

Abstract
Background/Aims
The Wnt/β-catenin signaling pathway has been reported to play an important role in liver fibrosis. This study was designed to investigate whether mesoderm-specific transcript homologue (Mest), a strong negative regulator of Wnt/β-catenin signaling, could inhibit liver fibrosis.
Methods
pcDNA-Mest was transfected into hepatic stellate cells (HSCs) and rats. Rats were randomly divided into four groups: normal group (normal saline), treatment group (pcDNA-Mest+CCl4), control group (pcDNA-neo+CCl4), and model group (normal saline+CCl4). Changes in liver pathology were evaluated by hematoxylin and eosin and Masson's trichrome staining. The levels of alanine transaminase, aspartate transaminase, lactic dehygrogenase, hyaluronic acid, and laminin in the serum and hydroxyproline in the liver were detected by biochemical examination and radioimmunoassay, respectively. The expression and distribution of β-catenin, α-smooth muscle actin (α-SMA), Smad3, and tissue inhibitor of metalloproteinase type I were determined, and the viability of the HSCs was tested.
Results
Our data demonstrate that Mest alleviated CCl4-induced collagen deposition in liver tissue and improved the condition of the liver in rats. Mest also significantly reduced the expression and distribution of β-catenin, α-SMA and Smad3 both in vivo and in vitro, in addition to the viability of HSCs in vitro.
Conclusions
We found that Mest attenuates liver fibrosis by repressing β-catenin expression, which provides a new therapeutic approach for treating liver fibrosis.
Keywords: Mesoderm-specific transcript homologue, Liver cirrhosis, Wnt/β-catenin, Hepatic stellate cell
INTRODUCTION
Liver fibrosis has been considered to be the wound-healing response of liver to various toxic stimuli, including hepatitis, alcohol, and immune compounds. Liver fibrosis is characterized by an excessive deposition of extracellular matrix (ECM) that type I collagen predominates. Hepatic stellate cell (HSCs) has been regarded as the main source of ECM. Inhibition of the activation and function of HSCs has become the most important treatment strategy for liver fibrosis.1
Wnt/β-catenin signaling pathway has been reported to play many cellular physiological functions including cell development, proliferation, and apoptosis.2 So far, four subgroups of the Wnt-mediated signaling pathway have been reported, Wnt/β-catenin (canonical) signaling, Wnt/planar cell polarity, Wnt/c-Jun N-terminal kinase, Wnt/calcium, and Wnt/Rho signaling pathway. Meanwhile, Wnt/β-catenin (canonical) signaling was demonstrated to play important roles in HSCs development and liver fibrogenesis.3,4 Inhibition of Wnt/β-catenin (canonical) signaling resulted in the downregulation of HSC activation.5 We have also reported that inhibition of Wnt/β-catenin signaling pathway could attenuate CCl4-induced liver fibrosis.6
Paternally expressed gene 1/mesoderm-specific transcript homologue (Mest) encodes an α/β hydrolase fold family enzyme. Its function remains poorly understood. However, Mest has been reported to inhibit Wnt/β-catenin signaling pathway.7 Whether Mest could attenuate liver fibrosis has not been studied. Therefore, the present study was designed to investigate the effects of Mest on hepatic fibrosis and found that Mest can suppress the development of liver fibrosis through inhibiting Wnt/β-catenin pathway.
MATERIALS AND METHODS
1. Materials
CCl4 (Wuhan Yafa Biological Technology Co., Ltd., Wuhan, China) was diluted into 40% (v/v) in olive oil before use. Enzyme-linked immunosorbent assay (ELISA) kits for determining serum alanine transaminase (ALT), aspartate transaminase (AST), and lactic dehygrogenase (LDH) were obtained from Wuhan Boster Biological Technology Co., Ltd. (Wuhan, China). Kits for serum hyaluronic acid (HA) and laminin (LN) were bought from Senxiong Co. (Shanghai, China). Kits for liver hydroxyproline (HYP) was provided by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Rabbit polyclonal antibodies against β-catenin and Mest were bought from Santa Cruz (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Rabbit polyclonal antibodies against α-smooth muscle actin (α-SMA) and β-actin, and antibodies of rabbit immunoglobulin G conjugated with horseradish peroxidase (HRP) were obtained from Wuhan Boster Biological Technology Co., Ltd.. TRIzol™ Reagent was purchased from Invitrogen (Carlsbad, CA, USA). Lipofectamine 2000 was presented by Invitrogen.
