

Abstract
Total internal reflection fluorescence microscopy (TIRFM) is becoming an increasingly common methodology to narrow the illumination excitation thickness to study cellular process such as exocytosis, endocytosis, and membrane dynamics. It is also frequently used as a method to improve signal/noise in other techniques such as in vitro single-molecule imaging, stochastic optical reconstruction microscopy/photoactivated localization microscopy imaging, and fluorescence resonance energy transfer imaging. The unique illumination geometry of TIRFM also enables a distinct method to create an excitation field for selectively exciting fluorophores that are aligned either parallel or perpendicular to the optical axis. This selectivity has been used to study orientation of cell membranes and cellular proteins. Unfortunately, the coherent nature of laser light, the typical excitation source in TIRFM, often creates spatial interference fringes across the illuminated area. These fringes are particularly problematic when imaging large cellular areas or when accurate quantification is necessary. Methods have been developed to minimize these fringes by modulating the TIRFM field during a frame capture period; however, these approaches eliminate the possibility to simultaneously excite with a specific polarization. A new, to our knowledge, technique is presented, which compensates for spatial fringes while simultaneously permitting rapid image acquisition of both parallel and perpendicular excitation directions in ∼25 ms. In addition, a back reflection detection scheme was developed that enables quick and accurate alignment of the excitation laser. The detector also facilitates focus drift compensation, a common problem in TIRFM due to the narrow excitation depth, particularly when imaging over long time courses or when using a perfusion flow chamber. The capabilities of this instrument were demonstrated by imaging membrane orientation using DiO on live cells and on lipid bilayers that were supported on a glass slide (supported lipid bilayer). The use of the approach to biological problems was illustrated by examining the temporal and spatial dynamics of exocytic vesicles.
Introduction
A variety of optical techniques are now being used to break the diffraction limit of optical resolution. One of the first to be widely applied is the use of total internal reflection (TIR) to generate an excitatory evanescent field that decays exponentially with a space constant roughly an order of magnitude smaller than the wavelength of light (1). In addition to improving axial illumination resolution, the thin field also enhances image contrast by minimizing out-of-focus excitation. The application of TIR to fluorescence microscopy has been widely used to study biological events that occur at or near the cell membrane, such as exocytosis, endocytosis, cell surface receptor kinetics, and membrane dynamics (for application examples see (2)). In addition, total internal reflection fluorescence microscopy (TIRFM) is frequently used for in vitro biochemical studies such as imaging actin and microtubule assembly, or single-molecules studies on molecular motor proteins (3–5). TIR is also a common illumination method for other super-resolution techniques such as photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM) (6,7).
Beyond improving image signal/noise, when coupled with polarized light, TIRFM can be used to study three-dimensional molecular orientation and dynamics. Polarized TIRFM (pTIRFM) has been applied to a variety of biological systems including plasma membrane structure (8), exocytosis (9–12), vesicle fusion on a supported lipid bilayer (13), membrane lipid order (14–16), and molecular motor protein movement (17). In pTIRFM two electric field polarizations are typically used, one parallel to the glass surface, in the plane spanned by the - axis, and another perpendicular (normal) to the glass surface, along the optical axis (Fig. 1 A). The field parallel to the glass surface is produced by TIR of light with its electric field perpendicular to the plane of incidence (the plane containing the incident Poynting vector and the glass surface normal) and is commonly identified as polarized light. The field perpendicular to the glass surface is produced by TIR of light with its electric field in the plane of incidence and is identified as polarized light. In reality this electric field is only fully perpendicular () when the incident ray is at the TIR critical angle (1). At sub- and supercritical angles the field also has a component parallel to the surface () (Fig. 1 A, left). At increasing supercritical incident angles the TIR illumination depth decreases (1), thus there is a tradeoff between minimizing the illumination depth and producing a field with minimal component.
Figure 1.
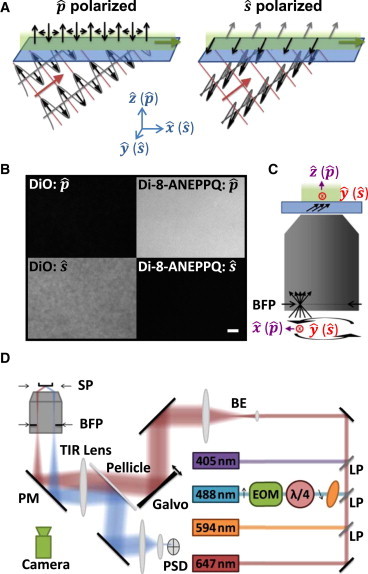
(A) Illustration of TIR of polarized light. The direction of the TIR electric field produced by incident light cycles between the optical axis () and the surface plane () (left figure). The TIR field from incident light is polarized along the surface plane () (right figure). (B) Supported lipid bilayer labeled with either DiO (left panels) or di-8-ANEPPQ (right panels) and imaged with either incident excitation light (top panels) or incident light (bottom panels). 1 μm scale bar. (C) Cartoon illustration of azimuthal scanning illumination. (D) Schematic of TIRFM illuminator. EOM, electrooptic modulator; λ/4, quarter-wave plate; LP, longpass filter; BE, beam expander; PM, polychroic mirror; BFP, back focal plane; SP, sample plane; PSD, position-sensitive detector.
Assuming a fluorophore in an excitation field is rotationally confined (relative to the time scale of its fluorescence lifetime), its orientation can be probed by determining the direction of the emission polarization, or by comparing the emission intensity when excited with orthogonal excitation polarizations. The second scenario can be used in TIR to probe orientation by comparing the emission intensity when excited with incident versus incident light. As an example, DiO is a dye that binds to lipid membranes such that the direction of its excitation dipole is preferentially aligned along the plane of the membrane (detailed dipole orientation study of similar DiI given in (18)). In a membrane labeled with DiO polarized light preferentially excites the molecule compared to incident light (Fig. 1 B, left). An alternative dye, di-8-ANEPPQ, associates to a lipid bilayer with its excitation dipole primarily perpendicular to the surface (19,20). In this case polarized light more favorably excites the fluorophore (Fig. 1 B, right).
