

Abstract
Cystic fibrosis (CF) is a life-shortening disease arising as a consequence of mutations within the CFTR gene. Novel therapeutics for CF are emerging that target CF transmembrane conductance regulator protein (CFTR) defects resulting from specific CFTR variants. Ivacaftor is a drug that potentiates CFTR gating function and is specifically indicated for CF patients with a particular CFTR variant, G551D-CFTR (rs75527207). Here, we provide therapeutic recommendations for ivacaftor based on preemptive CFTR genotype results.
Ivacaftor has recently been approved for the treatment of cystic fibrosis (CF) in patients with a known variant (G551D-CFTR) in the CF transmembrane conductance regulator protein (CFTR). The purpose of this guideline is to provide information to facilitate the interpretation of genotype tests in order to guide ivacaftor therapy. Detailed guidelines for the use of ivacaftor as well as analyses of cost-effectiveness are beyond the scope of this document. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines are periodically updated at http://www.pharmgkb.org.
Focused Literature Review
A systematic literature review focused on CFTR genotype and ivacaftor use (details in Supplementary Material online) was conducted.
Gene: CFTR
Background
CF is a life-shortening disease that affects >70,000 people across the globe and >30,000 individuals in the United States (Cystic Fibrosis Foundation registry statistics, 2011).1,2 CF is caused by autosomal recessive mutations in the gene coding for CFTR, located on chromosome 7q31.2. The CFTR protein is a member of the traffic adenosine 5'-triphosphatase (ATPase) family. It is characterized by two nucleotide-binding domains, nucleotide-binding domain-1 and -2, which bind to and hydrolyze ATP; and two transmembrane domains (transmembrane domain-1 and -2), which anchor the protein to the plasma membrane. The two nucleotide-binding domains of CFTR form a heterodimer complex to gate the chloride channel formed by the transmembrane domains.3,4,5 CFTR is unique among traffic ATPases in that it is regulated by cyclic adenosine monophosphate and protein kinase A–dependent phosphorylation of its unique regulatory (R) domain.
CFTR is a critical regulator of fluid and ion transports, and it also plays an important host defense role in the airways.1 CFTR transports chloride and bicarbonate6,7,8 and also functions as a regulator of other transporters (including other chloride channels), thiocyanate and glutathione transport, and the epithelial sodium channel.1,9,10,11,12 CFTR is expressed in numerous tissues, with direct relationships between its expression and CF pathology in the airways, the small and large intestines, pancreatic ducts, bile ducts, vas deferens, and sweat glands. CFTR expression is high in airway submucosal glands, and CFTR is moderately expressed on the surface epithelium of the small, medium, and large airways.13
