

Abstract
Recent discoveries on the presence and location of phosphoinositides in the eukaryotic cell nucleoplasm and nuclear membrane prompted us to study the putative interaction of chromatin components with these lipids in model membranes (liposomes). Turbidimetric studies revealed that a variety of histones and histone combinations (H1, H2AH2B, H3H4, octamers) caused a dose-dependent aggregation of phosphatidylcholine vesicles (large unilamellar vesicle or small unilamellar vesicle) containing negatively charged phospholipids. 5 mol % phosphatidylinositol-4-phosphate (PIP) was enough to cause extensive aggregation under our conditions, whereas with phosphatidylinositol (PI) at least 20 mol % was necessary to obtain a similar effect. Histone binding to giant unilamellar vesicle and vesicle aggregation was visualized by confocal microscopy. Histone did not cause vesicle aggregation in the presence of DNA, and the latter was able to disassemble the histone-vesicle aggregates. At DNA/H1 weight ratios 0.1–0.5 DNA- and PIP-bound H1 appear to coexist. Isothermal calorimetry studies revealed that the PIP-H1 association constant was one order of magnitude higher than that of PI-H1, and the corresponding lipid/histone stoichiometries were ∼0.5 and ∼1, respectively. The results suggest that, in the nucleoplasm, a complex interplay of histones, DNA, and phosphoinositides may be taking place, particularly at the nucleoplasmic reticula that reach deep within the nucleoplasm, or during somatic and nonsomatic nuclear envelope assembly. The data described here provide a minimal model for analyzing and understanding the mechanism of these interactions.
Introduction
Phosphoinositides are negatively charged phospholipids that are usually considered as minor components of cell membranes. For about three decades now they have been recognized as important second messengers but more recent studies involve also the lipids in structural roles related to the regulation of membrane dynamics (1–3). To our knowledge, a number of important novel discoveries concerning the structure of cell nuclei and nuclear envelopes (NE) are challenging the established view on chromatin-nuclear membrane interactions. In particular, the NE is no longer seen as a smooth sheet structure covering the chromatin, but as a dynamic complex morphology encompassing structures such as the nucleoplasmatic reticulum whose invaginations penetrate deep into the nucleoplasm (7,8). Thus, the phospholipids, particularly egg phosphatidylcholine (PC) and phosphatidylinositol (PI), that were known to exist in the nucleus but whose physical structure was unknown (9) may well exist as part of those invaginations. In an independent series of studies, Larijani and co-workers have described a role for phosphoinositides and their derivatives in both somatic and nonsomatic NE assembly. During the male pronucleus formation, the male pronucleus is formed from two main membranous compartments. This newly formed nuclear envelope is assembled from the oocyte’s nonendoplasmic-derived membrane vesicles (MV1) and the sperm nuclear envelope remnants. Both these membrane compartments are highly enriched in unsaturated polyphosphoinositides, and become incorporated into the newly formed NE at the poles of the sperm nuclei (10,11). Although a distinct nuclear inositol lipid signaling has been known for years (12), structural questions concerning the aggregation patterns of phosphoinositides in the nucleus have only recently been addressed (13,14).
Because of the presence of histones and other basic proteins in the nucleus it was reasonable to expect some interaction of these proteins with the phosphoinositides. Histones contain 25–35% basic amino acid residues (4) and even if the majority of them are bound to chromatin, thus unavailable for interaction with membrane lipids, it is also known that H1 variants are mobile molecules that interact with nucleosomes as part of a dynamic protein network (5,6) and the same may take place with other histones, so that an equilibrium between binding DNA and binding other negatively charged molecules may occur. In fact, previous authors (15–17) had studied the interaction of histones with phosphatidylserine and other negatively charged phospholipids virtually absent from the nuclear membrane. This work describes our experiments on the interaction of histones, mainly H1, with phosphoinositides. Phosphatidylinositol-4-phosphate (PIP) and phosphatidylinositol-4,5-bisphosphate (PIP2) have a higher affinity for H1 than PI. Histone-phosphoinositide interaction leads to liposome aggregation. Because histones are considered to exist mainly bound to DNA, the effect of a nonspecific DNA fraction on the histone-PIP aggregates was tested, and DNA was found to compete for histones with phosphoinositides. A combination of spectroscopic, microscopic, and calorimetric techniques reveals that in the eukaryotic nucleus phosphoinositides, DNA/RNA, and chromatin proteins may be binding in a multiple equilibrium, particularly under conditions of NE disassembly and reassembly, e.g., during open mitosis. Our in vitro studies could constitute a minimal model system for depicting the type of interactions, which may occur during the complex nuclear membrane reorganization.
Materials and Methods
Materials
PC, phosphatidylserine (PS), and phosphatidylglycerol (PG) were purchased from Lipid Products (South Nutfield, UK). PI, PIP, PIP2, cardiolipin (CL), and lissamine rhodamine PE (Rho-PE) were supplied by Avanti Polar Lipids (Alabaster, AL). Histones were obtained from chicken (Gallus gallus) erythrocyte chromatin, as described by Wang et al. (18). Alexa Fluor 488 Histone H1 (H1-Alexa488), Alexa Fluor 633 goat antimouse IgM, and 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD) were from Molecular Probes (Eugene, OR). BODIPY-TMR PIP and purified Anti-PI(4)P antibody mouse monoclonal IgM were purchased from Echelon (Salt Lake City, UT). DNA from herring sperm (partially degraded) was from Sigma-Aldrich (St. Louis, MO). All other materials (salts and organic solvents) were of analytical grade.