The use of TIRFM is often limited by spatial nonuniformities in the illumination field. These variations are introduced by constructive and destructive interference of coherent laser light when reflected and refracted from various optical components in the beam path, including back reflections, nonideal optics, and undesirable scattering objects (including dust). This optical interference is particularly problematic when a uniform field is necessary for quantitative studies. To overcome this challenge several labs have developed objective-based TIRFM systems that modulate the position of the excitation light over the period of a single image acquisition frame. This process effectively averages out nonuniformities. These methods include using a spinning wedge (21), acousto-optic deflector (AOD) (22), piezo mirror (23), and galvo mirrors (24). In these systems the focused excitation light is scanned in a circular motion around the periphery of the objective’s back focal plane (BFP) (Fig. 1 C), producing a roughly planar beam of light at the sample plane that rotates azimuthally around the optical axis. When the period of this azimuthal scan is shorter than the image acquisition time the irregularities in the excitation field are averaged out.
When linearly polarized laser light of a fixed orientation enters an illumination compensation system as described previously, the excitation field polarization continuously changes between and directions throughout each azimuthal scan. For example, assuming the incoming beam is polarized along the direction entering the objective BFP and that the beam is currently focused at a position along the x axis (Fig. 1 C, encircled red X), the resulting polarization in the sample plane is () (Fig. 1 A, right panel). The opposite is true when the beam is positioned along the y axis in the objective’s BFP (with the same incoming polarization), in which case () polarized light is produced at the sample. As the beam moves in a circle around the objective’s BFP, averaging out interference irregularities in the sample plane, the electric field polarization at the sample plane cycles between and . Therefore, although the spatial field is more uniform, this illumination method can no longer be used for polarization based experiments.
Here, we present a TIRFM illumination scheme in which the polarization state in the sample plane was maintained, whereas interference artifacts were simultaneously eliminated. In addition, the novel, to our knowledge, TIRFM illuminator contained a back reflection position sensitive detector (PSD) for facilitating quick alignment of the azimuthal beam, real-time observation of the TIR state, and focus drift compensation. To rotate the polarized state an electro-optic modulator (EOM) in combination with a quarter-wave plate was used. In addition, polarization-dependent phase shifts introduced by optical components were corrected with a Berek compensator. The azimuthal illumination and back reflection alignment methods are discussed first, followed by the polarization rotation technique, and the focus drift compensation scheme. The illuminator was used to image membrane orientation in live cells, revealing topological phenomena such as cellular protrusions in which there was tight membrane association with the glass surface. In addition, vesicle exocytosis was rapidly imaged to observe membrane orientation dynamics throughout fusion.
Materials and Methods
Optical configuration
405 nm (120 mW laser diode, Omicron Laserage, Rodgau-Dudenhofen, Germany), 488 nm (100 mW laser diode, Omicron), 594 nm (100 mW diode-pumped solid-state laser, Cobolt AB, Solna, Sweden), and 647 nm (140 mW diode laser, Omicron) lasers were combined with longpass filters (LaserMUX series, Semrock, Rochester, NY) and positioned to match beam diameters following a beam expander. Dielectric mirrors (BB1-E02, Thorlabs Newton, NJ) were used throughout the system to position the beams. Before being combined with the other laser beams, the polarization of the 488 nm beam was modulated with an EOM (#350-105 modulator with BK clamp option to minimize resonance and 302RM amplifier, Conoptics, Danbury, CT), followed by a zero-order quarter-wave plate (Thorlabs), and a Berek variable wave plate (Newport Corp., Irvine, CA) to compensate for polarization ellipticity introduced by optics downstream. Combined beams were expanded to ∼13 mm with a telescope (achromatic lenses from Thorlabs) before entering a 2-axis flexure galvo-based scan head (OFH-15 with QuantumDrive-4000 servo-amplifiers, Nutfield Technology, Hudson, NH) mounted to the rear port of an inverted microscope (IX81-ZDC2, Olympus, Shinjuku, Tokyo, Japan). A 170 mm focal length achromatic doublet lens (30 mm diameter, Ross Optical Industries, El Paso, TX) was mounted downstream from the scan-head and both were mounted to a one-dimensional translation stage (Thorlabs) to enable movement of the scan head and optics relative to the objective position. A custom quadruple band polychroic with flatness sufficient for TIR illumination (Chroma Technology, Bellows Falls, VT) was mounted in the fluorescence filter cube turret and a 100 X UAPON 1.49 NA TIRFM objective (Olympus) was used for establishing TIR and imaging emission. Emitted light was split based on wavelength or polarization with a TripleSplit (Cairn Research, Faversham, UK) and was collected with an ORCA-Flash 4.0 complementary metal oxide semiconductor (CMOS) camera (Hamamatsu Photonics, Hamamatsu, Japan). Back reflected light used for beam alignment and focus compensation was sampled with a pellicle beamsplitter (BP208, Thorlabs) mounted between the galvos and the TIR focusing lens and imaged onto a PSD (quadrant photodiode PDQ80A with TQD001 amplifier, Thorlabs). A bandpass filter holder was incorporated into the detection optics to enable selection of a laser line for tracking the glass surface focus, while simultaneously rejecting fluorescence excitation laser lines. To control environment temperature the entire microscope was enclosed and the air was heated with an Air-Therm Heater (World Precision Instruments, Sarasota, FL).
System control
Galvanometers, EOM, laser shutters, and camera exposure were controlled with digital and analog outputs from a multifunction data acquisition board (PCIe-6323, National Instruments, Austin, TX) with custom software written in LabView (National Instruments). TIR critical angle was determined by finding the maximum derivative of the reflected intensity, whereas the focused beam in the BFP was iteratively scanned along the x and y axes. The amplitude of the EOM sawtooth function was adjusted to ensure full polarization rotation during each azimuthal scan, and the phase was tuned to align the polarization with the beam position. To compensate for polarization ellipticity induced by the optics the Berek variable wave plate was adjusted to maximize extinction, as tested with a linear analyzer at the objective position, over all input polarization angles. To enable rapid and image acquisition the beam was azimuthally scanned at 200 Hz, the EOM at 400 Hz, and the camera exposure (collected from the center quadrant of the CMOS chip) was fixed at 12.5 ms with an external synchronous readout trigger of 12.5 ms. In sync with the camera shutter and galvo scanning, the EOM phase was shifted 90° every 12.5 ms, enabling successive imaging with and excitations. To eliminate rolling shutter artifacts and prevent mixed polarization excitation, the sample was exposed with laser light for 5 ms (a single 360° azimuthal beam scan at 200 Hz) during the 12.5 ms exposure period. Images were acquired with MetaMorph (Molecular Devices, LLC, Sunnyvale, CA) and analyzed in either MetaMorph or ImageJ (25). For focus compensation the position signal from one axis of the PSD was detected with a lock-in amplifier (#5210, Signal Recovery, Oak Ridge, TN), using one of the galvo sinusoidal position signals as the reference and an integration time of 30 ms. The lock-in amplifier signal, in combination with proportional-integral-derivative feedback in LabView, was used to compensate for focus shifts by adjusting the position of the objective with the microscope’s internal focus drive motor.