2. Plasmid pcDNA-Mest construction
A Mest expression vector, pCS2-myc-Mest was obtained from Pro. Eek-hoon Jho (Seoul) and the vector pcDNA 3.1 was from Invitrogen. The full-length cDNA for Mest was generated by polymerase chain reaction (PCR), using these primes: forward, 5'-GGT ACC ATG GTG CGC CGA GAT-3'; and reverse, 5'-GCG GCC GCT TAG TTG AGG AAG ACT-3'. The underlining sequences representing the recognition sites for Kpn I and Not I endonucleases respectively. The cDNA obtained from pCS2-myc-Mest was finally constructed into Kpn I-Not I digested multiple cloning site of pcDNA 3.1 to generate pcDNA-Mest encoding Mest protein in proper order as described previously.8 Plasmids including pcDNA 3.1 blank vector and pcDNA-Mest were purified and resuspended in ddH2O, then stored at -20℃. Purity and concentration of DNA were determined by ultraviolet spectrophotometry and agarose gel electrophoresis.
3. Animals
Male Wistar rats (Experimental Animal Center of Anhui Medical University, Hefei, China) weighing 200 to 300 g were included in this study. All animal procedures were performed under the guidelines set by Anhui Medical University Animal Care and Use Committee.
They were randomly divided into four groups (n=20 to 25 rats per group). Rats in model group were injected subcutaneously with CCl4 at dose of 3 mL/kg twice a week for 8 weeks. At the same time, rats in treatment group were given an injection of pcDNA-Mest using hydrodynamics-based gene delivery technique, via the caudal vein as described previously:9 400 µg pcDNA-Mest dissolved in a volume of phosphate-buffered saline (PBS) equivalent to 10% of the body weight was injected into the caudal vein within 10 to 15 seconds, every 3 days in combination with CCl4. Meanwhile rats in control group were treated with pcDNA 3.1-neo, while the normal group received normal saline instead of CCl4.
At the end of the experiment, rats were anesthetized with 10% chloral hydrate and sacrificed. Serum samples were collected from each rats and stored at -80℃ to determine the serum biochemical parameters ELISA kits. Livers were harvested 3 days after the last injection for three uses: 1) fixed with 10% formalin for histological examinations; 2) preserved at -80℃ for HYP kits; and 3) homogenized in Trizol for RNA isolation.
4. Biochemical determination
The levels of ALT, AST, and LDH in serum of rats were determined by ELISA kits, and the serum levels of HA and LN were detected by radioimmunoassay.
5. Histopathological examination
Liver tissues were fixed in 10% formalin, embedded in paraffin and sectioned at a thickness of 5 µm. Hematoxylin and eosin (H&E) staining was used to examine the changes in liver pathology. The collagen deposition in liver tissue was evaluated by Masson's trichrome staining. The scores of hepatic fibrosis grading were determined by two independent pathologists blindly according to the score system described by Chevallier et al.10
6. Immunohistochemical staining
Immunohistochemical staining was performed on paraffin embedded liver tissue sections of 5 µm thickness, which were deparaffinized, treated with 0.3% endogenous peroxidase blocking solution for 20 minutes. Sections were treated sequentially with 3% hydrogen peroxidase in methanol for 10 minutes at room temperature and washed with PBS for 5 minutes three times to block endogenous peroxidase activity. The liver sections were then incubated with rat anti-β-catenin antibody at a dilution of 1:200 for 1.5 hours at room temperature and then incubated with HRP-labeled goat-antirabbit secondary antibodies (diluted to 1:200). Samples were analyzed by light microscope (Olympus, Tokyo, Japan). The expression of α-SMA and collagen I in liver tissue was performed by the same method and measured by a positive index (PI).
| PI=mean optical density×positive area percentage. |
7. HYP content
Content of HYP in liver tissue was determined by spectrophotometric method according to the HYP assay kit's instruction manual. The data was expressed as HYP (µg)/protein (mg).
8. Cell culture
HSC-T6 cells were presented by Dr. Quan Wu, Key lab of Anhui Provincial Hospital, Anhui Medical University. HSC-T6 cells were maintained in RPMI 1640 medium (Gibco, New York, NY, USA) with 10% new bovine serum (Gibco) in 5% (v/v) CO2 humidified atmosphere at 37℃. Cells were divided into four groups: normal group, cells treated with RPMI 1640 without transfection; control group, cells transfected with pcDNA 3.1-neo; two experiment groups, 24 hours after transfected with pcDNA-Mest and 48 hours after transfected with pcDNA-Mest.
9. Transfection
The expression plasmids pcDNA-Mest and pcDNA 3.1-neo were transfected into cultured HSC-T6 cells by Lipofectamine 2000 (Invitrogen) respectively. HSC-T6 cells were cultured in 6-well plate at a density of 5×105 cells/mL in RPIM 1640 without bovine serum for 12 hours. Plasmid was added to Lipofectamine 2000 in the ratio of 1 to 5 (w/v, 2 µg plasmid each well) and then the concentration was added to RPIM 1640 without bovine serum (2 mL). The solution was mixed gently and added to the 6-cell plate covered by HSCs by 80% in area. The plate was incubated for 48 hours at 37℃ under a humidified 5% (v/v) CO2 atmosphere, the transfection efficiency in cells was calculated with fluorescence microscope. Cells without transfection were treated in parallel.