Liposome preparation
The appropriate lipids were mixed in organic solution, and the solvent was evaporated to dryness under a stream of N2. The sample was then kept under vacuum for 2 h to remove solvent traces. The lipids were swollen in 10 mM Hepes, 150 mM NaCl, pH 7.4 buffer. Small unilamellar vesicles (SUVs) were prepared from the swollen lipids by sonication with a probe tip sonicator (MSE Soniprep 150 (MSE, UK)) for 20 min (10 s on and off cycles) at 10–20 μm amplitude. The vials were kept on ice during the process to avoid overheating. When large unilamellar vesicles (LUVs) were required, swollen lipids were subjected to 10 freeze/thaw cycles and then extruded using 0.1-μm pore-size Nuclepore filters, as described by Mayer et al. (19) Vesicle size was checked by quasielastic light scattering, using a Malvern Zeta-Sizer 4 spectrometer (Malvern Instruments, Worcestershire, UK). LUVs had an average diameter of 100 nm and SUVs had an average diameter of 30 nm. Lipid concentration was determined by phosphate analysis (20).
Aggregation assays
All assays were carried out at room temperature with continuous stirring, in 10 mM Hepes, 150 mM NaCl buffer (pH 7.4). All experiments were performed at a vesicle concentration equivalent to 0.3 mM phosphate and 10 μg/ml H1 were used unless otherwise stated. Lipid aggregation was monitored in a Cary 3 Bio (Varian, Victoria, Australia) spectrophotometer as an increase in turbidity (absorbance at 400 nm) of the sample.
Giant unilamellar vesicle (GUV) preparation
GUVs were prepared using the electroformation method developed by Angelova et al. (21). For direct visualization under the microscope a homemade chamber was used (22). Transferred GUVs were formed in a PRETGUV 4 chamber supplied by Industrias Técnicas ITC (Bilbao, Spain). Stock solutions of lipids (0.2 mM total lipid containing either 0.2 mol % Rho-PE, 0.4 mol % DiD, or 0.3% BODIPY-TMR PIP) were prepared in chloroform:diethylether:methanol (4:5:1, v/v), 3 μl of the lipid stocks were added onto the surface of Pt electrodes and solvent traces were removed by evacuating the chamber under high vacuum for at least 2 h.
Direct visualization of GUVs
The Pt electrodes were covered with 400 μl of 10 mM Hepes, 150 mM NaCl, pH 7.4. The Pt wires were connected to an electric wave generator (TG330 function generator, Thurlby Thandar Instruments, Huntington, UK) under alternating current (AC) field conditions (500 Hz, 0.031 VRMS for 6 min; 500 Hz, 0.281 VRMS for 20 min, and 500 Hz, 0.623 VRMS for 1 h 30 min) at 37°C. After GUV formation, the chamber was placed on an inverted confocal fluorescence microscope (Nikon D-ECLIPSE C1, Nikon, Melville, NY).
The excitation wavelengths were 561 nm for Rho-PE and BODIPY-TMR PIP, and 637 nm for DiD. The images were collected using band-pass filters of 593 ± 20 nm for Rho-PE and BODIPY-TMR PIP, and a long-pass filter of 650 nm for DiD. Then, 100 μl of H1-Alexa488, at 40 μg/ml, were added to study the histone effect on the GUVs. In the latter case, an excitation light at 488 nm was used and images were collected using a band-pass filter of 515 ± 15 nm. All these experiments were performed at room temperature. Image treatment was performed using the EZ-C1 3.20 software (Nikon).
Observation of transferred GUVs
The Pt electrodes were covered with 500 μl of a 300 mM sucrose solution, previously heated at 37°C. The Pt electrodes were connected to a generator (TG330 function generator, Thurlby Thandar Instruments) under AC field conditions (10 Hz, 1 VRMS for 2 h, followed by 1 Hz, 1 VRMS, 10 min) at 37°C. Finally, the AC field was turned off and the vesicles (in 300 mM sucrose) were collected from the PRETGUV 4 chamber with a pipette and transferred to chambers pretreated with bovine serum albumin (BSA) (2 mg/ml) and containing an equiosmolar buffer solution of 10 mM Hepes, 150 mM NaCl, pH 7.4. Due to the different density between the two solutions, the vesicles were sedimenting at the bottom of the chamber, and this facilitated observation under the microscope.
For PIP detection, anti-PIP antibodies were added to the GUVs. After 1 h incubation with the vesicles the antibody was washed away. A secondary antibody was then added and incubated for 30 min before observation under the microscope. Finally H1-Alexa488 was added to study its effect on the GUVs.
The excitation wavelengths were 488 nm for H1-Alexa488, 561 nm for Rho-PE, and 635 nm for AlexaFluor 633 goat antimouse IgM; and the emission was collected using 515 ± 15 nm and 593 ± 20 nm band-pass filters and a long-pass filter of 650 nm, respectively. All these experiments were performed at room temperature. Image treatment was performed using the EZ-C1 3.20 software (Nikon).
