

Abstract
Cardiac myosin binding protein-C (cMyBP-C) is a cardiac-specific, thick-filament regulatory protein that is differentially phosphorylated at Ser273, Ser282, and Ser302 by various kinases and modulates contraction. In this study, phosphorylation-site-specific effects of cMyBP-C on myocardial contractility and cross-bridge kinetics were studied by sinusoidal analysis in papillary and trabecular muscle fibers isolated from t/t (cMyBP-C-null) mice and in their counterparts in which cMyBP-C contains the ADA (Ala273-Asp282-Ala302), DAD (Asp273-Ala282-Asp302), and SAS (Ser273-Ala282-Ser302) mutations; the results were compared to those from mice expressing the wild-type (WT) transgene on the t/t background. Under standard activating conditions, DAD fibers showed significant decreases in tension (∼50%), stiffness, the fast apparent rate constant 2πc, and its magnitude C, as well as its magnitude H, but an increase in the medium rate constant 2πb, with respect to WT. The t/t fibers showed a smaller drop in stiffness and a significant decrease in 2πc that can be explained by isoform shift of myosin heavy chain. In the pCa-tension study using the 8 mM phosphate (Pi) solution, there was hardly any difference in Ca2+ sensitivity (pCa50) and cooperativity (nH) between the mutant and WT samples. However, in the solutions without Pi, DAD showed increased nH and slightly decreased pCa50. We infer from these observations that the nonphosphorylatable residue 282 combined with phosphomimetic residues Asp273 and/or Asp302 (in DAD) is detrimental to cardiomyocytes by lowering isometric tension and altering cross-bridge kinetics with decreased 2πc and increased 2πb. In contrast, a single change of residue 282 to nonphosphorylatable Ala (SAS), or to phosphomimetic Asps together with the changes of residues 273 and 302 to nonphosphorylatable Ala (ADA) causes minute changes in fiber mechanics.
Introduction
Cardiac myosin binding protein-C (cMyBP-C) is a 140 kDa thick-filament-associated protein (1) that plays both structural and functional roles in sarcomeres (2,3). Mutations in the cMyBP-C gene (MYBPC3) cause >40% of familial hypertrophic cardiomyopathy (FHC) (4), the leading cause of sudden cardiac death in young adults. cMyBP-C differs from the skeletal isoforms in that it contains multiple phosphorylation sites. These are phosphorylated in a site-specific way by at least six kinases: protein kinase A (PKA) (5,6), protein kinase C (PKC) (5), protein kinase D (PKD) (7), Ca2+/calmodulin-dependent protein kinases II (CaMKII) (6), ribosomal s6 kinase (8), and glycogen synthase kinase 3β (GSK3β) (9). cMyBP-C is extensively phosphorylated under basal conditions (10), but its phosphorylation level decreases under pathological conditions (10–15). Despite extensive research on cMyBP-C, the physiological role of its phosphorylation is not well understood.
Three phosphorylation sites identified in mice and their homologous sites in humans are: Ser273, Ser282, and Ser302 (6,14,16). These residues are located in the M domain between domains C1 and C2. It was found that PKA phosphorylates all three sites, PKC phosphorylates Ser273 and Ser302, and CaMKII phosphorylates only Ser282 (17). Being a substrate of PKA and PKC (5,6), cMyBP-C is thought to be important in regulating myocardial contraction through a β-adrenergic signaling mechanism (18). Mutation of the three Ser residues to phosphorylation-deficient Ala results in impaired cardiac function, reduced contractile reserve, and cardiac hypertrophy in transgenic mice (19,20). In contrast, mutation of Ser to the phosphor-mimetic Asp does not impair cardiac function and results in a cardioprotective effect (21).
The phosphorylation sites in cMyBP-C are functionally unequal and hierarchically regulated (6). That is, the three phosphorylation sites do not act independently, but cooperatively (22). Among these three sites, Ser282 appears to play a central role, as evidenced by the inactivation of cMyBP-C phosphorylation at other sites when Ser282 is mutated to Ala (6,17). It may be the consequence of a conformational change that makes other sites inaccessible to kinases (6). In a previous investigation, we studied the importance of Ser282 phosphorylation on cardiac morphology and function, using the transgenic mouse model SAS (Ser272-Ala282-Ser302), in which Ser282 was mutated to Ala (17). At the same time, two additional mouse models, DAD (Asp273-Ala282-Asp302, in which Ser273 and Ser302 were mutated to the phosphorylation-mimetic Asp), and ADA (Ala273-Asp282-Ala302), were studied (17). It was found that the hearts of DAD mice, but not those of their ADA or SAS counterparts, showed significant hypertrophy, fibrosis, and myocyte disarray, mimicking the t/t (null) phenotype (17). Based on these observations, we hypothesized that DAD diminishes cardiac contractility and cross-bridge kinetics. The objective of this study was to determine the consequences of phosphorylation at different cMyBP-C sites for myocardial contractility and cross-bridge kinetics, using the ADA, DAD, and SAS mutant mice. The wild-type (WT) cMyBP-C transgene was introduced on the cMyBP-C null (t/t) background, and this cDNA was subjected to site-directed mutagenesis to generate ADA, DAD, and SAS transgenes. The results from each mouse model were compared with those from mice expressing the WT transgene. Our results indicate that the cross-bridge kinetics are significantly altered in mice carrying the DAD mutation, and that this results in a reduction of isometric tension.
Materials and Methods
Animal models
The transgenic mouse models used in this study were reported in a previous publication (17). The t/t mouse is a hypertrophic cardiomyopathy mutant in which modifications to the carboxyl end of the cMyBP-C protein cause it to be absent from sarcomeres (23). The strain referred to as WT is a transgenic line in which the WT cMyBP-C gene was introduced into the t/t background. Three mutant transgenes were also created, in which Ser residues of cMyBP-C at positions 273, 282, and 302 were either unchanged or mutated to Ala or Asp to produce a phosphorylation-deficient or phosphorylation-mimetic effect, respectively. In the mutant names, Ala is coded as A, Asp is coded as D, and Ser is coded as S, in the standard way, and each mutant is identified by the three-letter code: ADA (expressing Ala273-Asp282-Ala302), DAD (Asp273-Ala282-Asp302), and SAS (Ser273-Ala282-Ser302). Information on the number of mice used, including their age and gender, is listed in Table 1. These animal experiments were in accordance with the guidelines of the Institutional Animal Care and Use Committees at Loyola University in Chicago and The University of Iowa, as well as those listed in the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health.
Table 1.
Gender and age for the adult mice used in this study
| Mouse model | Gender | Age (months) |
|---|---|---|
| WT | Male | 5.8 |
| WT | Female | 4.5 |
| WT | Male | 2.3 |
| t/t | Male | 2.7 |
| t/t | Male | 5.1 |
| t/t | Male | 5.1 |
| t/t | Male | 2.4 |
| t/t | Male | 2.4 |
| ADA | Male | 5.0 |
| ADA | Male | 8.0 |
| ADA | Male | 8.0 |
| DAD | Male | 7.4 |
| DAD | Male | 5.5 |
| SAS | Male | 10.4 |
| SAS | Female | 4.4 |
| SAS | Male | 2.9 |
Muscle preparations
Each mouse was euthanized through intraperitoneal injection of Na pentobarbital at 50 mg/kg, and the heart was removed. The whole heart was perfused using the Langendorff system with perfusion buffer at 37°C for 5 min. The hearts were then moved sequentially to Na skinning solution for 15–30 min, K skinning solution for 30 min, and storage solution on ice overnight. On the next day, the storage solution was replaced once. The compositions of the solutions used have previously been reported (24). The hearts were then shipped on ice to Iowa City, where they were stored at −20°C until used for biophysical studies. The experiments were performed two days to four weeks after shipment. There were hardly any changes in the performance, with respect to either isometric tension or rate constants, during this period. In the cases of preparations for which a change in one of these parameters was observed, we disposed of the data rather than proceeding with further analysis. With these criteria, many of the preparations were usable beyond four weeks.