Dye-labeled supported lipid bilayer preparation
To form the lipid bilayer 500 μg of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, Alabaster, Alabama) in chloroform was added to a glass test tube, and a film was produced by evaporating the chloroform in the presence of argon while slowly rotating the tube. The film was then incubated for 5 min with 500 μl of HEPES buffer (25 mM HEPES, 100 mM KCl, pH 7.4 with KOH), vortexed for 10 s to form multilamellar vesicles, and sonicated (#G112SP1, Laboratory Supply,Hicksville, NY) until solution was clear (∼3 min) to form unilaminar vesicles. 0.1 ml of vesicles was mixed with 0.9 ml of HEPES buffer and then added to a Skyes-Moore Chamber (Belco Glass, Vineland, NJ) containing a cover glass (cleaned with 8 g/L of NaOH in MeOH followed by ddH20). Before imaging, the HEPES buffer was exchanged 10 times to remove free vesicles and either 1 μl of DiO (Vybrant DiO at 1 mM in DMF, Life Technologies, Grand Island, NY) or 1 μl of Di-8-ANEPPQ (1 mM in EtOH, Biotium, Hayward, CA) was added to the solution above the bilayer. Bright and dim phase separated regions were observed for both dyes, which we attribute to different concentrations of dye. Both regions had similar emission profiles when comparing versus excitation. Presented images are from bright regions.
DiO plasma membrane labeling
HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (#11965-092, Life Technologies) with 10% fetal bovine serum at 37°C in the presence of 5% C02. To label plasma membrane with DiO ∼106 cells were suspended (with trypsin) in 1 ml of DMEM. 5 μl of 1 mM Vybrant DiO was mixed with cells and incubated for 8 min at 37°C. Labeled cells were washed three times by pelleting at 400 × g for 5 min, removing DMEM and resuspending with warm DMEM between each spin. After the final spin cells were plated on a glass bottom culture dish (MakTek, Ashland, MA). Before imaging, ∼4 h later, the media was replaced with cell imaging media consisting of 10 mM HEPES (#H-3375, Sigma-Aldrich, St. Louis, MO), 9.7 g/L of Hanks BBS (#H-1387, Sigma-Aldrich), and NaOH to bring pH to 7.4. 1% fetal bovine serum was added following sterile filtration.
VSVG-mEGFP vesicle labeling
For vesicle endocytosis experiments HeLa cells were transfected with temperature-sensitive VSVG-mEGFP expression vector (kind gift of J. Lippincott-Schwartz lab). 1 μg of plasmid was incubated with 100 μl of Opti-MEM (Life Technologies) for 5 min, and then 5 μl of Fugene 6 (Promega, Madison, WI) was added to the solution. 5 min later solution was added to plated HeLa cells (∼30% confluency) in a 35 mm glass bottom culture dish (MakTek). Cells were grown for 12 h in 5% C02 at 37°C and then were transferred to 40°C for an additional 24 h. Before imaging the media was replaced with cell imaging media and the cells were incubated at 34°C for ∼2 h, allowing the VSVG-mEGFP to escape the endoplasmic reticulum and travel in vesicles to the plasma membrane.
Results and Discussion
Improving sample excitation field uniformity
To correct for spatial illumination nonuniformities in objective-based TIRFM we use a system similar to those described previously (26). Instead of focusing the fluorescence excitation light to a single point in the radial periphery of the objective’s BFP, as is commonly done in objective-based TIRFM, the position of the focused light is scanned in a circle around the periphery of the objective’s BFP (Fig. 1 C). To ensure that TIR is maintained at the glass-water interface, the focused beam must remain at a radial distance from the optical center that will produce an angle of incidence of the illumination beam on the interface that is equal or larger than that required for TIR. The TIR angle will depend on the refractive indices of the glass, 1.52 for BK7, and of the aqueous media: 1.33 for water, typically around 1.35–1.4 for cell cytoplasm (27–29). Ideally, the distance from the optical center should remain constant throughout scanning because the evanescent field penetration depth varies with distance.
We considered several beam scanning options, including galvanometer scan head, piezo mirror, voice-coil actuated tip-tilt mirror, and AODs. Each of these technologies has advantages and disadvantages in terms of resolution, speed, aperture size, deflection angle, and cost. AOD-based deflectors are suitable for high-speed applications, though typically at the sacrifice of smaller aperture as well as the potential for deflection-dependent intensity modulation. In the mirror-based beam steering devices, piezo systems typically have the highest resolution, but generally are also significantly more expensive. At similar aperture size and speed, galvanometers usually have a larger deflection angle compared to piezo-based tip-tilt deflection systems. From these considerations we decided to use a two-dimensional galvanometer scan head. Of note, even though the two mirrors are mounted close to each other in the scan head, the deflection point is not identical for the orthogonal directions, resulting in small lateral translation of the illumination field at the sample plane. This compromise has a potential benefit of creating a more uniform field throughout the scanning process. In choosing a deflection system we also considered mirror flatness, with thicker substrates typically being flatter. Unfortunately, the benefit of a thicker mirror comes at cost of slower scanning speed, therefore a compromise was necessary. Our galvanometers have mirrors that are ∼2.5 mm thick. We ensured the beam remained collimated with a shear plate interferometer following the galvanometer as well as after other mirrors in the system, including the dielectric coated polychroic.
The scan head was fixed to a linear translational stage that was mounted directly to the rear illumination port of an inverted microscope (Fig. 1 D). The translation stage also contains the lens that focuses collimated light deflected by the galvanometers onto the BFP of the objective. This TIR focusing lens is positioned one focal length from the objective and the galvanometer is positioned roughly one focal length from the focusing lens. Therefore, an angular deflection of collimated beam at the galvanometer results in a radial translation of the focus beam at the position of the objective’s BFP (26). The size of illuminated field at the sample plane is related to the demagnification of the beam by the combination of the TIR lens and the objective lens, and the system aperture determined by the galvo mirrors. Securely mounting the illumination system directly to the microscope ensures stability of the beam from short (∼ ms) to long durations (∼ h), making illumination more consistent throughout experiments and decreasing the frequency of needing to realign the optics. With this configuration TIR of excitation light was verified (Fig. S1 in the Supporting Material). In addition, while imaging acridine orange (5 μg/ml in ddH20, Life Technologies) nonspecifically attached to the surface of a glass slide with azimuthal scanning illumination, we observed a significant visible improvement in spatial field uniformity as compared to fixed position illumination (Fig. 2, A and B).