Isothermal titration calorimetry (ITC)
ITC was performed using a VP-ITC high-sensitivity titration calorimeter (MicroCal, Northampton, MA). In this study the calorimetric cell was filled with H1 solution at 8.33 μM. Lipid vesicles or DNA at a 2 mM concentration (as phosphate) were injected into the cell (1.43 mL) in 10 μl steps, i.e., leading to a 100- to 200-fold dilution of lipid vesicles or nucleotide. To minimize the contribution of dilution to the heat of partitioning, all samples were prepared in the same buffer and were degassed under vacuum immediately before use. Typically, the injections were made at 10 min intervals and at 2 s/μl. Constant stirring speed of 290 rpm was maintained during the experiment to ensure proper mixing after each injection. Titration experiments were performed at 25°C in Hepes buffer. Dilution heats of lipid vesicles into the buffer were determined in separate experiments and subtracted from experimental heats of binding. At each lipid injection, free H1 partitioned into the bilayer membrane and the corresponding heat of reaction was measured. The heat of reaction became smaller as less H1 remained free in solution. The integration of each calorimetric peak yielded a heat of reaction. These heats were plotted versus the lipid concentration.
The reaction heat for each injection is related to the calorimetric enthalpy of binding, ΔH. The binding isotherms, ΔH versus molar ratio, were analyzed using MicroCal Origin. A one set of sites model has been used, assuming that binding is virtually exclusively electrostatics-based. The fit of the binding curve yields the apparent stoichiometry N, the binding constant Ka (Kd = 1/Ka) and the enthalpy ΔH of the binding reaction. The Gibbs free energy of binding (ΔG°) and the entropy (ΔS°) are determined from the basic thermodynamic expression ΔG° = −RTlnKa = ΔH°-TΔS°, where R and T are the gas constant and the absolute temperature, respectively. We assume ΔH = ΔH°. Standard conditions are 25°C, 1 atm, 1M lipid, DNA, and histone, pH 7.0.
Results
Vesicle aggregation
As a first approach to test the interaction of histones with vesicles containing negatively charged phospholipids the aggregation of liposomes of different lipid compositions in the presence of H1 histones was monitored. In these experiments both SUV and LUV were tested. Aggregation can be monitored continuously in a spectrophotometer as an increase in sample turbidity (absorbance at 400 nm (23)). Histone H1 was observed to cause vesicle aggregation when PC-based vesicles contained 10 mol % phospholipids with two or more negative charges in the same molecule, such as PIP, PIP2, or CL. No aggregation was observed for vesicles with 10 mol % phospholipids with a single negative charge per molecule (PI, PG or PS) (Fig. 1, A and B). Aggregation occurred equally with SUV or LUV (Fig. 1, A and C).
Figure 1.
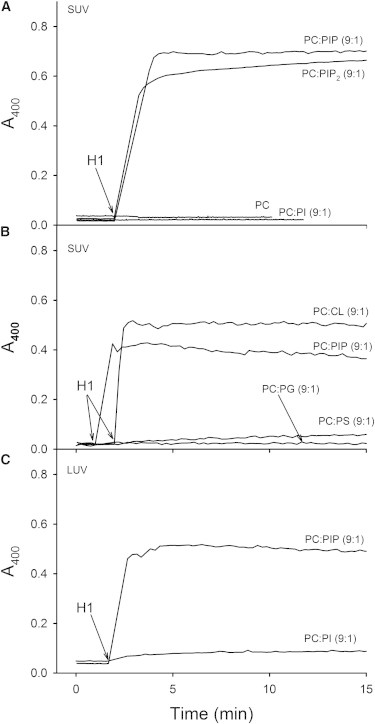
Time-course of H1-induced SUV and LUV aggregation for different lipid compositions. Arrows indicate time of H1 addition. (A and B) SUV. (C) LUV. Lipid compositions are indicated for each curve. Each curve is a representative of one of three or more experiments.
Similar aggregation experiments were performed varying independently lipid and protein concentrations. Aggregation was dependent on both SUV and H1 concentration (Fig. 2, A and B). The extent of aggregation (ΔA400) increased with SUV concentrations up to 0.2 mM phospholipid, and then a plateau was reached (Fig. 2 A). Aggregation increased linearly with H1 concentration, at least up to 10 μg/ml (Fig. 2 B).
Figure 2.
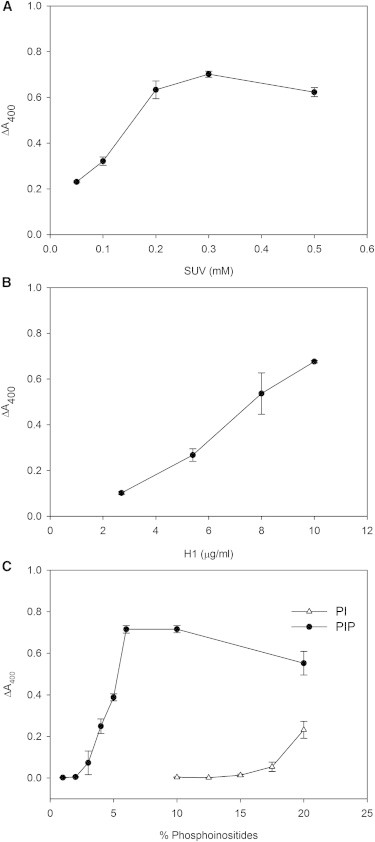
Extents of SUV aggregation, measured as H1-induced changes in turbidity (ΔA400). (A) Increasing concentrations of PC:PIP (9:1) SUVs were treated with 10 μg/ml H1. (B) Increasing concentrations of H1 were added to a fixed concentration of 0.3 mM PC:PIP (9:1) SUVs. (C) Vesicles (total concentration 0.3 mM) with increasing PIP or PI proportions were treated with 10 μg/ml H1. Each point represents the mean ± SE of three separated experiments.