The method used for the mechanical measurements has been described previously (25); the compositions of the solutions are found in our earlier report (24). In brief, papillary and trabecular muscle fibers from both ventricles ∼1 mm in length and 100 μm in diameter were dissected and mounted on the experimental apparatus by attaching their ends to two stainless steel wire hooks using nail polish. One hook was connected to a length driver, and the other to a tension transducer. Fibers were soaked for 3–5 min in 600 μL relaxing solution, followed by further skinning in the relaxing solution containing 1% Triton X-100 for 20 min. Fibers were then washed with the relaxing solution and stretched to remove the slack. This procedure generally resulted in a sarcomere length of 2.1–2.2 μm, as judged by optical diffraction using a He-Ne laser (wavelength: 0.6328 μm) (25). Fiber length (L0) and cross-sectional area were recorded at this time. All the solutions were applied twice (600 μl each) to ensure that significant amounts of the old solution did not remain.
Solutions
Each fiber was tested in the standard activating solution, followed by the pCa-tension and pCa-stiffness study. The standard activating solution contained (in mM) 6 K2CaEGTA, 6.1 Na2H2ATP, 15 Na2CP (creatine phosphate), 6.6 MgAc2 (Ac, acetate), 8 K1.5H1.5Pi (phosphate), 13 NaAc, 83 KAc, 10 MOPS, and 80 U/mL CK (creatine kinase); the ionic compositions of this solution were pCa (–log10[Ca2+]) 4.55, Mg2+ 1 mM, and MgATP2- 5 mM, pH 7.00, and ionic strength (IS) was 200 mM (adjusted by KAc). The pCa-tension and pCa-stiffness relations were studied in two sets of solutions, 8Pi and 0Pi. The 8Pi solution is our standard activating solution, in which the total Ca concentration was changed from 0.94 mM to 6 mM to produce pCa levels in the range 7.00–4.55, respectively. The 0Pi solution did not contain added Pi, and contained (in mM) 7 K2CaEGTA, 2 Na2H2ATP, 15 Na2CP, 3 MgAc2, 81 KAc, 10 MOPS, and 80 U/ml CK; the ionic compositions of this solution series were Mg2+ 0.78–0.89 mM and MgATP2− 1.62 mM, pH 7.00, with IS 150 mM (Table 1 of Bubb et al. (26)). pCa was adjusted to 7.00–4.40 by changing the total Ca concentration across the range of 1.1–7.0 mM, respectively. The 0Pi solution has been used by many other investigators (27–31), whereas we have been using the 8Pi solution (24,33–35).
Sinusoidal analysis
The sinusoidal analysis is complementary to step analysis, in which length changes are limited to a fraction of 1% of the total length; the details of this analysis method are described in Wang et al. (24). In brief, when tension reached a plateau, fiber length was oscillated in sine waves of varying frequencies in the range 0.5–140 Hz at a small amplitude (0.125%, corresponding to 1.3 nm at the half-sarcomere level). Both length (strain) and tension (stress) time courses were digitized simultaneously using two 16-bit A/D converters without filtration. The complex modulus Y(f) was defined as the ratio of the stress change to the strain change expressed in the frequency domain. Y(f) consists of two components: the viscous modulus (imaginary part of Y(f)) and the elastic modulus (real part of Y(f)). For cardiac muscle, Y(f) was fitted to Eq. 1 to resolve into two exponential processes, B and C (33):
| (1) |
Processes B and C reflect the active interaction of myosin cross-bridges with the thin filament, which leads to the transduction of chemomechanical energy, because they are absent in the rigor condition (33). Process B corresponds to phase 3 of step analysis, which is a medium frequency-exponential delay during which the muscle generates oscillatory work. Process C corresponds to phase 2, which is a high-frequency exponential advance during which the muscle absorbs work. In fast-twitch skeletal muscle fibers, an additional exponential process A was observed (33–35). In cardiac muscles, process A is observed only at temperatures ≥30°C (see Fig. 3, C, F, I, L and O in Lu et al. (36)). The study described here was performed at 20°C to result in two exponential processes, as shown by Eq. 1. The rate constants of delayed tension 2πb, and fast tension recovery 2πc, are the apparent rate constants of processes B and C, and B and C are their respective magnitudes (amplitudes); H is the 0(zero) frequency stiffness. These were deduced by fitting Y(f) to Eq. 1. The complex modulus Y(f) extrapolated to infinite (∞) frequency is: Y∞ = H – B + C. Y∞ corresponds to phase 1 of the step analysis, and is loosely called stiffness in muscle mechanics literature.
Figure 3.
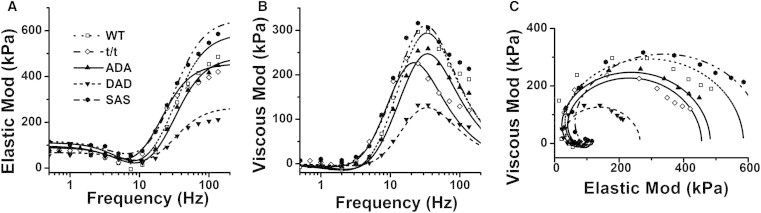
Averaged complex modulus data in the 8Pi solution. Data were plotted as elastic modulus (real Y(f)) versus frequency (f) (A), viscous modulus (imaginary Y(f)) versus frequency (B), and viscous modulus versus elastic modulus (Nyquist plot) (C). n = 31 for WT, 38 for t/t, 32 for ADA, 31 for DAD, and 27 for SAS. Solid lines represent Eq. 1 with best-fit parameters. In C, frequency increases in the clockwise direction; its value can be found in A and B.
pCa-tension and pCa-stiffness studies
Tension increased when the pCa was changed sequentially from high to low, as indicated in Fig. 1. The pCa-tension relationship was individually fitted to the Hill equation:
| (2) |
where Tmax is the maximum tension developed at saturating [Ca2+], Ca50 is the Ca2+ concentration at half-saturation (apparent Ca2+ dissociation constant), and nH is the Hill factor (describes cooperativity among contractile proteins, including troponin, tropomyosin, myosin, and cMyBP-C) based on tension. pCa50 = –log10Ca50 indicates Ca2+ sensitivity. The pCa-stiffness relationship was fitted to a similar equation:
| (3) |
where Smax is the maximum stiffness developed at saturating [Ca2+], Ca50S is the Ca2+ concentration at half-saturation, and nHS is the Hill factor (which describes cooperativity determined based on stiffness). pCa50S = –log10Ca50S indicates Ca2+ sensitivity based on measurement of stiffness.
Figure 1.