Figure 2.

Acridine orange on the surface of a glass slide. Imaged emission intensity ( + ) (A) without and (B) with azimuthal scanning. emission ratio (C) without and (D) with azimuthal scanning. A vertical and horizontal line scan is shown next to each image. Scale bar 5 μm.
A significant difficulty with azimuthal scanning TIR illumination is ensuring the focused beam in the objective’s BFP traverses a circular path at a constant radial distance from the optical axis, such that the incident excitation beam maintains a constant zenith incident angle at the glass-water interface. This circular path can be adjusted by modifying the amplitude and offset of a sinusoidal wave driving each galvo mirror, as well as modifying the relative phase between each sine wave (typically ∼90 degrees to trace a circular shape). One method to tune these parameters is to observe, either via the oculars or directly above the sample, fluorescent beads freely suspended in solution while rotating the beam near the critical TIR angle. When the parameters are tuned correctly the free beads will have similar excitation at all azimuthal angles (26). TIR is then achieved by increasing (in a scaled manner) the sine wave amplitudes driving the galvos.
Unfortunately, this tuning process is time-consuming and the alignment procedure likely has to be repeated whenever an optical component, such as a dichroic imaging mirror, is changed in the system. This method also requires the alignment be performed in a sample chamber that is different than the actual experimental chamber, which may introduce unanticipated errors from the glass-solution interface not having the same properties or not being mounted in exactly the same orientation. In addition, if alignment needs to be adjusted or verified during an experiment the sample chamber has to be completely removed.
Due to these challenges we incorporated a back reflection intensity detector into the TIR illuminator (Fig. 3 A). Between the galvanometers and TIR focusing lens a pellicle beamsplitter was installed to sample the light that transverses backward through the microscope following TIR at the glass-sample surface. The pellicle beamsplitter reflects roughly 8% of the light (exact amount is wavelength, polarization, and incident angle dependent), and was selected due to its thin profile, that minimizes aberrations when introduced in a converging or diverging optical beam. Using lenses following the beamsplitter the reflected light was imaged onto a PSD in an optical plane conjugate to the sample plane. An incident beam above the critical angle for TIR results in a significantly higher signal on the back reflection diode detector. Thus, the intensity of the signal at the diode provides an indication of the TIR state of the beam at the sample.
Figure 3.
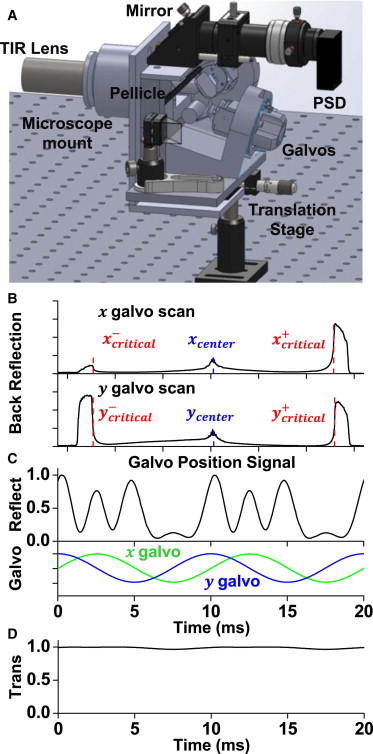
(A) Three-dimensional diagram of galvanometer/sensor assembly. (B) The back reflected laser intensity was monitored while each galvanometer was independently scanned with a linear ramp waveform, resulting in the focused beam translating across the diameter of the BFP of the objective. TIR (high intensity regions) occurred near the radial periphery of the objective's BFP. The critical angle (red dash line) and alignment center (blue dash line) were found from analysis of the intensity derivative. (C) Back reflected light intensity during azimuthal illumination scanning. The intensity variability was caused by changes in the light’s incident angle at the Pellicle beamsplitter throughout the azimuthal scan. (D) Illumination intensity at objective throughout scanning. To see this figure in color, go online.
With a sample mounted on the microscope each galvanometer mirror was scanned back and forth to find the positive () and negative () input signals that resulted in the system being at the critical angle (Fig. 3 B). From these endpoints center offsets ( and ) were determined as well as the necessary amplitude in each direction to reach the critical angle. The latter component is used to scale the relative amplitude between galvanometers, a necessity to compensate for slight deflection angle differences between the two galvanometers. The process of calibration, which is iterated to find the best center, is fully automated and requires only a few seconds to complete. This technique also enables, based on an average of the entire illumination area, calibration of regions where the refractive index is not well characterized, such as in the presence of adherent cells. For TIR experiments a deflection amplitude beyond the critical angle is used (Fig. 3 B), but smaller amplitudes can be used for other illumination profiles such as inclined planar illumination (22,30,31), as is sometimes used in photoactivated localization microscopy/stochastic optical reconstruction microscopy (32), or to conduct experiments in which the system is quickly cycled between TIR and epi-illumination (33,34).
The detected back reflected light intensity not only depends on the occurrence of TIR, but also the angle at which the beam reflects off of the pellicle beamsplitter. The pellicle itself is a thin film interference device, and therefore the amount of reflected versus transmitted light is sensitive to incident angle, wavelength, and polarization. This was illustrated by comparing the back reflected intensity when the galvanometers were scanned along the axis of the BFP of the objective versus along the axis. A scan along the axis (left ↔ right across sample) resulted in similar reflection intensities at the two critical angle positions () (Fig. 3 B). This similarity is because the beam was scanned in the sagittal plane, which has symmetric incident angles at ± y positions. This was not the case for a scan along the axis (Fig. 3 B), where the change of deflection angle was in the meridional plane of beamsplitter. The thin film interference experienced by the reflected light at the two critical positions () was different because of different incident angles on the pellicle itself (Fig. 3 B).
When the focused beam position was scanned in a circle on the BFP of the objective, the back reflected light sampled a variety of angles on the pellicle. Therefore, the detected reflected light intensity varied significantly throughout a single circular scan of the beam (Fig. 3 C). The angular reflection dependence of the beamsplitter also has the potential to affect the intensity of transmitted light that initially passes from the galvanometers to the sample plane. We measured the intensity of the transmitted light at the objective position during circular scanning and found the intensity only varied by a few percent (Fig. 3 D). This experiment was conducted with a fixed linear polarization beam state.