10. Cell viability determination
HSC-T6 cells were harvested at 48 hours after transfection for cell viability assay. HSC-T6 cells were plated in 96-well plate at the density of about 4×104 cells/mL in RPIM 1640 medium supplemented with 10% fetal bovine serum for 12, 24, 36, and 48 hours. Cell viability was determined using the CytoTox 96 Nonradioactive Cytotoxicity assay (Promega, Madison, WI, USA) by measurement of the lactate dehydrogenase activity in the cytoplasm according to the manufacturer's instructions. Each assay was performed in triplicate and each experiment was repeated at least three times. Cell-growth curves were calculated as optical density mean values of triplicates per group. Cells without transfection were treated in parallel.
11. Reverse transcription-polymerase chain reaction and quantitative real-time PCR
Total cellular RNA was extracted from collected cells and liver tissues of rats as described by the manufacturer's protocol (Trizol, Invitrogen), respectively. Single-strand cDNA was synthesized from 1ug of total RNA by reverse transcription according to the instructions (Toyobo, Tokyo, Japan). Reverse transcription (RT)-PCR and quantitative real-time PCR (qPCR) was performed as described previously.6 The primer was as follows:
1) β-Catenin (Invitrogen, Shanghai, China):
Forward, 5'-AGTGCGCACCATGCAGAATA-3'; reverse, 5'-CCACCACTGGCCAGAATGAT-3'.4
2) GAPDH (Toyobo, Japan):
Forward, 5'-ACCACAGTCCATGCCATCAC-3'; reverse, 5'-TCCACCACCCTGTTGCTGTA-3'.
3) Tissue inhibitor of metalloproteinase type I (TIMP-1) (Invitrogen):
Forward, 5'-TTTGCATCTCTGGCCTCTG-3'; reverse, 5'-AATGACTGTCACTCTCCAG-3'.
4) α-SMA (Invitrogen):
Forward, 5'-GATCACCATCGGGAATGAACGC-3'; reverse, 5'-CTTAGAAGCATTTGCGGTGGAC-3'.
12. Western blot assays
The total protein from liver tissue was extracted according to the instructions provided in the kits (Wuhan Yafa Biological Technology Co., Ltd.). Meanwhile, at 48 hours after transfection with plasmid pcDNA-Mest, cells were harvested and prepared for protein extraction. The extracted proteins were separated by a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis gel and transferred onto nitrocellulose membranes (Pierce, Rockfors, IL, USA). After incubated with 10% nonfat milk, the membranes were probed with polyclonal rabbit antirat β-catenin antibody (1:400) overnight at 4℃. After washing for 2×3 minutes, the membrane was incubated with HRP-labeled goat-antirabbit secondary antibodies for 1 hour at room temperature and colored by electrochemiluminescence. The membrane was scanned for the relative value of protein expression in gray scale by Image-Pro plus software 6.0 (Media Cybernetics, Silver Spring, MD, USA). The relative expressions were quantified according to the reference blots of β-actin.
13. Statistical analyses
All data were presented as mean±SD. Statistical analyses were performed using an independent t-test and one-way analysis of variance with the SPSS version 19.0 (IBM Co., Armonk, NY, USA). A p<0.05 was considered to be statistically significant.
RESULTS
1. Transfection efficiency of plasmid into HSCs and liver
A pEGFP-C1 blank vector was cotransfected with pcDNA-Mest to evaluate transfection efficiency by using immunofluorescence microscopy as stated in our previous studies.6 The transfection efficiency was calculated to be 85% (Fig. 1A-C).
Fig. 1.

(A-O) Transfection efficiency of pcDNA-mesoderm-specific transcript homologue (Mest; ×200) and pathological examination of rat liver (×200). (A, B, C) Recombinant plasmid pcDNA-Mest was transfected into hepatic stellate cells and (E) rat liver. Transfection was detected by (B) fluorescence microscopy and (E) light microscopy, respectively. The expression of Mest in liver tissue was determined in four groups. There was no Mest expression in the (D) normal group, (F) control group or (G) model group, (E) whereas there was considerable Mest expression in the liver tissue in the Mest treatment group. Changes in liver pathology were examined by (H, I, J, K) hematoxylin-eosin and (L, M, N, O) Masson's trichrome staining. (H, L) Liver samples from normal rats showed normal liver architecture and few inflammatory cells and collagen fibers. (K, O) Samples from model groups showed more infiltrated inflammatory cells and collagen fibers than those from the normal group, and (I, M) Mest treatment resulted in greater improvement of the liver condition than (J, N) that observed in the model group and pcDNA 3.1-neo control group.