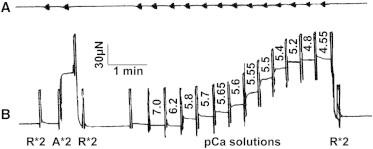
Slow time courses of length (A) and tension (B), demonstrating the experimental protocol. This example is from an ADA fiber preparation (0.9 mm in length and 87 μm in diameter). The amplitude of sinusoidal oscillation was kept at 0.125% L0 in all frequencies, but because the signal was filtered with a second-order low-pass filter (cutoff at 10 Hz), the amplitude looks attenuated at higher frequencies (to the left). R, relaxing solution; A, standard activating solution. ∗2 indicates that the solution was changed twice.
Identification of myosin heavy chain isoforms
To determine myosin heavy chain (MHC) isoform compositions, α- and β-isoforms were separated on a 6.25% acrylamide Hoefer gel (99:1 acrylamide:bis-acrylamide) by SDS-PAGE. Gels were fixed in 10% acetic acid followed by overnight staining with Sypro-Ruby. Gels were imaged and the percentage of myosin isoforms was determined using Image J software.
Statistics
In each experiment, >10 fibers were dissected from multiple hearts from each mouse model (as indicated in Table 1) and studied. All data are expressed as the mean ± SE. One-way ANOVA was performed to determine the significance of the difference among test groups. Post hoc tests (least significant difference) that allow pairwise multiple comparisons were applied to locate the difference among groups. The MHC expression level measured from SDS-PAGE was compared with Bonferroni post-hoc test (see Fig. 8). A significant difference is defined as p ≤ 0.05, and highly significant as p ≤ 0.01.
Figure 8.
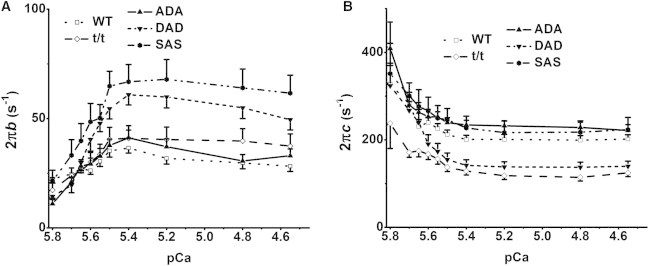
Apparent rate constants 2πb (A) and 2πc (B) plotted against pCa in the 8Pi solution. Values at pCa ≥ 6.0 are not included, because they were seriously contaminated with noise. n = 17 for WT, 13 for t/t, 14 for ADA, 18 for DAD, and 12 for SAS.
Results
All the values of biophysical parameters reported in this section are listed in Table S1.
Tension and stiffness during maximal Ca2+ activation
The cardiac fiber was soaked first in the relaxing solution and then in the standard activating solution. When the steady state in tension was achieved, the computer was triggered to perform sinusoidal analysis and to collect the fast-time-course data for length and tension signals as reported (37). The slow time courses were traced by a pen recorder, and typical records are shown in Fig. 1 A (length) and Fig. 1 B (tension). The fiber was subsequently relaxed. After this initial test, the fiber was activated in a series of solutions containing increasing amounts of Ca2+.
The above experiments were repeated for 11–38 fibers from each mouse model and the results were averaged. The complex modulus data were fitted to Eq. 1 to extract two exponential processes. Their parameters were used to calculate the elastic modulus Y∞, which is called stiffness. Isometric tension and stiffness are plotted for five groups of mice at pCa 4.55 in the 8Pi and 200IS solution (Fig. 2 A) and at pCa 4.40 in the 0Pi and 150IS solution (Fig. 2 B). These results demonstrate that tension and stiffness were significantly lower (by ∼48%) in the DAD group than in the WT group (p < 0.01) in both solutions. It was also noted that stiffness was significantly lower in the t/t group in the 8Pi solution (Fig. 2 A), and in the ADA group in the 0Pi solution (Fig. 2 B), than it was in the WT mice.
Figure 2.
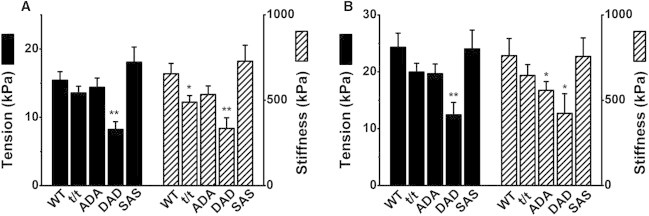
Isometric tension (Tmax in Eq. 2; left ordinate) and stiffness (Smax in Eq. 3; right ordinate) in 8Pi solution (A) and 0Pi solution (B) at saturating [Ca2+]. The 8Pi solution contained 8 mM Pi and its IS was 200 mM, whereas the 0Pi solution contained no added Pi and its IS was 150 mM. ∗∗p < 0.01 and ∗p < 0.05 compared to WT. The number of experiments (n) in A is 31 for WT, 38 for t/t, 32 for ADA, 31 for DAD, and 27 for SAS; in B, n = 14 for WT, t/t, ADA, and DAD, and n = 11 for SAS.
Tension was, as anticipated, 50–60% greater in the 0Pi solution (Fig. 2 B) than in the 8Pi solution (Fig. 2 A). This is due to the differences in both [Pi] and IS, as an increase in either can decrease isometric tension in skeletal (38,39) and cardiac (40,41) muscle fibers. Similarly, stiffness was 20–30% greater in the 0Pi solution than in the 8Pi solution.
Cross-bridge kinetics
During the tension plateau of the standard activation, sinusoidal analysis was performed as described in Materials and Methods, and the complex modulus data, Y(f), were collected and plotted as discrete points in Fig. 3. These data are equivalents of tension transients in response to step changes in length. Fig. 3 includes plots of the elastic modulus versus frequency (Fig. 3 A), the viscous modulus versus frequency (Fig. 3 B), and the viscous modulus versus the elastic modulus (Nyquist plots) (Fig. 3 C). Each Nyquist plot shows two contiguous semicircles. Hence, the complex modulus data can be resolved into two exponential processes, B and C, as shown in Eq. 1. Fig. 3 reveals that although the WT, t/t, ADA, and SAS groups had fairly similar complex modulus values, those of the DAD group were considerably smaller, as evidenced by the considerably smaller diameter of the semicircle in the Nyquist plot at frequencies >20 Hz (Fig. 3 C).
The complex modulus data were fitted to Eq. 1, and the best-fit data are shown in Fig. 3 as continuous curves. Fig. 3 A shows that the elastic modulus remained small in the low-frequency range (<7 Hz), reached its minimum value at ∼7 Hz (this frequency has been referred to as fmin by some investigators (42)), and increased significantly in the high-frequency range (>7 Hz). Fig. 3 B indicates that the viscous modulus had small negative values in the low-frequency range (<5 Hz), reached a shallow minimum at ∼2 Hz, increased steeply in the range 5–25 Hz, reached its maximum value at ∼30 Hz, and decreased again in the high-frequency range (50–140 Hz). The frequency at which the viscous modulus is at its minimum approximates the characteristic frequency, b, and that at which it is at its maximum approximates the characteristic frequency, c. In Fig. 3 B, it is evident that the frequency associated with the maximum viscous modulus is shifted to the left in the t/t group, indicating that 2πc is slower.