This azimuthal scanning illuminator reduces interference fringes and significantly eases alignment to find the beam center and the critical angle. The versatile system also enables rapid switching between TIR and planar illumination conditions, and makes it possible to correct for chromatic variations in evanescent field depth.
Modulating polarization
It is important to note that when using the azimuthal scanning illuminator the polarization of light at the sample plane, assuming fixed input polarization at the BFP, changes between and polarization throughout a single frame acquisition. This varying direction confounds polarization based TIRFM microscopy experiments. To maintain the same polarization throughout the acquisition of a single frame, the incoming linear polarization state must simultaneously be rotated in sync with the movement of the beam around the BFP. Several techniques to rotate polarization have been used in other microscopy applications including a liquid crystal polarization rotator (35), AOMs (36), motorized half-wave plate (37), and EOM (38). AOM and EOM based rotation provide faster modulation compared to a liquid crystal or motorized based rotator. AOMs have the advantage over EOMs in that the polarization state never has to be quickly flipped to cycle back to an initial polarization state; however, they are challenging to align and maintain in alignment. We used an EOM because of the potential for high-speed rotation and straightforward alignment. In our setup the EOM is configured as a fast variable wave plate, with its extraordinary axis offset 45° relative to the entering linearly polarized laser light. This arrangement facilitates a tunable phase shift between orthogonal components of the light, creating an elliptically polarized beam. The elliptical beam is then resolved into a rotated linearly polarized state with a quarter-wave plate (extraordinary axis aligned with the original laser polarization). Varying the relative phase between the orthogonal axes of the initial laser light changes the orientation of the linearly polarized beam following the quarter-wave plate. Of note, ellipticity introduced by other optics in the beam line can disrupt this linear state, which we corrected with a Berek compensator (see the Supporting Material Text, Fig. S2). A sawtooth waveform was used to drive the EOM in sync with the galvanometer during laser scanning. Because the electric field is symmetric the polarization only needs to be rotated 180° with the sawtooth waveform before it is flipped back to the initial polarization. By changing the relative phase between the sawtooth function and the galvanometer sinusoidal signals it is possible to switch between a sample plane polarization that is primarily or .
By fixing the position of the BFP focused beam above the critical angle along the x axis, (no azimuthal scanning), we imaged DiO in a supported lipid bilayer with both () polarization (Fig. 4 A, left) and primarily () polarization (Fig. 4 A, right). Emission from the polarized excitation was roughly three times greater than the emission from excitation, comparable to previous results with a similar dye (8). When the TIRFM excitation beam was azimuthally scanned with fixed polarization (mixture of and ) (Fig. 4 B, left) the emission was still roughly 3 times greater than azimuthally scanned () excitation (Fig. 4 B, right). Similar to acridine orange imaging (Fig. 2), with azimuthal scanning the spatial intensity nonuniformities were also reduced. A qualitative comparison of emission between excitation with and does not reveal a dramatic difference in the spatial intensity patterns (Fig. 4 A). A computed ratio image of reveals minor spatial variations with fixed beam excitation (Fig. 2 C), which disappear with azimuthal scanning (Fig. 2 D). For many polarization based experiments these minor variations would not dramatically affect direct comparisons between emission from and excitation. However, polarization based studies often also rely on information about total emission from and excitation (10) that would be affected by spatial nonuniformities.
Figure 4.
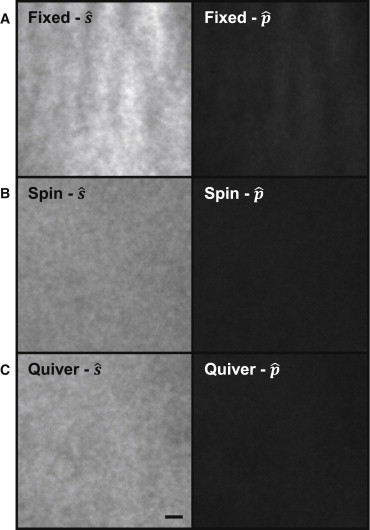
(A) DiO in a supported lipid bilayer was imaged with constant polarization (left) and constant polarization (right), whereas the excitation beam was fixed at (no azimuthal scanning). (B) DiO imaged with azimuthal scanning with polarization (left) and polarization (right). (C) DiO imaged with polarization (left) and polarization (right) during galvanometer quivering around . Scale bar 1 μm.
Fixed polarized light, in either the or direction, is useful for conducting molecular orientation anisotropy measurements in which fluorophore emission is compared along directions that are parallel and perpendicular to the excitation polarization. To fix the polarization direction while still correcting for spatial illumination nonuniformities, we tested small angle quivering of the focused beam in the BFP. The beam was quivered ∼10° around a fixed position () in the BFP while emission from DiO collected. During this small angle quivering the focused beam in the BFP was maintained equidistant from the optical center, ensuring evanescent field depth remained the same. Again, the emission from the excitation (Fig. 4 C, left) was roughly three times greater than emission from excitation (Fig. 4 C, right). With quivering the nonuniformities in the illumination field were also greatly diminished, although still providing fixed orientation polarization in the direction.
The pellicle beamsplitter used for detecting back reflections is not only sensitive to incident angle (see above), but also to incident polarization. incident light transmission intensity through the pellicle is ∼10% higher than incident light (Thorlabs spec). Thus, the intensity at the sample plane is expected to fluctuate throughout azimuthal scanning when the polarization is maintained. Conversely, the intensity of reflected light off of the pellicle is also polarization dependent, with incident light reflecting significantly incident light. These characteristics were verified by measuring the back reflection intensity (Fig. 5 A) during simultaneous azimuthal scanning with polarization rotation and by measuring the transmitted intensity at the position of the objective (Fig. 5 B). The intensity fluctuation at the sample plane is potentially a more significant problem because it affects fluorophore excitation. However, as long as each frame acquired by the camera has the same illumination characteristics, ensured by setting the galvanometer and EOM cycle rate to an integer number of azimuthal scans during each frame acquisition, the total intensity at the sample will remain constant. In situations where constant sample illumination intensity is necessary throughout azimuthal scanning the intensity of the laser can be modulated in phase with the polarization rotation. We verified this compensation by modulating the output power from our 488 nm diode laser with a sinusoidal signal synced with the galvanometers (Fig. 5 C).
Figure 5.