CF airway disease relates directly to disturbed CFTR regulation of ion transport in airway cells, which directly influences airway surface liquid (ASL) volume and the hydration of mucus.14 Surface airway epithelial cells tightly control the volume of a fluid compartment called the periciliary liquid layer (PCL, a subcomponent of the ASL). When the PCL volume is normal, it promotes normal ciliary function to drive effective clearance of the overlying mucus gel layer.15,16,17 This process helps maintain a clean airway without activating secondary host defenses. Under high ASL volume conditions, the epithelial sodium channel is active and drives sodium absorption, with passive flow of chloride (via CFTR and other chloride transporters) to maintain a “dry” mucosa. When the PCL volume approximates the height of extended cilia (~7 μm), fluid absorption matches secretion and homeostasis is achieved. If the PCL and ASL volumes are low, chloride secretion helps to increase the surface volume, and this is accomplished primarily through CFTR.18 Water follows passively, with the balance of water movement following the dominant ion flux.17,19
When CFTR is defective, cyclic adenosine monophosphate–dependent chloride transport is absent, and epithelial sodium channel activity (sodium absorption) is increased. This dehydrates the ASL and PCL, interrupts mucus clearance, and begins a chain of events, including mucus stasis, airway obstruction, infection, and inflammation. Over time, this produces bronchiectasis, respiratory failure, and ultimately death in young adulthood.1 Recent research also highlights a potential direct role of bicarbonate in CF disease because it controls normal mucin unpackaging and structure.8,20 Furthermore, recent studies have demonstrated that the ASL pH is low in CF (linked to defective bicarbonate transport), which interferes with the innate immune system's ability to kill bacteria.21 The buildup of thick, tenacious, and dehydrated mucus is a unifying feature of CF across many disease-affected organs. Additional evidence also points toward relationships between CFTR deficiency and excessive inflammation, with high levels of proinflammatory cytokines and neutrophil influx.22,23
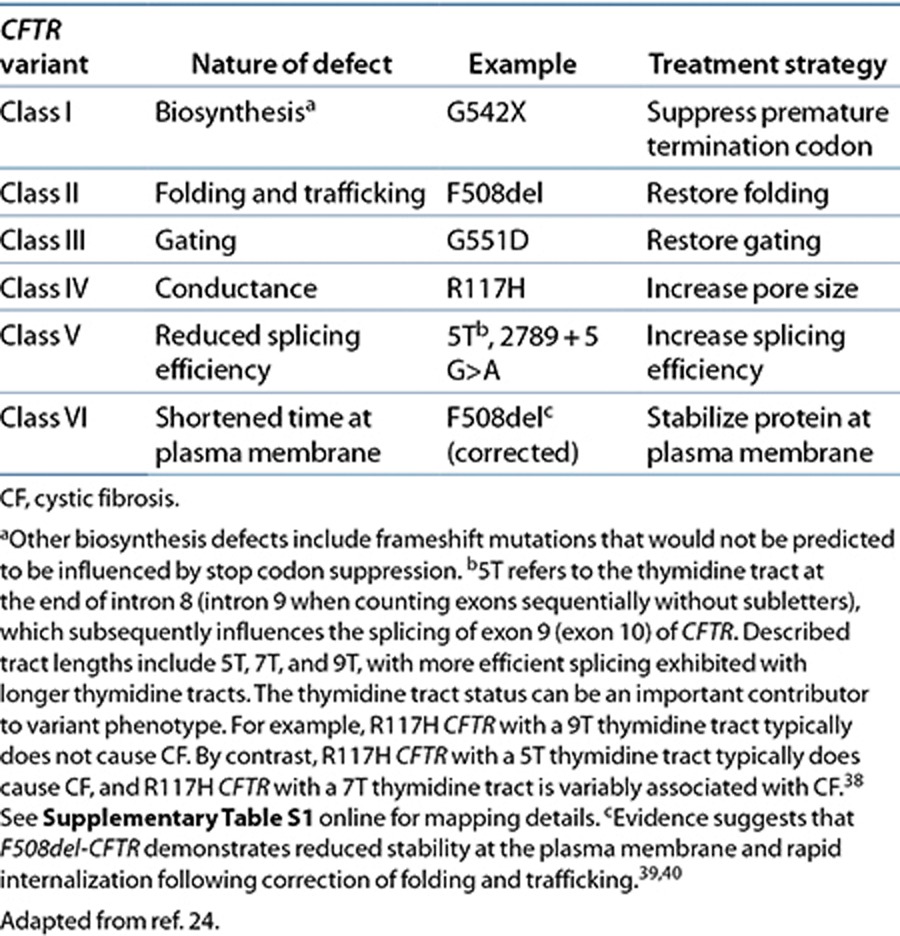
Approximately 1,900 disease-causing CFTR variants have been identified (http://www.genet.sickkids.on.ca/) (see Supplementary Table S1 online, selected CFTR variants) and have been extensively studied in multiple geographically, racially, and ethnically diverse CF patients (Supplementary Tables S2 and S3 online). Despite the large number of CFTR variants, most can be grouped into classes that have shared features (see Table 1 and Supplementary Table S1 online).24 Well-established CFTR variant classes include the following: impaired biosynthesis (class I); defective protein maturation and accelerated degradation (class II); defective regulation of CFTR at the plasma membrane (class III); defective chloride conductance (class IV); diminished CFTR transcription (class V); and accelerated turnover at the cell surface (class VI). Although many rare variants have been identified, a select few variants are more common and account for most CF clinical disease. Variants that fall into classes I, II, or III are considered nonfunctional and are associated with “severe” disease. This means that the vast majority of these patients are pancreatic insufficient and will require lifelong pancreatic enzyme replacement therapy. By contrast, patients who have at least one variant in classes IV, V, or VI retain some residual CFTR function, and the majority of these patients are pancreatic sufficient. Due to the many factors that contribute to pulmonary disease manifestations (environmental factors and the impact of modifier genes that influence the CF phenotype), it is more difficult to predict the pulmonary disease course based on the variant class of the patient.