To detect the expression of Mest in liver tissue, various tissues were stained for Mest (brown signal) at 48 hours after transfection. There was no Mest expression in normal group (Fig. 1D), control group (Fig. 1F), and model group (Fig. 1G). Our study showed that hydrodynamics-based transfection of Mest lead to significant expression of Mest in liver tissue (Fig. 1E).
2. Effects of Mest on CCl4-induced hepatic fibrosis in rats
As shown in Table 1, HA and LN levels in serum of rats in liver tissue significantly increased in model group compared with the normal group, but they were markedly decreased in treatment group compared with the model group (all p<0.05 respectively). Moreover, the serum levels of ALT, AST, and LDH in model group were much higher than in normal group, while the Mest treatment obviously reduced all the above parameters compared with the model group (all p<0.05 respectively). There were no significant differences between the control group and model group in the serum levels of HA, LN, ALT, AST, and LDH in liver tissue (p<0.05 respectively).
Table 1.
Serum Levels of Alanine Aminotransferase, Aspartate Tranfaminase, Lactic Dehydrogenase, Hyaluronic Acid and Laminin, and Hydroxyproline Content in Liver Tissue
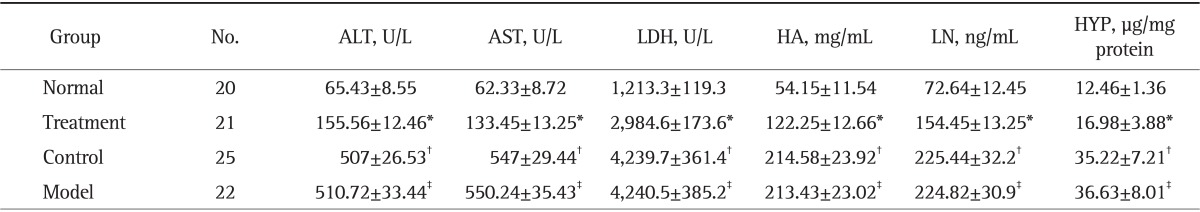
Data are presented as mean±SD.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactic dehydrogenase; HA, hyaluronic acid; LN, laminin; HYP, hydroxyproline.
*p<0.05 compared with normal group; †p<0.05 compared with treatment group; ‡p>0.05 compared with model group.
At the end of the experiment, liver tissue samples from normal rats showed normal lobular architecture with central veins as well as radiating hepatic cords (Fig. 1H and L). Liver tissue sections from model group showed that a markedly big number of inflammatory cells infiltrated into the liver tissues and more collagen fibers deposited in the hepatic lobules, separating them completely compared with normal group (Fig. 1K and O). On the contrary, pathological examination of the rat liver sections indicated that Mest remarkably ameliorated the adipose degeneration of hepatocytes and reduced the immigration of inflammatory cells compared with the model group (Fig. 1I). In addition, as showed by Masson, collagen fibers in model group (Fig. 1K) were obviously more than normal group (Fig. 1L), while they were markedly decreased by Mest (Fig. 1M). There was no significant difference in liver condition between control group and model group (Fig. 1J vs K, N vs O, respectively).
3. Effect of Mest on the expression and distribution of α-SMA, β-catenin, and collagen I in liver tissue
Immunohistochemistry method was used to examine the expression and distribution of α-SMA, β-catenin, and collagen I in liver tissue. As revealed in Fig. 2, there were few α-SMA and β-catenin positive regions in normal group (PI=0.0056±0.005 (Fig. 2A); PI=0.0049±0.004 (Fig. 2E), respectively). In contrast, considerable α-SMA and β-catenin positive regions can be seen around the periportal fibrotic band areas, central vein and fibrous septa elevated by CCl4 in model group (P1=0.0442±0.006 (Fig. 2D); PI=0.0572±0.009 (Fig. 2H), respectively), whereas Mest treatment sharply attenuated the expression of α-SMA and β-catenin compared with model group (PI=0.0254±0.003 (Fig. 2B); PI=0.0364±0.003 (Fig. 2F); p<0.05, respectively). There was no significant difference in the expression of α-SMA and β-catenin between control group and model group (PI=0.0449±0.004 (Fig. 2C); PI=0.0569±0.008 (Fig. 2G); p>0.05, respectively). The expression of collagen in liver tissue was consistent with that of α-SMA and β-catenin (PI=0.0024±0.002 (Fig. 2I); PI=0.0253±0.007 (Fig. 2J); PI=0.0512±0.010 (Fig. 2K); PI=0.0522±0.012 (Fig. 2L), respectively).
Fig. 2.