The fitting of the data in Fig. 3 to Eq. 1 yielded two apparent rate constants, 2πb and 2πc, and their respective magnitudes, B and C, as well as magnitude H, all of which are plotted in Fig. 4 for the 8Pi solution at pCa 4.55, and in Fig. 5 for the 0Pi solution at pCa 4.40. Fig. 4 A (left ordinate) reveals that the rate constant 2πb for the DAD and SAS groups was significantly larger than that for the WT mice, and 2πb of the t/t and ADA groups was similar to that of the WT group. The rate constant 2πc (Fig. 4 A, right ordinate) for the t/t and DAD groups was significantly less than that for the WT, but 2πc for the ADA and SAS groups was similar to that for the WT. The different directions of the two rate constants are not surprising, because each rate constant reflects a different event in the cross-bridge cycle (43,44). Fig. 4 B (left ordinate) indicates that magnitude B for the t/t and SAS groups was larger than that for the WT group (p < 0.05), and that B of the ADA and DAD groups did not differ significantly from that of WT. Magnitudes C and H were significantly smaller in the DAD group than in the WT group (p < 0.05), but were similar across the other groups (Fig. 4 B). In general, magnitude B was about half of magnitude C but twice magnitude H in the 8Pi solution.
Figure 4.
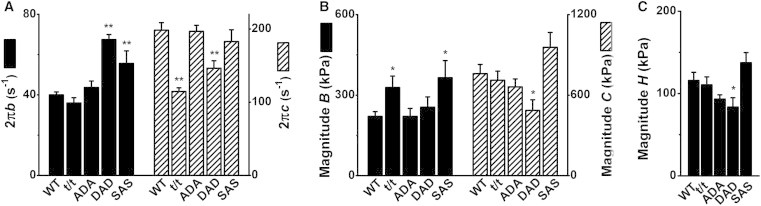
Parameters of exponential processes in the 8Pi solution (pCa 4.55). (A) Apparent rate constants 2πb (left ordinate) and 2πc (right ordinate). (B) Magnitudes of processes B (left ordinate) and C (right ordinate). (C) Magnitude H. Error bars represent the mean ± SE of 26–38 experiments carried out in each group (for numbers of mice, see Table 1). n = 31 for WT, 38 for t/t, 32 for ADA, 31 for DAD, and 27 for SAS. ∗∗p < 0.01 and ∗p < 0.05 compared to WT.
Figure 5.
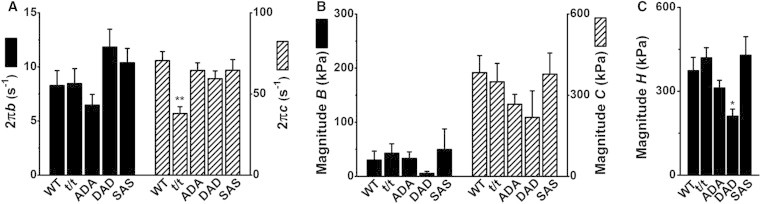
Parameters of exponential processes in the 0Pi solution (pCa 4.40). (A) Apparent rate constants 2πb (left ordinate) and 2πc (right ordinate). (B) Magnitudes of processes B (left ordinate) and C (right ordinate). (C) Magnitude H. n = 14 for WT, t/t, ADA, and DAD, and n = 11 for SAS. ∗∗p < 0.01 and ∗p < 0.05 compared to WT.
The apparent rate constants 2πb and 2πc deduced from the 0Pi solution (Fig. 5 A) were ∼1/5 and 1/3, respectively, of those deduced from the 8Pi solution (Fig. 4 A). Similarly, magnitude B was ∼1/10, and magnitude C ∼1/2, of the corresponding values in the 8Pi solution. Thus, the resolution of processes B and C was much less in the 0Pi solution than in the 8Pi solution. Because of this, the relative error bars plotted in Fig. 5 are larger than those in Fig. 4. Due to the small magnitude and large scatter of the data, the only statistical difference in the rate constants measured among the five groups was a decreased 2πc in the t/t group. The magnitudes B and C did not differ significantly among all groups, but were somewhat lower in the DAD group. Magnitudes H were significantly smaller in the DAD group than in the WT group (p < 0.05). In all groups, magnitude H was three times larger in the 0Pi solution than in the 8Pi solution.
pCa-tension and pCa-stiffness studies
pCa-tension and pCa-stiffness results are plotted in Fig. 6 (at 8Pi and 200IS) and Fig. 7 (at 0Pi and 150IS), as discrete points after proper normalization. The data for each preparation were fitted to Eq. 2 (tension) or Eq. 3 (stiffness), and the fitted parameters were averaged. The best-fit curves are shown by solid lines in Figs. 6 and 7. The averaged pCa50 and nH values are presented in Figs. 6, B and D, and 7, B and D.
Figure 6.
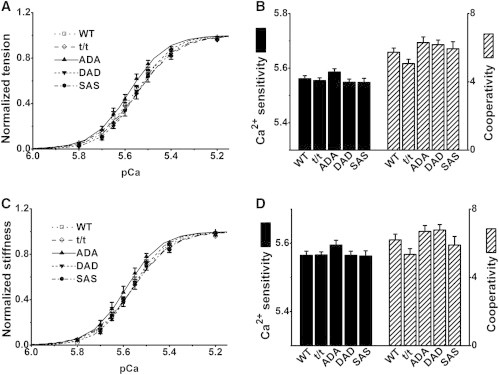
Effect of pCa in the 8Pi solution in five groups of mice. (A) pCa-tension curves normalized to Tmax. (B) pCa50 (left ordinate) and nH (right ordinate), as deduced by fitting the individual data to Eq. 2, and averaging. (C) pCa-stiffness curves normalized to Smax. (D) pCa50S (left ordinate) and nHS (right ordinate), deduced by fitting the data to Eq. 3, and averaging. In A and C, the data are shown by discrete points, and the best-fit curves are shown by solid lines. B and D do not reveal significant differences in any of the parameters measured. n = 17 for WT, 13 for t/t, 14 for ADA, 18 for DAD, and 12 for SAS.
Figure 7.
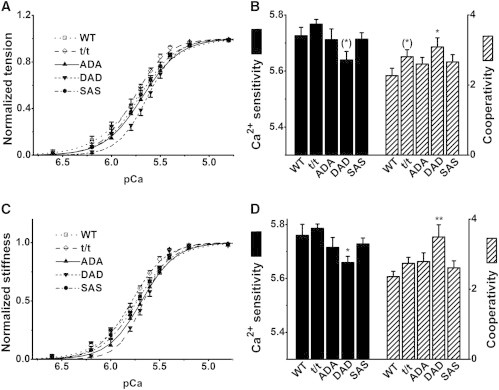
Effect of pCa in the 0Pi solution in five groups of mice. Nomenclatures and conventions used are as in Fig. 6. n = 14 for WT, t/t, ADA, and DAD, and n = 11 for SAS. ∗∗p ≤ 0.01, ∗0.01 < p ≤ 0.05, and (∗)0.05 < p ≤ 0.1 compared to WT.
In the 8Pi solution (Fig. 6 B, left ordinate), pCa50 and nH (Fig. 6 B, right ordinate) were centered at ∼5.56 and ∼6.0, respectively, based on tension, and neither differed significantly between the mutant and WT groups. Almost identical results were obtained based on stiffness: pCa50S was centered at ∼5.56, nHS was centered at ∼6.0, and the values from the five groups were not significantly different (Fig. 6 D).