Table 1. CFTR variant classes and strategies to restore function.
Genetic test interpretation
Patients with CF may have undergone prenatal screening or a genetic test to determine the underlying variants of their condition, and results of these genetic tests can be used to guide therapy for ivacaftor treatment. The current guidelines focus on the interpretation of genetic tests for the presence of G551D-CFTR and F508del-CFTR variants. For more information regarding screening for CF, see the Supplementary Material online.
The wealth of information regarding how various mutations disrupt CFTR function is beginning to help clinicians to categorize CF patients for viable therapeutic strategies. F508del-CFTR (Δ-F508, p.F508del, c.1521_1523delCTT (rs113993960), and c.1520_1522delTCT (rs199826652)) is the most common CFTR variant (see Supplementary Tables S1, S2, and S3 online). It is primarily a class II variant that is caused by the deletion of a phenylalanine at position 508 in the CFTR protein (Cystic Fibrosis Foundation, 2011 Patient Registry Report). The F508del-CFTR variant disrupts CFTR protein folding and processing, blocking maturation of the protein and localization to the plasma membrane.25 G551D-CFTR (p.G551D, c.1652G>A, rs75527207 allele A) is the third most common CFTR variant and is present in 4.4% of CF patients. It is a single-nucleotide change that causes a glycine to aspartic acid amino acid change at position 551 of the CFTR protein, resulting in a class III (gating) variant capable of localizing to the epithelial cell surface but with defects in ATP binding and hydrolysis (by the nucleotide-binding domains) such that G551D-CFTR fails to transport chloride normally (Cystic Fibrosis Foundation, 2011 Patient Registry Report).26
Available genetic test options
See the Supplementary Material online and http://www.PharmGKB.org for more information on commercially available clinical testing options.
Incidental findings
As noted previously, CF is an autosomal recessive disorder. The carrier frequency for CFTR variants in the US population is ~1:30.3 Two parents who are carriers may not have CF manifestations, but they do have a 25% chance of having a child with CF and a 50% chance of having a child who carries a CFTR variant. Thus, information regarding CF carrier status can be very important to extended family members of CF patients and families.
Other considerations
Not applicable.
Drug: Ivacaftor
Background
Current treatment for CF is supportive, targeting downstream manifestations that are caused by the dysfunctional CFTR protein. These treatments have led to steady advancements in patient survival but are accompanied by a high burden of care. A widely held belief is that restoring function to dysfunctional CFTR could dramatically alter the course of the disease and extend longevity.24 Therapeutic strategies that seek to restore function to mutant CFTR proteins have been reported for nearly 2 decades (see Table 1). The rationale behind these approaches is based on the determinants of total CFTR chloride channel activity, each of which is a potential mechanism to restore CFTR function. These include the amount of CFTR at the plasma membrane (N, which is influenced by both trafficking of the protein to the plasma membrane and how long it remains there), the gating of CFTR at the plasma membrane (or open-channel probability, i.e., Po), and CFTR's single-channel conductance (the ability of ions to permeate the open CFTR channel, i.e., G). Ivacaftor acts to increase the probability that the channel is open and is the first US Food and Drug Administration (FDA)–approved drug on the market that targets a specific CFTR defect.27 Before inclusion of ivacaftor in the CF therapeutic armamentarium, supportive therapies were the mainstay of practice and these targeted only secondary effects of CFTR dysfunction (e.g., pulmonary, gastrointestinal, hepatic, pancreatic, and reproductive abnormalities) rather than directly altering (or potentiating) CFTR function. Thus, ivacaftor and other treatment options that are currently being developed are leading a paradigm shift in the treatment of CF.
Originally identified by high-throughput screening of a large compound library (>200,000) followed by lead compound optimization, ivacaftor was subsequently shown to increase chloride ion flow in recombinant cells expressing the G551D-CFTR variant.27 Ultimately, it was discovered that for ivacaftor to be effective, two conditions must be present in the epithelium.