When the effect of different PIP proportions in the vesicles was explored (Fig. 2 C) a sigmoidal dependence of PIP concentration was found, with a half-maximal effect at ∼5 mol % PIP, and a maximum at ∼10 mol % PIP. When PI was used instead of PIP, the requirement of PI to observe aggregation was clearly larger, but still a sigmoidal dependence on PI concentration could be suggested from the data. The vesicle concentration data in Fig. 2 A are also likely to suggest a sigmoidal behavior, the common trends in the experiments in Fig. 2, A and C, confirming that aggregation increases sigmoidally with the concentration of negatively charged phospholipid present in the sample. The interaction of histones with vesicles containing high concentrations of PI had been observed by Zhao et al. (17). At the highest PIP concentrations in Fig. 2, A and C, the apparent aggregation decreases. This is probably due to the aggregates reaching a size that exceeds the Rayleigh condition, so that beyond a certain point a larger size leads to a smaller scattering (24).
The putative competition between negatively charged vesicles and DNA for histones was also explored. When H1 had been previously incubated with a nonspecific DNA no vesicle aggregation was observed (Fig. 3, A, curve 2). When SUV had been preincubated with DNA, addition of H1 did not cause aggregation either (Fig. 3 A, curve 3). When DNA was added once aggregation has started, the increase in turbidity stopped (Fig. 3 B, curve 5), and when added to the aggregates under steady-state conditions the aggregates appeared to disassemble (Fig. 3 B, curve 4). The capacity of DNA to disassemble H1/SUV aggregates increased linearly up to a DNA/H1 weight ratio = 0.8, and then a plateau was reached (Fig. 3 C).
Figure 3.
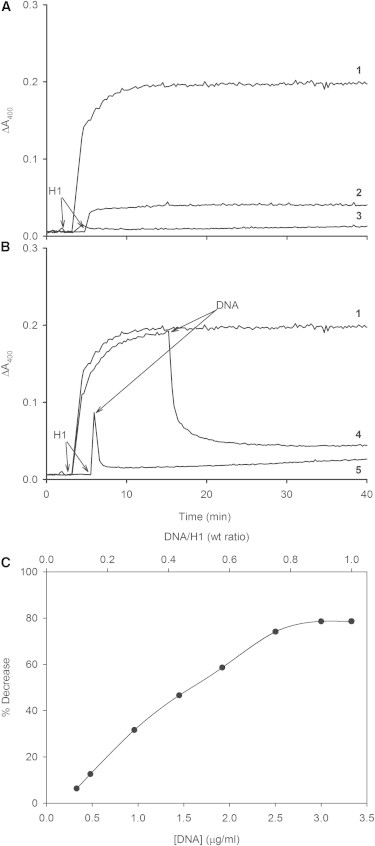
Competition aggregation assays with SUV [PC:PIP(9:1)], H1, and DNA. Arrows indicate time of H1 or DNA addition. All experiments were performed at 0.1 mM lipid concentration, 3.33 μg/ml H1, and 3.33 μg/ml DNA. (A) (1) Control. SUV aggregation caused by H1. (2) Addition of H1 pretreated with DNA. (3) Addition of H1 to a solution of SUVs and DNA. (B) (4) Addition of DNA to the SUV-H1 aggregates under steady-state conditions. (5) Addition of DNA when SUV-H1 aggregate is forming. (C) Effect of DNA/H1 wt ratio on the turbidity decrease observed in experiments as in curve 4 above (disassembly of SUV-H1 aggregates). Each curve is a representative one of three or more experiments.
Similar aggregation experiments were performed with core histones (H3H4, H2AH2B, and octamers) and aggregation was also observed (Fig. 4), although to a somewhat lower extent, on a per weight histone basis. Aggregation decreased in the order H1 > H2AH2B > octamer > H3H4.
Figure 4.
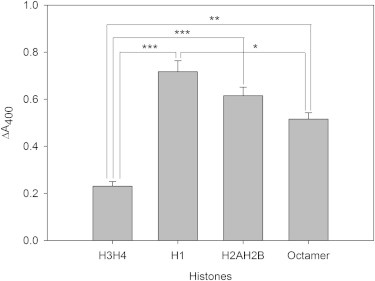
Effect of different histone classes on SUV aggregation. Bars represent mean ± SE of ΔA400. ∗∗∗P ≤ 0.001, ∗∗P ≤ 0.01, ∗P ≤ 0.05 of unpaired t-test.
Confocal microscopy
This technique enabled us to visualize H1 binding to vesicles. GUV of composition either PC:PI (9:1) or PC:PIP (9:1) labeled with the fluorescent probe Rho-PE were prepared, treated with H1, and directly visualized under the fluorescence microscope (Fig. 5). GUV containing PI was poorly labeled with H1-Alexa 488 (Fig. 5 A). In fact, the vesicles were seen as dark objects against a green (Alexa) background. However, for PIP-containing GUV the protein was clearly seen bound to the membranes (Fig. 5 B). The flattened areas between tightly aggregated GUV appeared enriched in H1-Alexa 488.