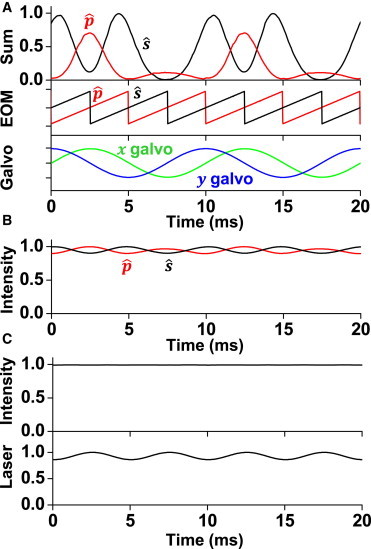
(A) Intensity at back reflection detector during simultaneous azimuthal scanning and polarization rotation ( maintained at sample plane). (B) Transmitted laser intensity at objective position during simultaneous galvanometer scanning and polarization rotation. (C) Diode laser power was modulated to maintain constant intensity at the objective position, whereas the polarization was rotated and galvanometers were scanned. To see this figure in color, go online.
This polarization based illuminator rotates the input excitation polarization as the TIR beam is scanned. This technique reduces spatial nonuniformities while simultaneously enabling control of sample plane polarization. With this method fluorophore emission from sequential and excitation can be compared to determine molecular orientation. In addition, with small angle galvonometer quivering we demonstrated the ability to create roughly fixed excitation polarization with simultaneous correction of spatial nonuniformities. With fixed excitation polarization molecular orientation and dynamics can be determined from emission anisotropy measurements.
Focus drift compensation
Temperature fluctuations in microscope components, relaxing of the mechanical focus drive, settling of immersion oil, and fluid forces in a flow chamber are just a few examples of sources that can cause the focus plane to drift in a fluorescence microscope. Drift is particularly apparent in TIRFM because the illumination plane, defined by the glass-solution surface, and the imaging plane, defined by the objective focus, is decoupled with respect to one another. Thus, even a small change in the glass surface positions (<0.5 μm) makes the sample seem out of focus. Microscope manufacturers, as well as third party vendors, have a variety of ways to compensate for this focal plane drift. Typically, these systems consist of either an image contrast detector or a glass surface position detector, sometimes achieved with light being reflected off of the glass surface. There are also systems that compensate by carefully measuring the position of the objective relative to the stage. We used the detector that was monitoring the back reflection of the TIR excitation to also compensate for focus drift. Because the PSD is in a conjugate plane to the sample, any changes in the axial height of the glass slide relative to the beam results in a lateral translation of the focus position on the PSD. For example, assuming the TIR illumination orientation is fixed (no azimuthal scanning), an axial shift of the glass slide will result in a translation of the position of the reflected beam on the glass surface. This movement results in a translation of the reflected beam on the PSD, creating a change in position signal on the detector. This change in signal can be used as a feedback reference signal for maintaining the proper relative height between the glass slide and objective focus. In practice either the sample surface height or the objective height can be adjusted to maintain the signal at the center of the PSD.
Focus compensation during azimuthal scanning is more complicated because as the sample goes out of focus the back reflection detector position signal rotates over the quadrants of the PSD diode. This rotation results in a sinusoidal position signal in phase with the scanning that cannot be linearly mapped to the distance between the slide and the objective. To overcome this challenge we detected the back reflected azimuthal scanning beam signal from the PSD with a lock-in amplifier. The X signal from the PSD was filtered with the lock-in using the position signal from the X galvanometer as a reference, resulting in a DC signal that quantified the relative distance. A plot of the sensitivity of the detected signal as a function of the height of the objective indicated a maximum sensitivity of ∼1.4 mV/nm near the center focus (Fig. 6 A). The noise of the detector (S.D.) near the center focus was ∼2 mV, equating to a measured focus accuracy of ∼2 nm.
Figure 6.
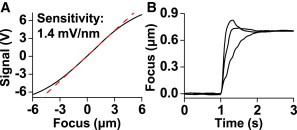
(A) Lock-in amplifier signal versus objective axial position. A focus sensitivity of 1.4 mV/nm was measured near the center focus. (B) Example of objective position response following step function change in focus set point. Focus position versus time illustrated for three feedback conditions; strong (fast with some overshoot, less stable), moderate (little overshoot), and weak (slow, more stable) response feedback parameters. To see this figure in color, go online.
The lock-in amplifier signal was used to create a feedback reference for maintaining focus. We used proportional-integral-derivative feedback to maintain the same signal at the z-height detector, while adjusting the focus by modulating the microscope’s motorized objective drive. This system allows for specifying feedback parameters based on the specific application. For experiments that might have a slow focal shift, such as thermal drift, we used feedback parameters that had relatively slow response, ensuring overshoot was minimized and stability was maintained (Fig. 6 B). To maintain focus during rapid focus drift, such as when using a flow chamber, it is often necessary to have faster focus compensation even at the expense of some overshoot. The microscope’s internal focus motor was sufficient for our experiments, but our feedback runs on top of the microscope’s focus drive feedback, thus is ultimately limited by its response rate. For faster response an independent piezo-based objective focus positioner or stage actuator independent of the focus drive could be used instead.
A challenge with our focus compensation system is that a laser needs to be continuously reflecting from the glass surface to generate a feedback signal, which may introduce unnecessary fluorophore bleaching in time-lapse experiments. We circumvent this problem by using an independent surface monitoring laser, typically at 647 nm, and distinguish it from the excitation lasers by incorporating a bandpass filter in the back detector optics. The system was arranged so the filter can easily be changed in case we desire to use a different laser for focus compensation.
With a single sensor our back reflection detector scheme enables TIR alignment and focus compensation, with simultaneous correction of spatial nonuniformities. This focus detection technique has an advantage over typical commercial systems because an additional glass flat is not inserted into the imaging path between the objective and fluorescence polychroic. A flat dichroic is in the microscope’s infinity space has no effect on imaging; however, it can introduce some distortion, such as astigmatism, into the TIR excitation beam because it is focused in the infinity space. Distortion of this form can be decreased by adding another slanted glass surface rotated 90° to the first, but the correction is not perfect. Our focus compensation system also enables user tuning of feedback parameters, allowing unique settings to satisfy the dynamics of each specific application. The combination of these features in this focal compensation system offers considerable advantages for use in quantitative pTIRFM imaging.
Imaging structure and dynamics of membrane orientation in live cells
We imaged two components of live cells, the structure of the plasma membrane and the dynamics of vesicle exocytosis, to verify the capability of the instrument to precisely control excitation polarization while simultaneously correcting for spatial intensity nonuniformities in the TIR field. To study the structure of the plasma membrane HeLa cells were labeled with DiO (see Materials and Methods), and the fluorescence emission was compared between excitation with and polarizations. The dipole of DiO preferentially aligns parallel to the cell membrane, thus regions of the plasma membrane along the glass surface were preferentially excited by polarization (Fig. 7 A, dark regions of ratio image), whereas regions where the membrane curves away from the surface (example: near cell edges) were preferentially excited by polarized light (bright regions).