First, CFTR must be expressed on the cell surface, meaning that mutations associated with improper folding or improper trafficking of protein to the cell surface are not ameliorated by ivacaftor. Second, the expressed protein must be capable of activation via normal intracellular signaling mechanisms, in particular, cyclic adenosine monophosphate/protein kinase A pathways including β-adrenergic or adenosine receptor stimulation. Although the precise mechanism by which ivacaftor potentiates the CFTR channel is unknown (possibly through ATP-independent direct activation of the protein),28 results of in vitro studies demonstrate an association between ivacaftor treatment and enhanced chloride secretion, increased apical surface fluid, and increased beating of cilia of cultured bronchial epithelial cells from a patient with the G551D/F508del-CFTR genotype.27
Linking CFTR genetic variants to ivacaftor effects in vitro and clinically
The evidence linking genetic variants in the CFTR gene to the effect of ivacaftor has been illustrated in vitro and in several clinical trials (Supplementary Table S4 online). Registrational trials demonstrated the efficacy of ivacaftor in two randomized, placebo-controlled clinical trials including patients with at least one G551D-CFTR variant. The primary end point was improvement in lung function (forced expiratory volume in 1 s) from baseline to 24 weeks. Both trials resulted in rapid improvements in forced expiratory volume in 1 s (>10%) from baseline and in comparison with the results in the placebo group.29,30 The larger, published phase III trial also demonstrated persistent improvements in forced expiratory volume in 1 s, weight, patient-reported outcomes, and risk of pulmonary exacerbations in the ivacaftor-treated group as compared with the placebo group over 48 weeks.29
Ivacaftor has also been studied in patients homozygous for the F508del-CFTR variant. A clinical trial failed to demonstrate benefit with ivacaftor monotherapy in comparison with placebo treatment, suggesting that ivacaftor does not potentiate CFTR function in F508del-CFTR homozygotes (probably due to inadequate CFTR expression at the cell membrane)31; however, the study was not powered to examine efficacy. In vitro experiments suggest that ivacaftor alone is not an effective potentiator in cells expressing F508del-CFTR32; however, if F508del-CFTR is rescued at the plasma membrane, ivacaftor potentiates its gating function.27,33 Clinical trials are currently under way in F508del-CFTR homozygotes and heterozygotes to investigate the efficacy of combining ivacaftor with an F508del-CFTR corrector (http://clinicaltrials.gov/). Speculatively, ivacaftor may be effective in combination with a suppressor in patients with CFTR variants that affect biosynthesis of CFTR (i.e., resulting in premature termination codons).24 Individuals with CFTR variants other than G551D-CFTR that also have a similar gating defect may respond to ivacaftor treatment; however, currently the only published studies addressing this aspect are based on in vitro models24,32,34 (Supplementary Table S4 online). Several clinical trials that aim to evaluate the use of ivacaftor in patients with other CFTR variants are currently under way or are recruiting participants.
Therapeutic recommendations
The evidence outlined in Supplementary Table S4 online provides the basis of the therapeutic recommendations detailed in Table 2 and Figure 1. Ivacaftor treatment is recommended only in CF patients who are either homozygous or heterozygous for the G551D-CFTR variant. This recommendation is based on the results of a small number of clinical trials that selected for patients with the G551D-CFTR variant29,30,35 and on recommendations by regulatory bodies. Furthermore, one study demonstrated a lack of clinical effect in patients with two copies of the F508del-CFTR variant; however, this study was underpowered to evaluate efficacy.31 This recommendation is also supported by the mechanism of action of ivacaftor, identified through drug screening as potentiating the gating function of mutant CFTR,27 and subsequent in vitro studies showing that ivacaftor enhances G551D-CFTR activity.
Table 2. Prescribing ivacaftor based on CFTR genotype for patients with cystic fibrosis.