(A-D) Expression of α-smooth muscle actin (α-SMA), (E-H) β-catenin, and (I-L) collagen I was determined by immunohistochemistry (×200). Lane 1, rats in the normal group (normal rats); lane 2, rats in the treatment group (pcDNA-mesoderm-specific transcript homologue [Mest]+CCl4); lane 3, rats in the control group (pcDNA-neo+CCl4); lane 4, rats in the model group (normal saline+CCl4). Considerable expression of α-SMA protein was observed in the periportal fibrotic areas, central vein, and fibrous septa in (D) the model group and (C) control group relative to (A) the normal group. (B) Mest treatment reduced the expression of α-SMA relative to the model group and control group. The expression of collagen I and β-catenin was consistent with the expression α-SMA.
4. Effect of Mest on the expression of α-SMA, β-catenin, Smad3, and TIMP-1 in liver tissue
To further elucidate the molecular mechanism involved in the antifibrosis effects of Mest and the role of β-catenin in the process of liver fibrosis, we tested the mRNA and protein expression of α-SMA, β-catenin, Smad3, and TIMP-1 in liver tissue by RT-PCR and western blot, respectively.
As revealed by Fig. 3, the mRNA and protein levels of α-SMA, β-catenin, Smad3, and TIMP-1 in liver tissue were markedly higher in model group than normal group (all p<0.05, respectively). While Mest apparently decreased all the above parameters compared with model group (p<0.05, respectively). Unfortunately, control group showed no significant difference from model group (all p>0.05, respectively) suggesting that Mest could remarkably reduce the expression of α-SMA which may be associated with decreased β-catenin activation.
Fig. 3.

Mesodermspecific transcript homologue decreased the mRNA and protein expression of β-catenin, Smad3, α-smooth muscle actin (α-SMA) and tissue inhibitor of metalloproteinase type I (TIMP-1) in the liver tissue. The mRNA and protein levels of β-catenin, Smad3, α-SMA, and TIMP-1 in liver tissue are shown by (A) reverse transcription-polymerase chain reaction and (B) Western blot, respectively. The relative expression levels were normalized to those of (C) glyceraldehyde-3-phosphate dehydrogenase (GADPH) and (D) β-actin, respectively. The data are representative of three independent experiments. Lane 1, rats in the normal group (normal rats); lane 2, rats in the treatment group (pcDNA-mesoderm-specific transcript homologue+CCl4); lane 3, rats in the control group (pcDNA-neo+CCl4); lane 4, rats in the model group (normal saline+CCl4).
*p<0.05 compared with the treatment group; †p<0.05 compared with the normal group; ‡p>0.05 compared with the model group.
5. Effects of Mest on the cell viability and activation of HSC-T6 cells
Wnt/β-catenin signal pathway has been reported to play important roles in HSC activation. To elucidate the role of Wnt/β-catenin in the activation process of HSC-T6 cells, we tested the viability of HSC-T6 cells. As shown in Fig. 4A, viability of HSC-T6 cells in normal group was significantly higher than treatment group (p<0.05), Mest treatment time-dependently decreased the viability of HSC-T6 cells. There was no significant difference in the cell viability between normal group and control group (p>0.05). Therefore, inhibition of Wnt/β-catenin signal pathway by Mest lead to the reduction of cell viability of HSC-T6 (Fig. 4A).
Fig. 4.

A) Effects of mesoderm-specific transcript homologue (Mest) on viability, (B) mRNA expression of α-smooth muscle actin (α-SMA) and (C, D) protein expression of α-SMA, Smad3, and β-catenin in hepatic stellate cell (HSC)-T6 cells. The cell viability was tested using the CytoTox 96 assay according to the manufacturer's instructions. The data are presented as the mean±SD of three individual experiments, and each experiment includes triplicate wells. (B) mRNA expression of α-SMA was determined by quantitative real-time polymerase chain reaction and (C, D) the protein expression of α-SMA, Smad3, and β-catenin of HSC-T6 cells was measured by western blotting. Data were presented as the mean±SD of three independent experiments. The relative expression was normalized to that of glyceraldehyde-3-phosphate dehydrogenase (β-actin). Lane 1, normal HSCs; lane 2, control HSCs (pcDNA-neo); lane 3, 24 hours after transfection with (pcDNA-Mest); lane 4, 48 hours after transfection with (pcDNA-Mest).
*p<0.05 compared with the normal group; †p>0.05 compared with the normal group; ‡p<0.05 compared with the control group.
It has been demonstrated that α-SMA was the key marker of HSCs activation. To further investigate the effects of Wnt/β-catenin signal pathway in the activation process of HSCs, we tested the mRNA expression of α-SMA in HSCs using qPCR. As indicated in Fig. 4B, mRNA expression of α-SMA in normal group was markedly higher than in the treatment group, Mest treatment decreased the mRNA expression of α-SMA of HSC-T6 cells in a time-dependent manner. There was no significant difference in the mRNA expression of α-SMA between normal group and control group (p>0.05). Therefore, inhibition of HSCs activation by down-regulating Wnt/β-catenin signal pathway by Mest was associated with the reduction of α-SMA expression.