Figure 7.

(A) ratio of DiO label HeLa cell. Image consists of an average of 40 frames (20 with excitation and 20 with excitation) collected over a total time of 500 ms. Regions outside of the cell have low signal and thus have significantly higher noise (including CMOS camera patterned noise). Scale bar 5 μm. (B) Time course of subregion of image in A (white box) illustrates dynamics of filopodia/lamellipodium. Scale bar 2 μm.
A complication with this technique was movement of DiO-labeled cellular structures, including plasma membrane and endocytosed vesicles, between the period it took to collect an image with each excitation polarization. Such movement created a false indication of membrane orientation within the cell. To overcome this challenge we rapidly imaged both polarizations (5 ms exposure, switching polarization every 12.5 ms). This was achieved by tightly synchronizing the galvomometer position, EOM polarization, camera exposure, and laser excitation (see Materials and Methods). Shutter duration of 12.5 ms was used for each acquisition, but during this period the sample was only excited by the laser for 5 ms. The 5 ms strobe ensured the entire frame was collected from same time period, overcoming the challenge of a digital camera with a rolling shutter. In addition, the 5 ms pulse ensured the excitation beam completed one azimuthal scan (200 Hz scan rate), enabling correction of spatial nonuniformities while simultaneously creating the same illumination profile for each frame. Using a short camera exposure with rapid polarization switching had the disadvantage that multiple images had to be averaged if improved signal/noise was desired. Averaging multiple short exposure images increased read noise over a single long exposure image, but it was a useful compromise to improve signal/noise while minimizing cell movement artifacts.
Using this averaging technique a computed image of a HeLa cell (Fig. 7 A) was created, which illustrates regions in which the cell was parallel to surface (dark regions), regions in which the membrane curved away from the glass (bright regions), and regions in which the cell had variable topology (moderate brightness regions). The image reveals prominent lamellipodia (dark regions) along the cell edge, likely a result of tight adherence to the glass surface. These lamellipodia were starkly different to the cell body that had significantly more varied topology. In addition, as indicated by bright edge lines between the lamellipodia and the cell body, this polarization technique highlights lamellipodia extending over the vertical edge of the cell body. The tubular structure of extending filopodia were also revealed, with bright edges indicative of the membrane curving away from the glass surface surrounding a dark core with membrane parallel to the surface. This technique provides strong contrast for visualizing the filopodia and for observing their movement, as well as their time-dependent expansion and contraction (Fig. 7 B).
We imaged secretory vesicle exocytosis at the plasma membrane by tagging the exterior of vesicles with the GFP-labeled transmembrane protein vesicular stomatitis virus G (VSVG-mEGFP). A temperature sensitive folding mutant of VSVG (ts045) was used, which traps the protein in the endoplasmic reticulum at temperatures above 40°C. At 32°C VSVG moves to the Golgi (39) before being transported to the plasma membrane in vesicles. Fusion to the plasma membrane typically occurs a few hours after the temperature is reduced (40). Exocytosis is identifiable as a vesicle that first approaches the plasma membrane, followed by fast (∼1 s) radial diffusion of the GFP fluorescence (41). Using rapid polarization switching a combined image was collected every 25 ms to minimize cell movement between images and multiple images were averaged to improve signal/noise. Fig. 8 A illustrates narrow (<250 nm), diffraction-limited stable tubular structures, which have a preference toward excitation (dark region - ratio: ∼1.5) relative to VSVG-GFP in the surrounding cell membrane ( ratio: ∼1.5). The >1 ratio for the plasma membrane indicates the GFP dipole was preferential aligned perpendicular to the membrane, which is consistent with the known orientation of the GFP excitation dipole (42) in relation to its amino terminal attachment to VSVG. We attribute the lower ratio to the tubular edges whose surface is perpendicular to the glass surface. Of note, around the tubular structures the was slightly greater than the surrounding plasma membrane, potentially a result of the vesicle flattening of the local plasma membrane.
Figure 8.
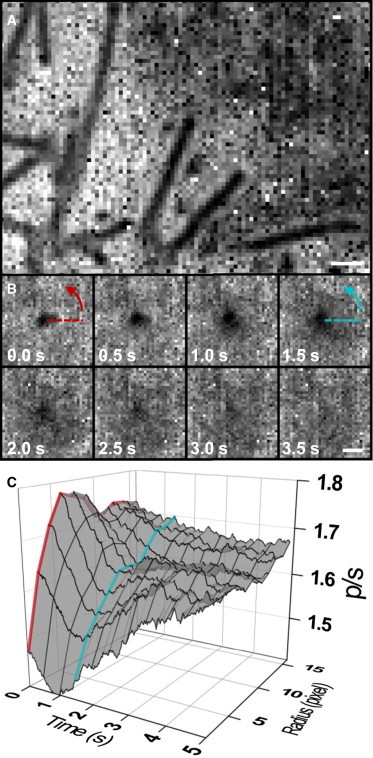
(A) Membrane orientation in stationary tubular structures (VSVG-GFP labeled) was visualized by averaging 1000 images collected over a 25 s period. The dark regions indicate the GFP dipole preferentially orients perpendicular to the membrane. (B) ratio time course during secretory vesicle exocytosis illustrates membrane dynamics. Each ratio image is an average of 80 images (1 s). (C) Radial profile from B of intensity with respect to time. The profile is calculated from the average value within concentric circles around the center of the fusing vesicle, and displays a transition from round to flattened structure. Red line and blue line correspond to specific profiles found at times t = 0.0 s and t = 1.5 s. Scale bars 0.5 μm.
Occasionally vesicles, both round and elongated in shape, were observed to fuse with the membrane (Fig. 8 B). Like the long tubular structures, the dark signal was believed to be produced by the edges of the vesicle being perpendicular to the glass surface. A radial profile (Fig. 8 C), calculated from an average of values within concentric circles from the center, illustrates the dark vesicle surrounded by a slightly brighter region relative to the rest of the plasma membrane. During the fusion event the vesicle’s ratio decreased and then increased, eventually approaching the plasma membrane level. The ratio of the locally surrounding brighter region dropped as fusion occurred (Fig. 8 C), though it never approached the low signal in the center of fusion. This drop may signify a transition of the membrane from being more parallel (flat) to the glass to being more perpendicular (vertical), potentially caused by the expansion of an already fused, hemispherical-like, structure. Similar to a previous report (10), complete return of the membrane to prefused topology was typically much slower, often lasting >10 s.