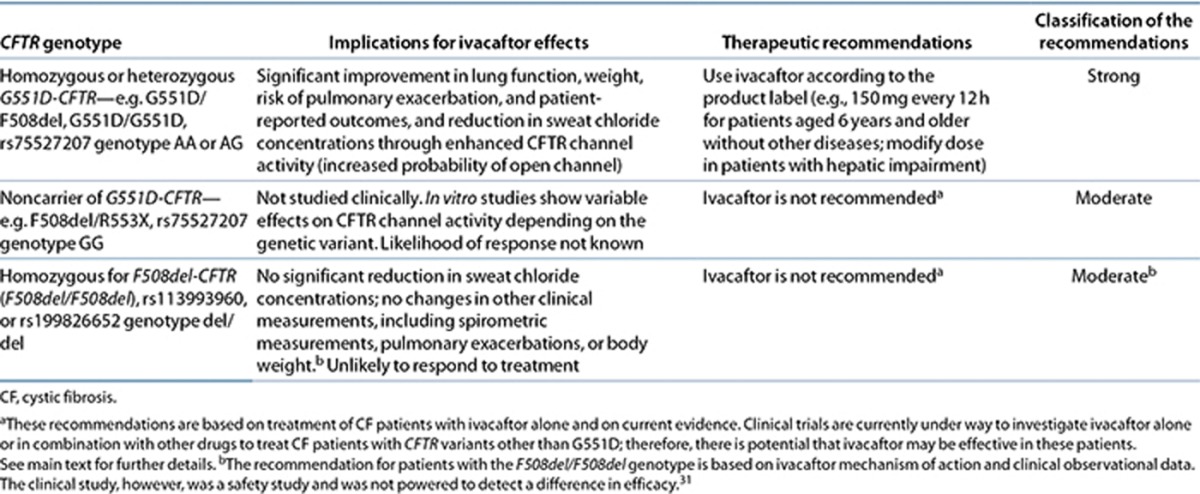
Figure 1.

Treatment algorithm for clinical use of ivacaftor for cystic fibrosis patients based on CFTR genotype.
These recommendations are consistent with the FDA and European Medicines Agency labeling for ivacaftor (see http://www.pharmgkb.org/drug/PA165950341?tabType=tabDrugLabels), in which the drug is indicated for use in CF patients who have a G551D-CFTR variant on at least one allele and is not recommended for those without a G551D-CFTR variant, including those homozygous for the F508del-CFTR variant. Both labels state that if genotype is unknown, accurate genetic testing of CFTR should be carried out before treatment with ivacaftor. In addition, the European Medicines Agency drug label states that the drug should be prescribed only by physicians who have experience in CF treatment.
Clinical trials of ivacaftor in patients with the G551D-CFTR variant were carried out in CF patients who were (i) 12 years and older29 and (ii) aged 6–11 years.30 The safety of the drug has not been established in children younger than 6 years of age; therefore, the recommendations in Table 2 are inclusive of pediatric patients aged 6 years and older, as well as in adults.
Recommendations for incidental findings
The G551D-CFTR variant is associated with CF and pancreatic insufficiency (when a nonfunctional CFTR variant is found on the other allele) (http://www.CFTR2.org).24 Patients should discuss their genotype results with a CF physician and/or a genetic counselor. Furthermore, genetic counseling is important for families with children diagnosed with CF and also for families of children who are carriers for CF variants, so that they can understand their risks of CF in future pregnancies.
Other considerations
There are no data regarding whether ivacaftor can replace other established CF therapies. The cost of ivacaftor treatment is high (>$300,000/year) relative to other CF therapies and due to the relatively small number of G551D-CFTR (and F508del/F508del) CF patients who have been treated with ivacaftor in clinical trials (<400), long-term safety data are limited. Ivacaftor may have the potential to be used in the treatment of diseases other than CF that display defects in CFTR function, for example, smoking-related lung disease such as chronic obstructive pulmonary disease36; however, this is beyond the scope of these guidelines. A patient with a G551D-CFTR variant typically has a second CFTR variant that could influence the efficacy of ivacaftor, for example, class I or class II variants that result in decreased expression of CFTR at the cell surface. Other CFTR variants with primary gating defects can also be activated by ivacaftor in vitro, and clinical trials examining patients with these variants are currently under way32 (http://clinicaltrials.gov/).
In vitro and clinical studies indicate that ivacaftor is primarily metabolized by cytochrome P450 (CYP)3A. However, to date, there are no data regarding the impact of CYP3A polymorphisms on the pharmacokinetics of ivacaftor.
