum in patients with gastrointestinal cancer. Cancer. 1994;73(2):253–258. doi: 10.1002/1097-0142(19940115)73:2<253::aid-cncr2820730204>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 18.Xu L, Betker J, Yin H, Anchordoquy TJ. Ligands located within a cholesterol domain enhance gene delivery to the target tissue. J Control Release. 2012;160(1):57–63. doi: 10.1016/j.jconrel.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Bradshaw-Pierce EL, DeLille A, Gustafson DL, Anchordoquy TJ. In vivo comparative study of lipid/DNA complexes with different in vitro serum stability: effects on biodistribution and tumor accumulation. J Pharm Sci. 2008;97(1):237–250. doi: 10.1002/jps.21076. [DOI] [PubMed] [Google Scholar]
- 20.Xu L, Anchordoquy TJ. Effect of cholesterol nanodomains on the targeting of lipid-based gene delivery in cultured cells. Mol Pharm. 2010;7(4):1311–1317. doi: 10.1021/mp100097b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee AL, Wang Y, Pervaiz S, Fan W, Yang YY. Synergistic anticancer effects achieved by co-delivery of TRAIL and paclitaxel using cationic polymeric micelles. Macromol Biosci. 2011;11(2):296–307. doi: 10.1002/mabi.201000332. [DOI] [PubMed] [Google Scholar]
- 22.Mita AC, Denis LJ, Rowinsky EK, et al. Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15(2):723–730. doi: 10.1158/1078-0432.CCR-08-0596. [DOI] [PubMed] [Google Scholar]
- 23.Pivot X, Koralewski P, Hidalgo J, et al. A multicenter phase II study of XRP6258 administered as a 1-h iv infusion every 3 weeks in taxane-resistant metastatic breast cancer patients. Ann Oncol. 2008;19(9):1547–1552. doi: 10.1093/annonc/mdn171. [DOI] [PubMed] [Google Scholar]
- 24.Oudard S. TROPIC: phase III trial of cabazitaxel for the treatment of metastatic castration-resistant prostate cancer. Future Oncol. 2011;7(4):497–506. doi: 10.2217/fon.11.23. [DOI] [PubMed] [Google Scholar]
- 25.Paller CJ, Antonarakis ES. Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des Devel Ther. 2011;5:117. doi: 10.2147/DDDT.S13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Deng Y, Chen D, Hong W, Lu Y, Dong X. Esterase-catalyzed dePEGylation of pH-sensitive vesicles modified with cleavable PEG-lipid derivatives. J Control Release. 2008;130(3):238–245. doi: 10.1016/j.jconrel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Song H, Geng H, Ruan J, et al. Development of polysorbate 80/phospholipid mixed micellar formation for docetaxel and assessment of its in vivo distribution in animal models. Nanoscale Res Lett. 2011;6(1):1–12. doi: 10.1186/1556-276X-6-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu D, Liu Z, Wang L, Zhang C, Zhang N. Nanostructured lipid carriers as novel carrier for parenteral delivery of docetaxel. Colloids and Surfaces B: Biointerfaces. 2011;85(2):262–269. doi: 10.1016/j.colsurfb.2011.02.038. [DOI] [PubMed] [Google Scholar]
- 29.Ananthapadmanabhan K, Goddard E, Turro N, Kuo P. Fluorescence probes for critical micelle concentration. Langmuir. 1985;1(3):352–355. doi: 10.1021/la00063a015. [DOI] [PubMed] [Google Scholar]
- 30.Kohori F, Yokoyama M, Sakai K, Okano T. Process design for efficient and controlled drug incorporation into polymeric micelle carrier systems. J Control Release. 2002;78(1):155–163. doi: 10.1016/s0168-3659(01)00492-8. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Jackson JK, Burt HM. Development of amphiphilic diblock copolymers as micellar carriers of taxol. Int J Pharm. 1996;132(1):195–206. [Google Scholar]
- 32.Sant VP, Smith D, Leroux J-C. Novel pH-sensitive supramolecular assemblies for oral delivery of poorly water soluble drugs: preparation and characterization. J Control Release. 2004;97(2):301–312. doi: 10.1016/j.jconrel.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 33.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188(6):759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo X, Shi C, Wang J, Di S, Zhou S. pH-triggered intracellular release from actively targeting polymer micelles. Biomaterials. 2013;34(18):4544–4554. doi: 10.1016/j.biomaterials.2013.02.071. [DOI] [PubMed] [Google Scholar]
- 35.Kim K-S, Park S-J. Effect of porous silica on sustained release behaviors of pH sensitive pluronic F127/poly (acrylic acid) hydrogels containing tulobuterol. Colloids Surf B Biointerfaces. 2010;80(2):240–246. doi: 10.1016/j.colsurfb.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 36.Gao Z-G, Fain HD, Rapoport N. Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. J Control Release. 2005;102(1):203–222. doi: 10.1016/j.jconrel.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 37.Lee AL, Venkataraman S, Sirat S, Gao S, Hedrick JL, Yang YY. The use of cholesterol-containing biodegradable block copolymers to exploit hydrophobic interactions for the delivery of anticancer drugs. Biomaterials. 2012;33(6):1921–1928. doi: 10.1016/j.biomaterials.2011.11.032. [DOI] [PubMed] [Google Scholar]
- 38.Simoes S, Tapadas J, Marques C, Cruz M, Martins M, Cevc G. Permeabilisation and solubilisation of soybean phosphatidylcholine bilayer vesicles, as membrane models, by polysorbate, Tween 80. Eur J Pharm Sci. 2005;26(3):307–317. doi: 10.1016/j.ejps.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Sausville EA, Burger AM. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006;66(7):3351–3354. doi: 10.1158/0008-5472.CAN-05-3627. [DOI] [PubMed] [Google Scholar]
- 40.Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol. 2006;16(1):3–15. doi: 10.1016/j.semcancer.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Maeda H, Bharate G, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]