6. Effect of Mest on the protein expression of α-SMA, β-catenin, and Smad3 in HSC-T6 cells
In order to investigate the molecular mechanism in the inhibitory effects of Mest on HSCs activation, we examined the protein expressions of α-SMA, β-catenin, and Smad3 in HSC-T6 cells.
As shown in Fig. 4C, when treated by Mest, the protein levels of α-SMA, β-catenin, and Smad3 were markedly decreased in comparison with normal group (p<0.05, respectively). There was no significant difference in the protein expression of α-SMA, β-catenin, and Smad3 between normal group and control group (p>0.05, respectively). This study suggested that Mest could suppress HSCs activation through inhibiting the protein expression of β-catenin and Smad3.
DISCUSSION
Liver fibrosis has been widely accepted as a wound-healing response of liver to many liver injuries. It was elicited by various stimuli, such as viral hepatitis, alcohol, copper, drug, immune compounds, and so on. Liver fibrosis has also been reported to be a chronic inflammation associated disease, which includes the infiltration of many inflammatory cells and interplay of many cytokines and signal molecules.11 Many reports suggested that reducing the release of cytokines and infiltration of inflammatory cells could prevent and reverse liver fibrosis.12
Liver fibrosis is also characterized by a distortion of liver tissue architecture and an excessive deposition of ECM proteins that type I collagen predominates.13 In the process of fibrosis, ECM deposition exceeds the resolution of ECM. So far, many reports suggested that matrix metalloproteinases (MMPs) contributes to the resolution of ECM in the liver, while it is counteracted by TIMP-1.14 Meanwhile, as the main source of accumulated ECM, activated HSCs have been considered to be the predominant cell type responsible for excessive collagen deposition during liver fibrosis. HSCs activation and proliferation has been demonstrated to be the key event of liver fibrosis. Reducing the MMPs activation and suppressing the apoptosis of HSCs has become one of the treatment strategies for liver fibrosis.15
Increasing evidences demonstrated that Wnt/β-catenin signaling pathway plays a pivotal role in the process of fibrosis, such as lung fibrosis, liver fibrosis, and so on.16 The canonical wnt signal pathway has been demonstrated to promote the release of cytokines and recruit of inflammatory cells.17 β-Catenin induced signaling pathway also regulates many biological functions including enhancing the function of HSCs and TIMP-1 and reducing the activation of MMPs which leads to the deterioration of liver fibrosis.18 Mest, a negative mediator of canonical wnt signaling pathway, has been reported to down-regulate the β-catenin induced signaling pathway by inhibiting LRP5/6, the receptor of Wnt protein.7 However, there is little known about the antifibrosis effects of Mest.
CCl4-induced liver fibrosis has long been used to be an animal model for liver fibrosis. Using this method, therapeutic effects against liver fibrosis have been greatly improved.19 Hydrodynamics-based gene delivery, via the caudal vein has also been accepted as a successful method to treat liver diseases especially liver fibrosis.8,20 In the present study, we tested the anti-fibrosis effect of Mest on CCl4-induced liver fibrosis using this technique and found that pcDNA vector drove the expression of Mest in rat liver (Fig. 1E). In this study, our data showed that Mest significantly reduced the serum levels of ALT, AST, and LDH in rats after treatment with CCl4. Histological examination also demonstrated that a large number of inflammatory cells infiltrated into the intralobular and interlobular regions together with increased collagen fibers and fatty degenerated hepatocytes in CCl4 treated rats compared with normal rats. On the contrary, Mest treatment remarkably reduced the immigration of inflammatory cells and the deposition of collagen fibers compared with rats in model group treated with CCl4. In addition, Mest also reduced the expression and distribution of β-catenin protein in liver tissue compared with CCl4 treated group. Therefore, Mest attenuated the development of liver fibrosis which may be associated with reduced β-catenin expression. Moreover, increasing evidence indicated that Wnt/β-catenin signaling pathway played important roles in hepatocellular carcinoma (HCC). Enhanced β-catenin activation aggravated the development of HCC,21 while inhibition of β-catenin expression lead to the necrosis of HepG2 cells.21 For confirming whether Mest treatment leads to the development of HCC, we examined the AFP expression in liver tissue by immunohistochemical staining and found that no AFP expression was detected in liver tissue (data were not shown). Moreover, no carcinoma cell was found in liver tissue (Fig. 1E-H). These results suggested that inhibition of β-catenin expression by Mest for 8 weeks would not result in the development of HCC.
HA and LN levels in serum are the important indices reflecting the degree of liver fibrosis. In the present study, the serum levels of HA and LN were much higher in model group than in normal group, whereas Mest markedly reduced the levels of HA and LN compared with model group. Collagen content was also determined by the measurement of HYP in liver tissue. As shown in Table 1, CCl4 treated rats showed a high level of HYP in liver tissue, indicating that Mest could attenuate CCl4-induced liver fibrosis.