Potential Benefits and Risks for the Patient
Benefits include identifying patients who are eligible for and could benefit from ivacaftor treatment. This genotype information may also contribute to understanding a patient's pathophysiology and therapeutic options. Patients with the G551D-CFTR variant are still at risk of not responding to ivacaftor treatment, and treatment outcomes vary between patients. Unfortunately, CFTR biomarkers (sweat chloride levels and nasal potential difference) did not correlate with clinical response in the phase II and phase III trials of ivacaftor in G551D-CFTR patients (forced expiratory volume in 1 s, weight, risk of pulmonary exacerbations, or patient-reported outcomes).29,35 For patients without the G551D-CFTR variant, ivacaftor is not indicated. However, several drugs targeting other CFTR variants are currently under development or in clinical trials and may benefit these patients in the future.24,37
Caveats: Appropriate use and/or Potential Misuse of Genetic Tests
Errors may occur in CFTR genotyping or in the reporting of results. If so, a patient may be subjected to ineffective ivacaftor treatment or may be omitted from potentially effective treatment with ivacaftor. The FDA label states that an FDA–cleared test should be carried out if a patient's genotype is unknown. Genetic test results should include coverage for positions rs75527207 (G551D-CFTR) and rs113993960 and rs199826652 (F508del-CFTR) in order to follow the CPIC ivacaftor guidelines.
Other genetic and environmental factors may affect a patient's response to ivacaftor. Although traditionally considered a monogenic disease, CF has a complex phenotype.24,37 CFTR itself has many roles, defects in other genes may be involved, and many physiological pathways affect a CF patient's life span and response to therapy.24,37 Ivacaftor targets the gating defect of CFTR found in patients carrying G551D-CFTR. These patients are still likely to require concomitant medications to treat the other accompanying disease manifestations and associated infections of CF. Drug–drug interactions may occur that affect ivacaftor efficacy and/or toxicity when taken in combination with other medications, in particular CYP3A inhibitors or inducers (http://www.pharmgkb.org/drug/PA165950341?tabType=tabDrugLabels), and conversely, ivacaftor may affect the efficacy and/or toxicity of concomitant therapies.
Acknowledgments
We acknowledge the critical input of members of the Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network, particularly Mary V. Relling (St Jude Children's Research Hospital), funded by the National Institutes of Health (NIH). This work is funded by NIH grants U19 GM61390 (K.M.G. and S.W.Y.), U01 GM092666, and U01 HL0105918. J.P.C. receives grant support from the NIH (NHLBI-FOA-HL 12–035) and the Cystic Fibrosis Foundation (R457-CR11, AMIN09YO). The PharmGKB is supported by NIH grant R24 GM061374.
Clinical Pharmacogenetics Implementation Consortium guidelines reflect expert consensus based on clinical evidence and peer-reviewed literature available at the time they are written and are intended only to assist clinicians in decision making and to identify questions for further research. New evidence may have emerged since the time a guideline was submitted for publication. Guidelines are limited in scope and are not applicable to interventions or diseases not specifically identified. Guidelines do not account for all individual variations among patients and cannot be considered inclusive of all proper methods of care or exclusive of other treatments. It remains the responsibility of the health-care provider to determine the best course of treatment for the patient. Adherence to any guideline is voluntary, with the ultimate determination regarding its application to be made solely by the clinician and the patient. The CPIC assumes no responsibility for any injury to persons or damage to property related to any use of CPIC guidelines, or for any errors or omissions.
J.P.C.'s institution (Cincinnati Children's Hospital Medical Center) receives contract research support from Vertex Pharmaceuticals for the conduct of CF clinical trials. He has served as a scientific advisor to Vertex Pharmaceuticals and has provided educational presentations regarding the appropriate use of Kalydeco. The other authors declared no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/cpt
Supplementary Material
References
- Rowe S.M., Miller S., Sorscher E.J. Cystic fibrosis. N. Engl. J. Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- Rogan M.P., Stoltz D.A., Hornick D.B. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest. 2011;139:1480–1490. doi: 10.1378/chest.10-2077. [DOI] [PubMed] [Google Scholar]
- Rommens J.M., et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- Riordan J.R., et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Riordan J.R. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- Anderson M.P., et al. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Welsh M.J., Smith J.J. cAMP stimulation of HCO3- secretion across airway epithelia. JOP. 2001;2:291–293. [PubMed] [Google Scholar]