In the 0Pi solution (Fig. 7, B and D), pCa50 was larger and nH was smaller compared to their values in the 8Pi solution. pCa50 and pCa50S centered at ∼5.68, and nH and nHS centered at ∼2.7. Thus, it can be said that cardiac muscle fibers are more sensitive to Ca2+ in the 0Pi solution than in the 8Pi solution (by 0.12 pCa unit), but cooperativity in the 0Pi solution is about half that in the 8Pi solution. These results are consistent with our recent study on myosin regulatory light chain mutations in mice (24). In addition, values among the five groups varied more extensively in the 0Pi solution, and differed significantly in the 0Pi versus the 8Pi solution. The cooperativity was significantly larger in the DAD group than in the WT group with respect to both tension (p < 0.05) and stiffness (p < 0.01). Ca2+ sensitivity was less in the DAD group than in the WT group with respect to both tension (p = 0.054) and stiffness (p < 0.05).
Cross-bridge kinetics during partial activation
As indicated in Fig. 1, sinusoidal analysis was also performed during the pCa study with 8Pi series solutions. The rate constants 2πb and 2πc are plotted in Fig. 8. At lower levels of activation (pCa ≥ 6.0), the signal-to-noise ratio was too small to resolve the cross-bridge kinetics. Between pCa 5.8 and 5.4, 2πb increased steeply and 2πc decreased steeply. Between pCa 5.4 and 4.5, 2πb decreased slightly, and 2πc remained approximately the same. At all pCa points, the order of 2πb and 2πc values among all mouse groups remained the same as that shown in Fig. 4 A, with larger 2πb in the DAD and SAS groups, and smaller 2πc in the t/t and DAD groups.
Myosin α- and β-isoform expression
Because it has been known that MHC isoforms affect the apparent rate constant 2πc (45) and the shifts in isoform distribution have been observed in some transgenic mice (46), we assessed the MHC isoform contents as described in Materials and Methods. A representative gel is shown in Fig. 9 A, and the analysis of the averaged data from three gels is shown in Fig. 9 B. This analysis demonstrated that WT mouse hearts contained 95.2 ± 0.2% α-MHC (and 4.8% β-MHC), that ADA contained 86 ± 6% α-MHC, and that DAD contained 84 ± 2% α-MHC; the values for the latter two groups were not significantly different from that of WT. In contrast, α-MHC was reduced to 57 ± 4% in t/t mice and to 74 ± 3% in SAS mice, and β-MHC increased significantly to 43 ± 4%, and 26 ± 3%, respectively.
Figure 9.
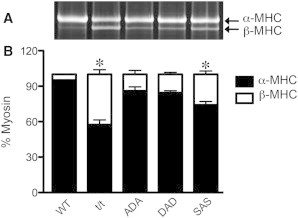
Expression of myosin α- and β-isoforms in each mouse group. (A) A representative experiment. Isoforms detected using specific antibodies after Sypro stained SDS-PAGE analysis. The molecular mass of α-MHC is 223.565 kDa, and that of β-MHC is 222.849 kDa. (B) Fractions of α- and β-MHC, as averaged from three gels (n = 3), similar to that represented in A, for WT and mutant cMyBP-C. Error bars are drawn for each fraction. ∗p ≤ 0.05.
Results excluding female mice
There may be a gender difference. Since we mixed results from 14 male mice and two female mice (Table 1), we constructed plots for the males only (Figs. S2–S7) to examine whether this makes a difference. The figure numbers, panel designations, and plotting conventions are the same as those in the main text. When the data were limited only to males, there were no substantial changes in the results, but the error bars became larger because of the smaller population. The exceptions were the larger active tension (Figs. S2 A) and magnitudes B, C, and H (Fig. S4) in the SAS group, in which the increase in tension and magnitudes C and H became significant (p ≤ 0.05), and B became more significant (p ≤ 0.01), compared to WT.
Discussion
The purpose of this study was to characterize the biophysical functions of cardiac muscle fibers that carry ADA, DAD, SAS, and null (t/t) forms of cMyBP-C, and to thereby advance our understanding of the role of cMyBP-C phosphorylation. Tension and stiffness, together with the apparent rate constants (2πb and 2πc) and the magnitudes (B and C) of two exponential processes were studied. Rate constants 2πb and 2πc are closely associated with the elementary steps of the cross-bridge cycle (25), including the binding of ATP and the release of Pi and ADP. These parameters can be used to characterize contractile properties and cross-bridge functions. pCa studies have been performed to establish the Ca2+ sensitivity and cooperativity.
In this report, two sets of solutions were used. The first is termed 8Pi solution and contains 5 mM MgATP2−, 8 mM Pi, and 200 mM IS, close to the composition in the cytosol of contracting cardiomyocytes, which is reported to be 6–9 mM [Pi] (47), 4–6 mM [MgATP2−] (47), and 215 mM IS (48). The [ATP] and [Pi] data were obtained using the chemical method in working hearts with adequate oxygenation (47). Given that the physiological ionic strength of cardiomyocytes has not been reported, the [IS] is based on resting skeletal muscle fibers (48). The second solution is termed 0Pi solution, and it has been used by many other investigators to characterize pCa-tension relationships (27–30,49,50). This solution does not have any added Pi, [MgATP2-] is 1.6 mM, and IS is 150 mM. Because of the contaminating Pi (mostly via ATP and phosphocreatine) and the liberation of Pi from ATP hydrolysis, the actual [Pi] in the 0Pi solution is in the range of 0.15–0.8 mM (51–53).
The tension values in Fig. 2 (8Pi solution) do not differ from those previously reported in cardiac fibers (54) or from those discussed in our previous work (24). The [Pi] significantly influenced muscle contraction in many aspects. The increased [Pi] caused less active tension and stiffness (Fig. 2 A versus Fig. 2 B), larger rate constants and magnitudes B and C (Fig. 4, A and B, versus Fig. 5, A and B), but less magnitude H (Fig. 4 C versus Fig. 5 C); nH became larger and pCa50 became smaller (Fig. 6, B and D, versus Fig. 7, B and D) regardless of mouse genotype in this study. The differences between the mouse groups in either tension or stiffness under 0Pi and 8Pi solutions were similar (Fig. 2). Most of the significant changes in the cross-bridge kinetics occurred in the 8Pi solution (Fig. 4). The trend was similar in the 0Pi solution (Fig. 5), although this was not a significant difference owing to the smaller magnitudes B and C, which made the rate-constant data more scattered. In contrast, with the pCa-tension study, the genotype difference was more clear in the 0Pi solution (Fig. 7, B and D) than in the 8Pi solution (Fig. 6, B and D). The kinetics of the cross-bridge cycle appear to be modified through the thin filament activation mechanism; therefore, in partial activation (pCa > 5.4), 2πb became slower and 2πc became faster compared to the full activation (pCa ≤ 5.0) (Fig. 8). Rate constant 2πc became significantly larger than 2πb (up to 20 times). These changes are similar to those found in skeletal muscle fibers (55). The ∼75% increase in 2πb and the ∼25% decrease in 2πc in DAD compared to WT (Fig. 4 A) demonstrate the profound effect of DAD on the elementary steps of the cross-bridge cycle.