Activated HSCs were characterized by a high expression of α-SMA. As revealed by Fig. 2, considerable protein expression of α-SMA and β-catenin was observed among the periportal fibrotic areas, central vein, and fibrous septa in rat liver in model group compared with normal group consistent with collagen I, while Mest obviously reduced all the above parameters suggesting that Mest alleviated CCl4-induced liver fibrosis by inhibiting Wnt/β-catenin pathway.
α-SMA has been also regarded as the marker of HSCs activation. In this study, Mest decreased the cell viability of HSCs in a time-dependent manner compared with normal group (Fig. 4A). For confirming whether Mest affects the activation of HSCs, we designed other assays and found that Mest time-dependently reduced the mRNA expression of α-SMA in HSCs (Fig. 4B) and down-regulated the protein expression of α-SMA and β-catenin in HSCs (Fig. 4C), indicating that inhibition of HSCs activation by Mest is associated with its suppressive effects on Wnt/β-catenin signaling pathway.
From the standpoint of liver disease status, HSC cell transition has been regarded to play key roles in liver fibrosis and Wnt/β-catenin signaling pathway has been considered to be the predominant cytokines responsible for cell transition in the process of chronic liver disease and liver fibrosis. Cheng et al.3 observed that HSCs played important roles in the regulation of retinoid metabolism and ECM remodeling and this physiological process could be prohibited by the inhibition of Wnt antagonists. In the process of liver fibrogenesis, myofibroblasts (activated HSCs) was the main source of ECM, characterized by high expression of α-SMA. When exposed to liver toxic stimuli, quiescent HSCs undergo activation and change their function and morphology to myofibroblast-like cells. Activated HSC lose vitamin A droplets and in contrast increase the expression of α-SMA as well as ECM components.22 The transition from quiescent HSCs to myofibroblasts (activated HSCs) has been reported to be the key event.22 Some researchers have reported that hepatocytes, cholangiocyte, and endothelial cells could differentiate into myofibroblastes (activated HSCs) increasing the deposition of ECM in liver, named epithelial-mesenchymal transition (EMT).23 However, some studies have demonstrated that EMT did not exist in the process of liver fibrosis.24 So, the molecular mechanism involved in HSC activation needs to be further investigated.
Over the past decades, many reports have suggested that transforming growth factor (TGF)-β1/Smads played a key role in the progress of liver fibrosis including cell development, HSC activation, collagen deposition, and ECM remodeling.25 However, accumulating documents suggested that canonical Wnt/β-catenin signaling is necessary for TGF-β-mediated fibrosis, establishing the critical role of Wnt/β-catenin in fibrogenesis.16 We have also reported that inhibition of Wnt/β-catenin signaling pathway could attenuate CCl4-induced liver fibrosis.6 In this study, our data showed that Mest treatment significantly reduced the protein expression of α-SMA and Smad3, suggesting that antifibrosis effects of Mest were related to the suppression of TGF-β1/Smads signaling pathway.
In conclusion, we demonstrated that Mest significantly inhibited CCl4-induced liver fibrosis, and its antifibrosis maybe related to its inhibitory effects on the activation of HSCs by down-regulating the Wnt/β-catenin singnaling pathway. Since hepatic fibrosis is a very complicated process, the molecular mechanism involved in therapeutic effects of Mest on hepatic fibrosis needs to be further explored.
ACKNOWLEDGEMENTS
This work was supported by Anhui Provincial Natural Science Foundation (No.11040606Q09), National Natural Science Foundation of China (No.81271713; No.81001046).