- Tang L., Fatehi M., Linsdell P. Mechanism of direct bicarbonate transport by the CFTR anion channel. J. Cyst. Fibros. 2009;8:115–121. doi: 10.1016/j.jcf.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Schwiebert E.M., Benos D.J., Egan M.E., Stutts M.J., Guggino W.B. CFTR is a conductance regulator as well as a chloride channel. Physiol. Rev. 1999;79:S145–S166. doi: 10.1152/physrev.1999.79.1.S145. [DOI] [PubMed] [Google Scholar]
- Hudson V.M. Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic. Biol. Med. 2001;30:1440–1461. doi: 10.1016/s0891-5849(01)00530-5. [DOI] [PubMed] [Google Scholar]
- Moskwa P., et al. A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007;175:174–183. doi: 10.1164/rccm.200607-1029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts M.J., et al. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Engelhardt J.F., Zepeda M., Cohn J.A., Yankaskas J.R., Wilson J.M. Expression of the cystic fibrosis gene in adult human lung. J. Clin. Invest. 1994;93:737–749. doi: 10.1172/JCI117028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo N.S., Irokawa T., Robbins R.C., Wine J.J. Hyposecretion, not hyperabsorption, is the basic defect of cystic fibrosis airway glands. J. Biol. Chem. 2006;281:7392–7398. doi: 10.1074/jbc.M512766200. [DOI] [PubMed] [Google Scholar]
- Matsui H., et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- Matsui H., Randell S.H., Peretti S.W., Davis C.W., Boucher R.C. Coordinated clearance of periciliary liquid and mucus from airway surfaces. J. Clin. Invest. 1998;102:1125–1131. doi: 10.1172/JCI2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher R.C. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- Agúndez J.A., Golka K., Martínez C., Selinski S., Blaszkewicz M., García-Martín E. Unraveling ambiguous NAT2 genotyping data. Clin. Chem. 2008;54:1390–1394. doi: 10.1373/clinchem.2008.105569. [DOI] [PubMed] [Google Scholar]
- Donaldson S.H., Boucher R.C. Sodium channels and cystic fibrosis. Chest. 2007;132:1631–1636. doi: 10.1378/chest.07-0288. [DOI] [PubMed] [Google Scholar]
- Gustafsson J.K., et al. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012;209:1263–1272. doi: 10.1084/jem.20120562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzulo A.A., et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner K.H., et al. Highlights of a workshop to discuss targeting inflammation in cystic fibrosis. J. Cyst. Fibros. 2009;8:1–8. doi: 10.1016/j.jcf.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagel S.D., Chmiel J.F., Konstan M.W. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proc. Am. Thorac. Soc. 2007;4:406–417. doi: 10.1513/pats.200703-044BR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J.P., Jain M. Personalized medicine in cystic fibrosis: dawning of a new era. Am. J. Respir. Crit. Care Med. 2012;186:593–597. doi: 10.1164/rccm.201204-0785PP. [DOI] [PubMed] [Google Scholar]
- Pilewski J.M., Frizzell R.A. Role of CFTR in airway disease. Physiol. Rev. 1999;79:S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- Logan J., et al. Cystic fibrosis transmembrane conductance regulator mutations that disrupt nucleotide binding. J. Clin. Invest. 1994;94:228–236. doi: 10.1172/JCI117311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F., et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford P.D., Li C., Ramjeesingh M., Bear C.E. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 2012;287:36639–36649. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey B.W., et al. VX08-770-102 Study Group A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.C., et al. VX08-770-103 (ENVISION) Study Group Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013;187:1219–1225. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flume P.A., et al. VX 08-770-104 Study Group Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142:718–724. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012;11:237–245. doi: 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Van Goor F., et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda S., et al. A young Hispanic with c.1646G>A mutation exhibits severe cystic fibrosis lung disease: is ivacaftor an option for therapy. Am. J. Respir. Crit. Care Med. 2012;186:694–696. doi: 10.1164/ajrccm.186.7.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso F.J., et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane P.A., et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS ONE. 2012;7:e39809. doi: 10.1371/journal.pone.0039809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubamba B., Dhooghe B., Noel S., Leal T. Cystic fibrosis: insight into CFTR pathophysiology and pharmacotherapy. Clin. Biochem. 2012;45:1132–1144. doi: 10.1016/j.clinbiochem.2012.05.034. [DOI] [PubMed] [Google Scholar]
- Kiesewetter S., et al. A mutation in CFTR produces different phenotypes depending on chromosomal background. Nat. Genet. 1993;5:274–278. doi: 10.1038/ng1193-274. [DOI] [PubMed] [Google Scholar]
- Jurkuvenaite A., et al. Functional stability of rescued delta F508 cystic fibrosis transmembrane conductance regulator in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2010;42:363–372. doi: 10.1165/rcmb.2008-0434OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., et al. Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013;27:536–545. doi: 10.1096/fj.12-216119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.