Electron microscopy studies (56,57) suggest that cMyBP-C is present only in the central 53% of the thick filaments (9 stripes out of the total 17: C-zone), and absent in the remaining 47% (8 stripes out of 17) (57). Therefore, cross-bridges can be considered as a mixture of two kinds—one-half (53%: C-zone) with cMyBP-C, and the other half (47%) without cMyBP-C—arranged in parallel. The role of cMyBP-C in modulating cardiac contractility appears to be localized in the C-zone as shown by in vitro motility assay (58). Our results demonstrate that isometric tension and stiffness are significantly reduced (by ∼48%) in the DAD fibers compared to the WT fibers (Fig. 2). Assuming that the cross-bridges outside of the C zone (∼47%) function normally, the cross-bridges containing cMyBP-C DAD protein (∼53%) must be almost inactive, or their function is extremely diminished to ∼5% (= 53% − 48%). However, it is also true that the cross-bridges that form with the DAD protein must be partially effective, as evidenced by the altered kinetics of the rate constants 2πb and 2πc in the 8Pi solution (Fig. 4 A). Therefore, it is possible that the DAD protein influences the cross-bridges beyond the C-zone.
In mouse cardiac muscles, PKA phosphorylates all three sites (Ser273, Ser282, and Ser302), whereas PKC phosphorylates only two sites (Ser273 and Ser302) (17). The significantly compromised tension-generating capacity and the reduced number of cross-bridges in DAD mice, but not in ADA and SAS observed in this study (Fig. 2), lead us to propose that PKC-mediated phosphorylation of cMyBP-C at Ser273 and Ser302 either serves as a compensatory mechanism that protects cardiomyocytes from overexertion of the already dysfunctional heart, or results in maladaptive contraction that ultimately compromises cardiac function. Our biophysical data from DAD mice are consistent with the cardiac hypertrophy, myocyte disarray, and interstitial fibrosis previously observed (17). A recent study investigated three additional cMyBP-C transgenic lines in mice: DAA, AAD, and SDS, together with AAA, ADA, and t/t (22). It was found that DAA and AAD mice were significantly affected, showing more severe cardiac pathology than t/t mice, with left ventricle dilation, interstitial fibrosis, irregular cardiac rhythm, and sudden cardiac death (22). Also, the sarcomere structure was seriously damaged in DAD fibers (17). When these data and our results are combined, we can conclude that a nonphosphorylatable Ala282 with phosphomimetic Asp273 and/or Asp302 (in AAD, DAA, and DAD) are detrimental to cardiac structure and function. We further conclude that the combination of the phosphorylation pattern is critical in the function of cMyBP-C.
An increase in active tension was previously found after partial (60–70%) extraction of cMyBP-C (59); some investigators have reported accelerated cross-bridge kinetics and increased power output in t/t compared to WT mice (60–63). We are not certain about the reasons for these discrepancies, but several differences in the experimental conditions may have contributed. First, differences in the methods used and the parameters investigated may account for the differences. In previous studies, the skinned fibers were produced by homogenization of the thawed frozen heart (60,61,63), but we generated skinned fibers from fresh hearts. It is also worth noting that the accelerated cross-bridge kinetics reported in previous studies were found to be significant in partial activation (50% or 25% of maximal force) in the absence of added Pi and in the 180 mM IS solution (60–63), whereas our standard solution contained 8 mM Pi and 200 mM IS. In addition, previous studies measured the rate constants of stretch-induced force decay (krel) and delayed force development (kdf) in stretch activation (60,61), and the rate constant of force redevelopment (ktr) through large release-restretch experiments (62,63). We measured the rate constants of delayed tension, 2πb, and of fast tension recovery, 2πc, through sinusoidal analysis under near-isometric conditions.
Compared to the DAD group, the ADA and SAS groups exhibited little change from the WT. In contrast to DAD, 100% phosphomimetic Asp282 within the context of nonphosphorylation at positions 273 and 302 (ADA) appears to have little effect other than a decreased stiffness in the 0Pi solution (Fig. 2 B). Because Ser282 plays an important role in the phosphorylation of Ser273 and Ser302 (6,17), it is reasonable to postulate that in the SAS mutation (with phosphorylation-deficient Ala282), the phosphorylation of Ser273 and Ser302 is significantly reduced. The smaller changes in SAS compared to DAD suggest that when residue 282 cannot be phosphorylated, 100% phosphomimetic Asp273 and Asp302 (DAD) may be detrimental for cardiac function. The SAS actually exhibited somewhat opposite effects to DAD in active tension, and in magnitudes B and H; these changes became significant when the data of one female SAS mouse was excluded (Figs. S2 and S4). This does not mean, however, that these parameters are lower in female SAS mice, because there were not adequate data in females.
Several studies found that a 13–19% increase in β-MHC expression (and a concomitant decrease in α-MHC expression) occurs in t/t mouse hearts (10,46,61). It has also been observed that a 100% shift from α- to β-MHC reduces 2πc by ∼75% without changing the isometric tension (45). Thus, our observations on 2πc may be confounded by two factors: a mutation in cMyBP-C and an MHC isoform shift. For this reason, we studied the MHC isoform content (Fig. 9). We found that our conclusions on DAD and ADA are not significantly affected by the isoform shift, because the β-MHC contents of DAD and ADA were small and did not differ significantly from that of WT (Fig. 9 B). In the case of t/t, there was a significant (43 ± 4%) decrease of the α-MHC content (Fig. 9 B), which could account for much of the observed decrease in 2πc (42% in Fig. 4 A and 33% in Fig. 5 A), because 75% × 43% = 32%. The absence of cMyBP-C causes a problem in cardiac muscles, as demonstrated by disorganized and damaged sarcomere structures in t/t mice (17) and disordered thick filaments with varying myosin heads extending from the backbone of the thick filament, as well as an ∼30% reduction in maximal force after partial (29 ± 3%) extraction of cMyBP-C (64). The reduced 2πc in t/t mouse hearts (Fig. 4 A) is consistent with the diminished left ventricle function based on in vivo measurements (61,65,66), depressed cross-bridge kinetics (67), slower shortening velocity, and reduced power output (68,69). All of these lines of evidence support the hypothesis that the absence of cMyBP-C results in impaired myocardial contractility.
In the case of SAS, there was a significant (26 ± 3%) decrease in α-MHC contents (Fig. 9 B), which predicts a 20% (75% × 26%) decrease in 2πc. Because the actual decrease in 2πc was ∼8% (Figs. 4 A and 5 A), the effect of mutation in SAS may be to increase this rate constant by ∼12% (20% − 8%), and the two antagonizing effects may compensate each other. Although there are no reports on the effects of MHC isoforms on 2πb, it is possible that the isoform content does not affect this parameter, given that little change was observed in 2πb of t/t in two different solutions (Figs. 4 A and 5 A). The increase in 2πb in DAD must be due to this mutant form of cMyBP-C, because there is no significant isoform shift (Fig. 9 B). The involvement of isoform shifting does not modify our conclusions on tension, stiffness, or magnitude parameters, because tension is not sensitive to the shifting (45), and parameters Y∞, B, C, and H are usually scaled with tension.