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Bona S, Filippin LI, Di Naso FC, et al. Effect of antioxidant treatment on fibrogenesis in rats with carbon tetrachloride-induced cirrhosis. ISRN Gastroenterol. 2012;2012:762920. doi: 10.5402/2012/762920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu CI, Hoffman JA, Shy BR, et al. Function of Wnt/β-catenin in counteracting Tcf3 repression through the Tcf3-β-catenin interaction. Development. 2012;139:2118–2129. doi: 10.1242/dev.076067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng JH, She H, Han YP, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G39–G49. doi: 10.1152/ajpgi.00263.2007. [DOI] [PubMed] [Google Scholar]
- 4.Nusse R. Wnt signaling. Cold Spring Harb Perspect Biol. 2012;4:a011163. doi: 10.1101/cshperspect.a011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kordes C, Sawitza I, Häussinger D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem Biophys Res Commun. 2008;367:116–123. doi: 10.1016/j.bbrc.2007.12.085. [DOI] [PubMed] [Google Scholar]
- 6.Li W, Zhu C, Chen X, Li Y, Gao R, Wu Q. Pokeweed antiviral protein down-regulates Wnt/β-catenin signalling to attenuate liver fibrogenesis in vitro and in vivo. Dig Liver Dis. 2011;43:559–566. doi: 10.1016/j.dld.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Jung H, Lee SK, Jho EH. Mest/Peg1 inhibits Wnt signalling through regulation of LRP6 glycosylation. Biochem J. 2011;436:263–269. doi: 10.1042/BJ20101512. [DOI] [PubMed] [Google Scholar]
- 8.Zhu C, Li Y, Li W, Wu Q, Gao R. Gene transfer of c-met confers protection against D-galactosamine/lipopolysaccharide-induced acute liver failure. Dig Dis Sci. 2012;57:925–934. doi: 10.1007/s10620-012-2052-4. [DOI] [PubMed] [Google Scholar]
- 9.Maruyama H, Higuchi N, Kameda S, Miyazaki J, Gejyo F. Rat liver-targeted naked plasmid DNA transfer by tail vein injection. Mol Biotechnol. 2004;26:165–172. doi: 10.1385/mb:26:2:165. [DOI] [PubMed] [Google Scholar]
- 10.Chevallier M, Guerret S, Chossegros P, Gerard F, Grimaud JA. A histological semiquantitative scoring system for evaluation of hepatic fibrosis in needle liver biopsy specimens: comparison with morphometric studies. Hepatology. 1994;20:349–355. [PubMed] [Google Scholar]
- 11.Tsuda M, Zhang W, Yang GX, et al. Deletion of interleukin (IL)-12p35 induces liver fibrosis in dominant-negative TGFβ receptor type II mice. Hepatology. 2013;57:806–816. doi: 10.1002/hep.25829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty JB, Oakley F, Walsh MJ. Mechanisms and biomarkers of apoptosis in liver disease and fibrosis. Int J Hepatol. 2012;2012:648915. doi: 10.1155/2012/648915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding J, Yu J, Wang C, et al. Ginkgo biloba extract alleviates liver fibrosis induced by CCl in rats. Liver Int. 2005;25:1224–1232. doi: 10.1111/j.1478-3231.2005.01169.x. [DOI] [PubMed] [Google Scholar]
- 14.Wu LM, Wu XX, Sun Y, Kong XW, Zhang YH, Xu Q. A novel synthetic oleanolic acid derivative (CPU-II2) attenuates liver fibrosis in mice through regulating the function of hepatic stellate cells. J Biomed Sci. 2008;15:251–259. doi: 10.1007/s11373-007-9216-9. [DOI] [PubMed] [Google Scholar]
- 15.Woodhoo A, Iruarrizaga-Lejarreta M, Beraza N, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology. 2012;56:1870–1882. doi: 10.1002/hep.25828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akhmetshina A, Palumbo K, Dees C, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;3:735. doi: 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitazawa M, Cheng D, Tsukamoto MR, et al. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer's disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma B, van Blitterswijk CA, Karperien M. A Wnt/β-catenin negative feedback loop inhibits interleukin-1-induced matrix metalloproteinase expression in human articular chondrocytes. Arthritis Rheum. 2012;64:2589–2600. doi: 10.1002/art.34425. [DOI] [PubMed] [Google Scholar]
- 19.Smith GP. Animal models of cutaneous and hepatic fibrosis. Prog Mol Biol Transl Sci. 2012;105:371–409. doi: 10.1016/B978-0-12-394596-9.00011-1. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Liu P, Gao X, Qian W, Xu K. rAAV2-TGF-β(3) decreases collagen synthesis and deposition in the liver of experimental hepatic fibrosis rat. Dig Dis Sci. 2010;55:2821–2830. doi: 10.1007/s10620-009-1119-3. [DOI] [PubMed] [Google Scholar]
- 21.Shan X, Miao Y, Fan R, et al. MiR-590-5P inhibits growth of HepG2 cells via decrease of S100A10 expression and inhibition of the Wnt pathway. Int J Mol Sci. 2013;14:8556–8569. doi: 10.3390/ijms14048556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawada N. Evolution of hepatic fibrosis research. Hepatol Res. 2011;41:199–208. doi: 10.1111/j.1872-034X.2011.00776.x. [DOI] [PubMed] [Google Scholar]
- 23.Brenner DA, Kisseleva T, Scholten D, et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S17. doi: 10.1186/1755-1536-5-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taura K, Miura K, Iwaisako K, et al. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology. 2010;51:1027–1036. doi: 10.1002/hep.23368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roderburg C, Luedde M, Vargas Cardenas D, et al. miR-133a mediates TGF-β-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. J Hepatol. 2013;58:736–742. doi: 10.1016/j.jhep.2012.11.022. [DOI] [PubMed] [Google Scholar]