Figure 5.
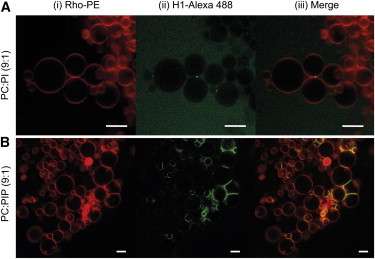
Confocal microscopy of representative GUVs. Composition is given at the left-hand side. (i) Membrane labeled with Rho-PE or (ii) with H1-Alexa 488. (iii) Colocalization of both probes. Scale bars: 10 μm.
Even though no heterogeneities were observed in the Rho-PE-stained lipid mixtures (Fig. 5) we checked whether PIP patches were induced when H1 was present. For this purpose we used Bodipy-labeled PIP (Fig. 6 A). The labeled lipid was homogeneously distributed throughout the vesicle membranes indicating the absence of macroscopic PIP-enriched domains (Fig. 6 A). Again flattened intervesicle contact surfaces were observed, which were enriched in H1.
Figure 6.
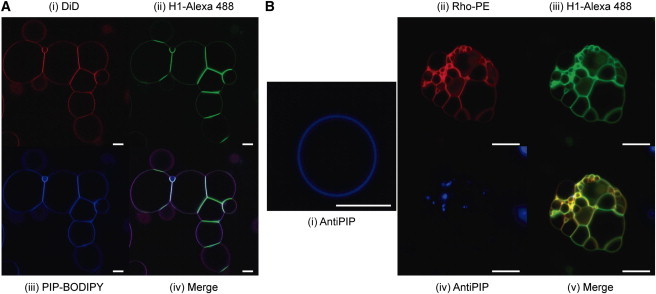
GUVs consisting of PC:PIP (9:1). Scale bars: 10 μm. (A) GUV labeled with DiD (i), or with 0.3% PIP-Bodipy (iii). After H1-Alexa488 addition (ii). Colocalization of probes (iv). (B) GUV stained with anti-PIP antibodies (i). GUV labeled with Rho-PE (ii) and H1-Alexa488 (iii). PIP is labeled with antibodies but when aggregation is produced antibodies are excluded from the aggregate (iv). Colocalization of probes (v).
To visualize native (i.e., unlabeled) PIP distribution in the vesicles anti-PIP antibodies were added that were subsequently labeled with a fluorescent secondary antibody. Native PIP was also homogeneously distributed throughout the membrane in isolated, transferred GUV according to these observations (Fig. 6 B, left panel). However when H1-Alexa 488 was added to aggregated GUV it appeared to displace the antibodies (Fig. 6 B).
Microscopic examination of vesicles allowed the observation of H1-induced aggregation, as shown in Fig. 7. Histone addition caused aggregation of the vesicle at the lower right-hand and more importantly, what were contact points in the absence of H1 became flattened contact surfaces in the presence of the histone. Moreover, addition of DNA at 8 μg/ml displaced the histone and greatly diminished and the contact surfaces between GUVs (Fig. 7, right panel).
Figure 7.

H1-induced GUV aggregation and reversal by DNA. PC:PIP (9:1) vesicles stained with Rho-PE (i). Vesicle aggregation after H1-Alexa 488 addition (ii). Histone displacement after DNA addition (iii). Scale bars: 10 μm.
Calorimetric studies
ITC is an ideal technique for measuring ligand binding to macromolecules. In our case it was applied to compare the binding affinities of H1 for vesicles containing PI or PIP. Small amounts of LUV were progressively added to a solution of H1 and measurements of heat exchanges allowed the calculation of ΔH°, ΔS°, and ΔG° of the process. When LUVs were composed of pure PC, in the absence of negatively charged lipids, no measurable heats of interaction were observed. Reliable measurements were obtained however with LUV containing 10% PI, or 10% PIP. The calorimetric data could be fitted to a model of equivalent binding sites. Representative experiments are shown in Fig. 8, A–D. The experimental values are summarized in Table 1. The association constant (Ka) was about one order of magnitude higher for PIP- than for PI-containing vesicles, indicating that the histone-lipid equilibrium is more displaced toward complex formation for PIP than for PI, in agreement with the turbidimetric and microscopic observations. The lipid:histone stoichiometries (N) that best fit the calorimetric data are ∼0.5 for PIP and ∼1 for PI, indicating that both negative charges of PIP are involved in histone binding, in agreement with the higher potency of PIP-containing vesicles to induce formation of vesicle-histone aggregates. ΔH° represents approximately the heat exchange upon binding, for both lipids PI and PIP ΔH° is negative, implying an exothermic process. The entropy change ΔS° is also negative in both cases, it would also be expected that entropy decrease when two molecules get associated into one. (Note however that changes in the properties of water adjacent to the binding partners often play a key role in the thermodynamic profile). For reasons that are probably associated to the different chemical nature of both lipids, PI and PIP, ΔH° is somewhat more negative for the former, but ΔS° is also clearly more negative. As a result, ΔG° is about one-half for PI than for PIP binding, being negative in both cases, i.e., predicting a spontaneous process under standard conditions. In any case, it should be noted that our calorimetric measurements correspond to both the binding and the aggregation events. Interpretation of the calorimetric data requires that although initially only one-half of the PI or PIP molecules will be accessible to the histones, i.e., in the vesicle outer monolayer, vesicle-histone interaction may induce flip-flop, or transmembrane lipid motions (our unpublished observation), so that inner leaflet lipids may also be (partly) available for interaction with histones.