Conclusion
This study investigated cardiac fiber contractility and cross-bridge kinetics in transgenic mouse models carrying cMyBP-C mutations that affect three different phosphorylation sites in its M-domain. Our investigation revealed that a lack of cMyBP-C diminishes the rate constant of fast tension recovery, 2πc, owing to the isoform shifting of MHC. Phosphomimetic Asp273 and Asp302 with a nonphosphorylatable Ala substituted for residue 282 (DAD) significantly diminishes active tension and altered cross-bridge kinetics by increasing the rate constant of delayed tension (2πb), and decreasing the rate constant of fast tension recovery (2πc). However, neither the single substitution of a nonphosphorylatable residue 282 (SAS), nor the combination of nonphosphorylatable residues at 273 and 302 and the presence of a 100% phosphomimetic Asp282 (ADA), causes significant changes in fiber tension and cross-bridge kinetics. In conclusion, cMyBP-C influences cardiac contractility and cross-bridge kinetics in a manner specific to the phosphorylation sites, and the nonphosphorylatable residue 282 combined with phosphomimetic residues 273 and/or 302 is detrimental to cardiomyocytes.
Acknowledgments
The authors thank Drs. Christine E. Seidman and Jonathan G. Seidman, Harvard Medical School, Boston, MA, for providing the cMyBP-C (t/t) mouse model of dilated cardiomyopathy and Dr. Jeffrey Robbins, Cincinnati Children’s Hospital, Cincinnati, OH, for providing the cMyBP-C WT, ADA, DAD, and SAS mouse models. In addition, we thank Dr. Suresh Govindan at Loyola University, Chicago, for his excellent technical assistance with mouse breeding and genotyping. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the funding organizations.
This work was supported by the National Institutes of Health (grants HL070041 to M.K. and R01HL105826 and K02HL114749 to S.S.), National Scientist Development Grant 0830311N (to S.S.), and the American Heart Association (13GRNT16810043 to M.K. and 11PRE7240022 to D.B.).
Supporting Material
References
- 1.Offer G., Moos C., Starr R. A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization. J. Mol. Biol. 1973;74:653–676. doi: 10.1016/0022-2836(73)90055-7. [DOI] [PubMed] [Google Scholar]
- 2.Winegrad S. Myosin-binding protein C (MyBP-C) in cardiac muscle and contractility. Adv. Exp. Med. Biol. 2003;538:31–40. doi: 10.1007/978-1-4419-9029-7_3. discussion 40–41. [DOI] [PubMed] [Google Scholar]
- 3.Bennett P.M., Fürst D.O., Gautel M. The C-protein (myosin binding protein C) family: regulators of contraction and sarcomere formation? Rev. Physiol. Biochem. Pharmacol. 1999;138:203–234. doi: 10.1007/BFb0119628. [DOI] [PubMed] [Google Scholar]
- 4.Richard P., Charron P., Komajda M., EUROGENE Heart Failure Project Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 5.Mohamed A.S., Dignam J.D., Schlender K.K. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch. Biochem. Biophys. 1998;358:313–319. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 6.Gautel M., Zuffardi O., Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bardswell S.C., Cuello F., Avkiran M. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J. Biol. Chem. 2010;285:5674–5682. doi: 10.1074/jbc.M109.066456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuello F., Bardswell S.C., Avkiran M. Novel role for p90 ribosomal S6 kinase in the regulation of cardiac myofilament phosphorylation. J. Biol. Chem. 2011;286:5300–5310. doi: 10.1074/jbc.M110.202713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuster D.W., Sequeira V., van der Velden J. GSK3β phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication. Circ. Res. 2013;112:633–639. doi: 10.1161/CIRCRESAHA.112.275602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadayappan S., Gulick J., Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Armouche A., Pohlmann L., Carrier L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J. Mol. Cell. Cardiol. 2007;43:223–229. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 12.van Dijk S.J., Dooijes D., van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 13.Decker R.S., Nakamura S., Winegrad S. The dynamic role of cardiac myosin binding protein-C during ischemia. J. Mol. Cell. Cardiol. 2012;52:1145–1154. doi: 10.1016/j.yjmcc.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Yuan C., Guo Y., Murphy A.M. Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts— evidence for novel phosphorylation sites. Proteomics. 2006;6:4176–4186. doi: 10.1002/pmic.200500894. [DOI] [PubMed] [Google Scholar]
- 15.El-Armouche A., Boknik P., Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–680. doi: 10.1161/CIRCULATIONAHA.106.636845. [DOI] [PubMed] [Google Scholar]
- 16.Sadayappan S., de Tombe P.P. Cardiac myosin binding protein-C: redefining its structure and function. Biophys. Rev. 2012;4:93–106. doi: 10.1007/s12551-012-0067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadayappan S., Gulick J., Robbins J. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 2011;109:141–150. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McClellan G., Weisberg A., Winegrad S. cAMP can raise or lower cardiac actomyosin ATPase activity depending on α-adrenergic activity. Am. J. Physiol. 1994;267:H431–H442. doi: 10.1152/ajpheart.1994.267.2.H431. [DOI] [PubMed] [Google Scholar]
- 19.Tong C.W., Stelzer J.E., Moss R.L. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ. Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagayama T., Takimoto E., Kass D.A. Control of in vivo left ventricular [correction] contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 2007;116:2399–2408. doi: 10.1161/CIRCULATIONAHA.107.706523. (Erratum in Circulation. 117:e11.) [DOI] [PubMed] [Google Scholar]
- 21.Sadayappan S., Osinska H., Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta M.K., Gulick J., Robbins J. Functional dissection of myosin binding protein C phosphorylation. J. Mol. Cell. Cardiol. 2013;64:39–50. doi: 10.1016/j.yjmcc.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McConnell B.K., Jones K.A., Seidman J.G. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J. Clin. Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. (Erratum in J. Clin. Invest. 104:1771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L., Muthu P., Kawai M. Diversity and similarity of motor function and cross-bridge kinetics in papillary muscles of transgenic mice carrying myosin regulatory light chain mutations D166V and R58Q. J. Mol. Cell. Cardiol. 2013;62:153–163. doi: 10.1016/j.yjmcc.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L., Muthu P., Kawai M. Characterizations of myosin essential light chain’s N-terminal truncation mutant Δ43 in transgenic mouse papillary muscles by using tension transients in response to sinusoidal length alterations. J. Muscle Res. Cell Motil. 2013;34:93–105. doi: 10.1007/s10974-013-9337-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubb M.R., Senderowicz A.M., Korn E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 27.Kerrick W.G., Kazmierczak K., Szczesna-Cordary D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2009;23:855–865. doi: 10.1096/fj.08-118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mettikolla P., Calander N., Borejdo J. Cross-bridge kinetics in myofibrils containing familial hypertrophic cardiomyopathy R58Q mutation in the regulatory light chain of myosin. J. Theor. Biol. 2011;284:71–81. doi: 10.1016/j.jtbi.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muthu P., Mettikolla P., Borejdo J. Single molecule kinetics in the familial hypertrophic cardiomyopathy D166V mutant mouse heart. J. Mol. Cell. Cardiol. 2010;48:989–998. doi: 10.1016/j.yjmcc.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borejdo J., Szczesna-Cordary D., Calander N. Familial hypertrophic cardiomyopathy can be characterized by a specific pattern of orientation fluctuations of actin molecules. Biochemistry. 2010;49:5269–5277. doi: 10.1021/bi1006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kazmierczak K., Xu Y., Szczesna-Cordary D. The role of the N-terminus of the myosin essential light chain in cardiac muscle contraction. J. Mol. Biol. 2009;387:706–725. doi: 10.1016/j.jmb.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reference deleted in proof.