Figure 8.
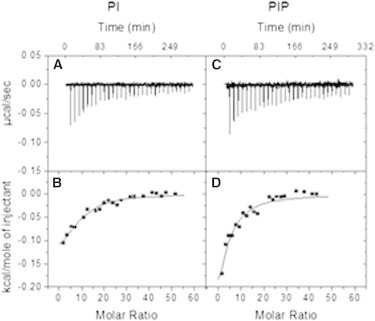
Representative experiments of liposome:histone binding as measured by isothermal titration calorimetry. Results obtained from injecting PI-containing vesicles (A and B), or PIP-containing vesicles (C and D), into a H1 histone suspension are shown. The observed heat exchanges after repeated injections (A and C) are transformed into heat/mole injectant (B and D) then fitted to a binding model (see Methods for details).
Table 1.
Thermodynamic parameters for the interaction of H1 with unilamellar vesicles of various lipid compositions
| PC:PI (9:1) | PC:PIP (9:1) | |
|---|---|---|
| N | 0.99 ± 0.17 | 0.53 ± 0.13 |
| Ka | 1.08 ± 0.3·104 | 9.31 ± 0.44·104 |
| ΔH° | −4.25 ± 1.3·106 cal/mol | −1.41 ± 0.27·106 cal/mol |
| ΔS° | −1.42 ± 0.45·104 cal/K·mol | −4.71 ± 0.91·103 cal/K·mol |
| ΔG° | −2.99 ± 0.32·103 cal/mol | −7.9 ± 0.35·103 cal/mol |
Results in cal/mol injectant (PC + PI or PC + PIP). Average values ±SD (n = 3,4).
Discussion
The two main observations in this contribution are i), the capacity of histones to bind and aggregate phospholipid vesicles containing negatively charged lipids, particularly when the lipids bear more than one negative charge per molecule, and ii), the displacement of histones and reversal of aggregation by a nonspecific DNA. Each aspect will be discussed separately.
Histone-induced vesicle aggregation
The phenomenon has an obvious electrostatic origin, and can be interpreted as a particular case of polyelectrolyte-induced aggregation of colloidal particles (27,28). The same electrostatic effect has been used to encapsulate electrically charged drugs or other compounds in liposomes (29), including the entrapping of plasmid DNA or siRNA in liposomes or nanoparticles containing positively charged lipids (30,31). Several authors (15–17) described the interaction of H1 with vesicles containing negatively charged phospholipids. This study uses phosphoinositides as negatively charged lipids, and this has important physiological implications, as discussed below. Moreover, we show (Fig. 2 C) that the requirement of negatively charged phospholipids decreases by one order of magnitude when the lipid has at least two negative charges per headgroup. This suggests that each of the charged phosphates can bind a separate histone molecule that will in turn bind a different vesicle. The two charges of PIP appear to be undistinguishable from the point of view of histone binding, because the calorimetric data can be fitted to a single population of sites both for PI and for PIP. Interestingly, in the binding of H1 to PI, the entropic factor all but compensates the large enthalpic component (Table 1), the decreased entropy being probably due to clustering of several PI molecules by H1. The double charge of PIP means that a smaller number of lipid molecules are required to compensate for the positive charges of histones, hence smaller clusters and smaller entropy decrease. The possibility of polyphosphoinositide clusters interacting with polybasic residues in proteins has been suggested (32).
Phospholipid displacement by DNA
The turbidimetry and microscopy data (Figs. 3 and 7) show that DNA prevents H1 from binding PIP vesicles and even displaces H1 from H1-vesicle aggregates. A direct application of thermodynamic data to the processes observed in Fig. 3 cannot be made in the simple terms that would be used for a chemical reaction in solution. Both DNA and vesicles are in some sense solid-state particles, thus the chemical activities that would be required for a proper quantitative treatment of the process cannot be known with certainty. It is interesting however that at least for a DNA/H1 wt ratio 0.1–0.5 a substantial proportion of PIP-H1 complex appears to exist (Fig. 3 C). Results pointing in this direction, with DNA or RNA competing for histones with negatively charged phospholipids, have been published (16,33). This means that, even if the precise conditions in the cell nucleoplasm cannot be defined at present, the situation with DNA-PIP-H1 multiple equilibrium should not be discarded.
Physiological implications
The fact that strong interactions are observed in the previous experiments between histones and polyphosphoinositides does not imply a biological specificity. Other positively charged chromatin proteins could interact as well, and cardiolipin can substitute PIP (Fig. 1 B). The physiological relevance of this work does not arise solely from specificity, but from the proof that such interactions are feasible. Understanding the thermodynamics and types of interactions between nucleic acids, positively charged chromatin proteins and phosphoinositides, in a simple model aids us to have a more complete comprehension of the interaction dynamics in a complex environment of the nucleoplasm. Among the various negatively charged phospholipids that can interact electrostatically with histones, the polyphosphoinositides are particularly important because of their presence at the nuclear membrane and nucleoplasm. Phosphoinositides are important lipid messengers and their presence in the nucleoplasm, together with enzymes responsible for their metabolism, has been described (9). Their role in nuclear signaling has been reviewed by Barlow et al., (34). These authors mentioned as well that nuclear phosphoinositides appeared to be compartmentalized, although their distribution into functionally specific locations remained a mystery. Important steps in the elucidation of this enigma have come from Larijani’s laboratory. Garnier-Lhomme et al. (35) studied the lipid composition of the nuclear envelope remnants, membraneous domains of the sperm nuclear envelope that remain, upon the sperm nuclear membrane disassembly at the initial stages of fertilization and are subsequently incorporated to reform the new nuclear envelope known as the male pronucleus. These membrane domains are enormously enriched in phosphoinositides (51% of all phospholipids distributed among 18% PI, 12% PIP, 12% PIP2, 9% PIP3). This is in contrast to the average <5% of phosphoinositides in cell membranes. The data provide support to the idea of nuclear membrane domains highly enriched in negatively charged phospholipids. More recently (11) the same group has provided evidence that localized and acute depletion of PIP2 results in incomplete nuclear envelope reformation during mitosis. Moreover, using correlative light and electron microscopy, they show that the nuclear membrane, far from being a smooth, quasispherical structure, may actually consist of a convoluted lamellar and tubular system penetrating the nuclear core. This is in agreement with previous morphological observations by Fricker et al. (13) and Echevarria et al. (14) who showed invaginations of the nuclear envelope in live intact nuclei. Irvine (9) suggested that those invaginations could be highly enriched in PIP2. The correct assembly of the nuclear membrane and the maintenance of its morphology is crucial as the various components of the nucleoplasm rely on interacting with the inner bilayer of the NE. Investigations by Domart et al. (11) have illustrated that the malformation of the NE due to acute depletion of PIP2 derivative, have resulted in the daughter cells not being able to go through mitosis. Moreover, the work by Vaux and co-workers (7,8) has highlighted the presence and putative roles of the nucleoplasmic reticulum, including its relevance in a series of pathological conditions, e.g., cell malignancy, laminopathies, or viral access to the nuclear interior. To our knowledge, these novel views of the nuclear membrane structure immediately evoke complex and unforeseen possibilities of phosphoinositide interaction with chromatin proteins and DNA/RNA, for which the data in this work may constitute a minimal model system, in line with the reductionist biophysical approach to cell biology.
Acknowledgments
The authors are grateful to Dr. S. Taneva for her help with the analysis of calorimetric data.
This work was supported in part by grants from the Spanish Ministerio de Economia (BFU 2012-36241 to FMG, BFU 2011-28566 to AA) and from the Basque Government (grants No. IT 838-13 to A.A., No. IT 849-13 to F.M.G.). M.G.L. and N.F.-R. were predoctoral students supported by the Basque Government.
References
- 1.Mima J., Hickey C.M., Wickner W. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J. 2008;27:2031–2042. doi: 10.1038/emboj.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch K.L., Gerona R.R., Martin T.F. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol. Biol. Cell. 2008;19:5093–5103. doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.James D.J., Khodthong C., Martin T.F. Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J. Cell Biol. 2008;182:355–366. doi: 10.1083/jcb.200801056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramos I., Prado A., Ausió J. Nucleoplasmin-mediated unfolding of chromatin involves the displacement of linker-associated chromatin proteins. Biochemistry. 2005;44:8274–8281. doi: 10.1021/bi050386w. [DOI] [PubMed] [Google Scholar]