- 33.Kawai M., Brandt P., Orentlicher M. Dependence of energy transduction in intact skeletal muscles on the time in tension. Biophys. J. 1977;18:161–172. doi: 10.1016/S0006-3495(77)85605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawai M., Brandt P.W. Effectof MgATP on stiffness measured at two frequencies in Ca2+-activated muscle fibers. Proc. Natl. Acad. Sci. USA. 1977;74:4073–4075. doi: 10.1073/pnas.74.9.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawai M. Head rotation or dissociation? A study of exponential rate processes in chemically skinned rabbit muscle fibers when MgATP concentration is changed. Biophys. J. 1978;22:97–103. doi: 10.1016/S0006-3495(78)85473-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X., Tobacman L.S., Kawai M. Temperature-dependence of isometric tension and cross-bridge kinetics of cardiac muscle fibers reconstituted with a tropomyosin internal deletion mutant. Biophys. J. 2006;91:4230–4240. doi: 10.1529/biophysj.106.084608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawai M., Brandt P.W. Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J. Muscle Res. Cell Motil. 1980;1:279–303. doi: 10.1007/BF00711932. [DOI] [PubMed] [Google Scholar]
- 38.Kawai M. The role of orthophosphate in crossbridge kinetics in chemically skinned rabbit psoas fibres as detected with sinusoidal and step length alterations. J. Muscle Res. Cell Motil. 1986;7:421–434. doi: 10.1007/BF01753585. [DOI] [PubMed] [Google Scholar]
- 39.Fink R.H., Stephenson D.G., Williams D.A. Potassium and ionic strength effects on the isometric force of skinned twitch muscle fibres of the rat and toad. J. Physiol. 1986;370:317–337. doi: 10.1113/jphysiol.1986.sp015937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kentish J.C. The inhibitory effects of monovalent ions on force development in detergent-skinned ventricular muscle from guinea-pig. J. Physiol. 1984;352:353–374. doi: 10.1113/jphysiol.1984.sp015296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai F., Weis A., Kawai M. Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin’s HCM mutations. Biophys. J. 2011;100:1014–1023. doi: 10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossmanith G.H., Hoh J.F., Ludowyke R.I. Mechanism of action of endothelin in rat cardiac muscle: cross-bridge kinetics and myosin light chain phosphorylation. J. Physiol. 1997;505:217–227. doi: 10.1111/j.1469-7793.1997.217bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawai M., Zhao Y. Cross-bridge scheme and force per cross-bridge state in skinned rabbit psoas muscle fibers. Biophys. J. 1993;65:638–651. doi: 10.1016/S0006-3495(93)81109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawai M., Halvorson H.R. Force transients and minimum cross-bridge models in muscular contraction. J. Muscle Res. Cell Motil. 2007;28:371–395. doi: 10.1007/s10974-008-9131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Tanner B.C., Palmer B.M. Cardiac myosin isoforms exhibit differential rates of MgADP release and MgATP binding detected by myocardial viscoelasticity. J. Mol. Cell. Cardiol. 2013;54:1–8. doi: 10.1016/j.yjmcc.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer B.M., Sadayappan S., Maughan D.W. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophys. J. 2011;101:1661–1669. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Opie L.H., Mansford K.R., Owen P. Effects of increased heart work on glycolysis and adenine nucleotides in the perfused heart of normal and diabetic rats. Biochem. J. 1971;124:475–490. doi: 10.1042/bj1240475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Godt R.E., Maughan D.W. On the composition of the cytosol of relaxed skeletal muscle of the frog. Am. J. Physiol. 1988;254:C591–C604. doi: 10.1152/ajpcell.1988.254.5.C591. [DOI] [PubMed] [Google Scholar]
- 49.Song W., Dyer E., Marston S.B. Molecular mechanism of the E99K mutation in cardiac actin (ACTC Gene) that causes apical hypertrophy in man and mouse. J. Biol. Chem. 2011;286:27582–27593. doi: 10.1074/jbc.M111.252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolska B.M., Keller R.S., Solaro R.J. Correlation between myofilament response to Ca2+ and altered dynamics of contraction and relaxation in transgenic cardiac cells that express β-tropomyosin. Circ. Res. 1999;84:745–751. doi: 10.1161/01.res.84.7.745. [DOI] [PubMed] [Google Scholar]
- 51.Tesi C., Colomo F., Poggesi C. The effect of inorganic phosphate on force generation in single myofibrils from rabbit skeletal muscle. Biophys. J. 2000;78:3081–3092. doi: 10.1016/S0006-3495(00)76845-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai M., Halvorson H.R. Two step mechanism of phosphate release and the mechanism of force generation in chemically skinned fibers of rabbit psoas muscle. Biophys. J. 1991;59:329–342. doi: 10.1016/S0006-3495(91)82227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dantzig J.A., Goldman Y.E., Homsher E. Reversal of the cross-bridge force-generating transition by photogeneration of phosphate in rabbit psoas muscle fibres. J. Physiol. 1992;451:247–278. doi: 10.1113/jphysiol.1992.sp019163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stelzer J.E., Patel J.R., Moss R.L. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J. Gen. Physiol. 2006;128:261–272. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawai M., Cox R.N., Brandt P.W. Effect of Ca ion concentration on cross-bridge kinetics in rabbit psoas fibers. Evidence for the presence of two Ca-activated states of thin filament. Biophys. J. 1981;35:375–384. doi: 10.1016/S0006-3495(81)84796-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Craig R., Offer G. The location of C-protein in rabbit skeletal muscle. Proc. R. Soc. Lond. B Biol. Sci. 1976;192:451–461. doi: 10.1098/rspb.1976.0023. [DOI] [PubMed] [Google Scholar]
- 57.Luther P.K., Bennett P.M., Moss R.L. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J. Mol. Biol. 2008;384:60–72. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Previs M.J., Beck Previs S., Warshaw D.M. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hofmann P.A., Hartzell H.C., Moss R.L. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J. Gen. Physiol. 1991;97:1141–1163. doi: 10.1085/jgp.97.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stelzer J.E., Dunning S.B., Moss R.L. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ. Res. 2006;98:1212–1218. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- 61.Desjardins C.L., Chen Y., Stelzer J.E. Cardiac myosin binding protein C insufficiency leads to early onset of mechanical dysfunction. Circ Cardiovasc Imaging. 2012;5:127–136. doi: 10.1161/CIRCIMAGING.111.965772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korte F.S., McDonald K.S., Moss R.L. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 63.Stelzer J.E., Fitzsimons D.P., Moss R.L. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys. J. 2006;90:4119–4127. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kulikovskaya I., McClellan G., Winegrad S. Effect of extraction of myosin binding protein C on contractility of rat heart. Am. J. Physiol. Heart Circ. Physiol. 2003;285:H857–H865. doi: 10.1152/ajpheart.00841.2002. [DOI] [PubMed] [Google Scholar]
- 65.Harris S.P., Bartley C.R., Moss R.L. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 66.Palmer B.M., Georgakopoulos D., Kass D.A. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circ. Res. 2004;94:1249–1255. doi: 10.1161/01.RES.0000126898.95550.31. [DOI] [PubMed] [Google Scholar]
- 67.Rundell V.L., Manaves V., de Tombe P.P. Impact of β-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H896–H903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- 68.Herron T.J., Korte F.S., McDonald K.S. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H1217–H1222. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- 69.Korte F.S., Herron T.J., McDonald K.S. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H801–H812. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.