- 5.Bustin M., Catez F., Lim J.H. The dynamics of histone H1 function in chromatin. Mol. Cell. 2005;17:617–620. doi: 10.1016/j.molcel.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Collepardo-Guevara R., Schlick T. The effect of linker histone’s nucleosome binding affinity on chromatin unfolding mechanisms. Biophys. J. 2011;101:1670–1680. doi: 10.1016/j.bpj.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malhas A., Goulbourne C., Vaux D.J. The nucleoplasmic reticulum: form and function. Trends Cell Biol. 2011;21:362–373. doi: 10.1016/j.tcb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Goulbourne C.N., Malhas A.N., Vaux D.J. The induction of a nucleoplasmic reticulum by prelamin A accumulation requires CTP:phosphocholine cytidylyltransferase-α. J. Cell Sci. 2011;124:4253–4266. doi: 10.1242/jcs.091009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irvine R.F. Nuclear inositide signalling—expansion, structures and clarification. Biochim. Biophys. Acta. 2006;1761:505–508. doi: 10.1016/j.bbalip.2006.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larijani B., Barona T.M., Poccia D.L. Role for phosphatidylinositol in nuclear envelope formation. Biochem. J. 2001;356:495–501. doi: 10.1042/0264-6021:3560495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domart M.C., Hobday T.M., Larijani B. Acute manipulation of diacylglycerol reveals roles in nuclear envelope assembly & endoplasmic reticulum morphology. PLoS ONE. 2012;7:e51150. doi: 10.1371/journal.pone.0051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cocco L., Gilmour R.S., Irvine R.F. Synthesis of polyphosphoinositides in nuclei of Friend cells. Evidence for polyphosphoinositide metabolism inside the nucleus which changes with cell differentiation. Biochem. J. 1987;248:765–770. doi: 10.1042/bj2480765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fricker M., Hollinshead M., Vaux D. Interphase nuclei of many mammalian cell types contain deep, dynamic, tubular membrane-bound invaginations of the nuclear envelope. J. Cell Biol. 1997;136:531–544. doi: 10.1083/jcb.136.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Echevarría W., Leite M.F., Nathanson M.H. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 2003;5:440–446. doi: 10.1038/ncb980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira L.F., Marco F.M., Subiza J.L. Histones interact with anionic phospholipids with high avidity; its relevance for the binding of histone-antihistone immune complexes. Clin. Exp. Immunol. 1994;97:175–180. doi: 10.1111/j.1365-2249.1994.tb06064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kõiv A., Palvimo J., Kinnunen P.K. Evidence for ternary complex formation by histone H1, DNA, and liposomes. Biochemistry. 1995;34:8018–8027. doi: 10.1021/bi00025a007. [DOI] [PubMed] [Google Scholar]
- 17.Zhao H., Bose S., Kinnunen P.K. Interactions of histone H1 with phospholipids and comparison of its binding to giant liposomes and human leukemic T cells. Biochemistry. 2004;43:10192–10202. doi: 10.1021/bi049758b. [DOI] [PubMed] [Google Scholar]
- 18.Wang X., Moore S.C., Ausió J. Acetylation increases the alpha-helical content of the histone tails of the nucleosome. J. Biol. Chem. 2000;275:35013–35020. doi: 10.1074/jbc.M004998200. [DOI] [PubMed] [Google Scholar]
- 19.Mayer L.D., Hope M.J., Cullis P.R. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 20.Böttcher C.J.F., van Gent C.M., Fries C. A rapid and sensitive sub-micro phosphorus determination. Anal. Chim. Acta. 1961;24:203–204. [Google Scholar]
- 21.Angelova M.I., Tsoneva I. Interactions of DNA with giant liposomes. Chem. Phys. Lipids. 1999;101:123–137. doi: 10.1016/s0009-3084(99)00060-2. [DOI] [PubMed] [Google Scholar]
- 22.Fidorra M., Duelund L., Bagatolli L.A. Absence of fluid-ordered/fluid-disordered phase coexistence in ceramide/POPC mixtures containing cholesterol. Biophys. J. 2006;90:4437–4451. doi: 10.1529/biophysj.105.077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goñi F.M., Villar A.V., Alonso A. Interaction of phospholipases C and sphingomyelinase with liposomes. Methods Enzymol. 2003;372:3–19. doi: 10.1016/S0076-6879(03)72001-1. [DOI] [PubMed] [Google Scholar]
- 24.Viguera A.-R., Alonso A., Goñi F.M. Liposome aggregation induced by poly (ethylene glycol). Rapid kinetic studies. Colloids Surf. B Biointerfaces. 1995;3:263–270. [Google Scholar]
- 25.Reference deleted in proof.
- 26.Reference deleted in proof.
- 27.Bordi F., Sennato S., Truzzolillo D. Polyelectrolyte-induced aggregation of liposomes: a new cluster phase with interesting applications. J. Physics. Condens. Matter. 2009;21:203102. doi: 10.1088/0953-8984/21/20/203102. [DOI] [PubMed] [Google Scholar]
- 28.Cametti C. Polyion-induced aggregation of oppositely charged liposomes and charged colloidal particles: the many facets of complex formation in low-density colloidal systems. Chem. Phys. Lipids. 2008;155:63–73. doi: 10.1016/j.chemphyslip.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Ulrich A.S. Biophysical aspects of using liposomes as delivery vehicles. Biosci. Rep. 2002;22:129–150. doi: 10.1023/a:1020178304031. [DOI] [PubMed] [Google Scholar]
- 30.Fenske D.B., Chonn A., Cullis P.R. Liposomal nanomedicines: an emerging field. Toxicol. Pathol. 2008;36:21–29. doi: 10.1177/0192623307310960. [DOI] [PubMed] [Google Scholar]
- 31.Ozaki S., DeWald D.B., Prestwich G.D. Intracellular delivery of phosphoinositides and inositol phosphates using polyamine carriers. Proc. Natl. Acad. Sci. USA. 2000;97:11286–11291. doi: 10.1073/pnas.210197897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhendre V., Grélard A., Dufourc E.J. Key role of polyphosphoinositides in dynamics of fusogenic nuclear membrane vesicles. PLoS ONE. 2011;6:e23859. doi: 10.1371/journal.pone.0023859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H., Fukami K., Takenawa T. Phosphatidylinositol 4,5-bisphosphate reverses the inhibition of RNA transcription caused by histone H1. Eur. J. Biochem. 1998;251:281–287. doi: 10.1046/j.1432-1327.1998.2510281.x. [DOI] [PubMed] [Google Scholar]
- 34.Barlow C.A., Laishram R.S., Anderson R.A. Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends Cell Biol. 2010;20:25–35. doi: 10.1016/j.tcb.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garnier-Lhomme M., Byrne R.D., Larijani B. Nuclear envelope remnants: fluid membranes enriched in sterols and polyphosphoinositides. PLoS ONE. 2009;4:e4255. doi: 10.1371/journal.pone.0004255. [DOI] [PMC free article] [PubMed] [Google Scholar]