The orientation sensitivity of these GFP-labeled vesicles, as measured by the signal from bright regions (perpendicular to the glass) relative to dark regions (parallel to glass), was significantly lower than the sensitivity from DiO label cells, making it more challenging to ascertain orientation due to noise and requiring additional averaging. The orientation of the GFP excitation dipole relative to VSVG has not been carefully characterized, but based on these results it is likely the dipole is not strongly aligned directly perpendicular to the membrane. In addition, flexibility in the peptide linker between GFP and VSVG and the presence of a single membrane anchor, as opposed to two anchors in DiO, may both result in decreased sensitivity. However, GFP labeling does provide distinct advantages over DiO because the fluorophore can be targeted to specific cellular components. Furthermore, some of the drawbacks of synthetic dyes, such as aggregation as a result of poor solubility and membrane disruption due to exogenous molecule presence, are avoided. Future sensitivity improvements may be achieved by optimizing both the linker and the fluorescent protein such that the excitation dipole is oriented distinctly parallel or perpendicular to the membrane.
The spatial field corrected polarization based TIRFM illuminator presented here provides a number of advantages. First, it automates beam alignment and provides focus stabilization. It also offers the added benefit of enabling measurements of anisotropy and excitation by selective polarization with a spatially even excitation field. In addition to the work presented here on cell membrane structure and quantitative exocytosis membrane dynamics, this instrument can also be used to quantitatively study three-dimensional orientation in a wide variety of biological systems such as endocytosis, cytoskeleton structure, virus assembly, and membrane protein orientation.
Acknowledgments
The authors thank S. Kirov and Y. Li for engineering and design support, and thank M. Bleck for assistance with producing supported lipid bilayers and GFP-labeled vesicles.
This work was supported by NSF CBET-1126312, NIH R01AI089844 and NIH P50GM103297 to S.M.S.
Footnotes
A. Mattheyses’ current address is: Department of Cell Biology, Emory University School of Medicine, Atlanta, Georgia
Supporting Material
References
- 1.Axelrod D., Burghardt T.P., Thompson N.L. Total internal reflection fluorescence. Annu. Rev. Biophys. Bioeng. 1984;13:247–268. doi: 10.1146/annurev.bb.13.060184.001335. [DOI] [PubMed] [Google Scholar]
- 2.Axelrod D. Chapter 7: Total internal reflection fluorescence microscopy. Methods Cell Biol. 2008;89:169–221. doi: 10.1016/S0091-679X(08)00607-9. [DOI] [PubMed] [Google Scholar]
- 3.Amann K.J., Pollard T.D. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. USA. 2001;98:15009–15013. doi: 10.1073/pnas.211556398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner M.K., Charlebois B.D., Odde D.J. Rapid microtubule self-assembly kinetics. Cell. 2011;146:582–592. doi: 10.1016/j.cell.2011.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yildiz A., Tomishige M., Selvin P.R. Kinesin walks hand-over-hand. Science. 2004;303:676–678. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]
- 6.Betzig E., Patterson G.H., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 7.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sund S.E., Swanson J.A., Axelrod D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys. J. 1999;77:2266–2283. doi: 10.1016/S0006-3495(99)77066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zenisek D., Steyer J.A., Almers W. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron. 2002;35:1085–1097. doi: 10.1016/s0896-6273(02)00896-6. [DOI] [PubMed] [Google Scholar]
- 10.Anantharam A., Onoa B., Axelrod D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J. Cell Biol. 2010;188:415–428. doi: 10.1083/jcb.200908010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anantharam A., Axelrod D., Holz R.W. Polarized TIRFM reveals changes in plasma membrane topology before and during granule fusion. Cell. Mol. Neurobiol. 2010;30:1343–1349. doi: 10.1007/s10571-010-9590-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anantharam A., Bittner M.A., Holz R.W. A new role for the dynamin GTPase in the regulation of fusion pore expansion. Mol. Biol. Cell. 2011;22:1907–1918. doi: 10.1091/mbc.E11-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiessling V., Domanska M.K., Tamm L.K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys. J. 2010;99:4047–4055. doi: 10.1016/j.bpj.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson N.L., McConnell H.M., Burhardt T.P. Order in supported phospholipid monolayers detected by the dichroism of fluorescence excited with polarized evanescent illumination. Biophys. J. 1984;46:739–747. doi: 10.1016/S0006-3495(84)84072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oreopoulos J., Yip C.M. Probing membrane order and topography in supported lipid bilayers by combined polarized total internal reflection fluorescence-atomic force microscopy. Biophys. J. 2009;96:1970–1984. doi: 10.1016/j.bpj.2008.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oreopoulos J., Epand R.F., Yip C.M. Peptide-induced domain formation in supported lipid bilayers: direct evidence by combined atomic force and polarized total internal reflection fluorescence microscopy. Biophys. J. 2010;98:815–823. doi: 10.1016/j.bpj.2009.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beausang J.F., Shroder D.Y., Goldman Y.E. Tilting and wobble of myosin V by high-speed single-molecule polarized fluorescence microscopy. Biophys. J. 2013;104:1263–1273. doi: 10.1016/j.bpj.2013.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Axelrod D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 1979;26:557–573. doi: 10.1016/S0006-3495(79)85271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gasecka A., Han T.J., Brasselet S. Quantitative imaging of molecular order in lipid membranes using two-photon fluorescence polarimetry. Biophys. J. 2009;97:2854–2862. doi: 10.1016/j.bpj.2009.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kress A., Ferrand P., Brasselet S. Probing orientational behavior of MHC class I protein and lipid probes in cell membranes by fluorescence polarization-resolved imaging. Biophys. J. 2011;101:468–476. doi: 10.1016/j.bpj.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattheyses A.L., Shaw K., Axelrod D. Effective elimination of laser interference fringing in fluorescence microscopy by spinning azimuthal incidence angle. Microsc. Res. Tech. 2006;69:642–647. doi: 10.1002/jemt.20334. [DOI] [PubMed] [Google Scholar]
- 22.van ’t Hoff M., de Sars V., Oheim M. A programmable light engine for quantitative single molecule TIRF and HILO imaging. Opt. Express. 2008;16:18495–18504. doi: 10.1364/oe.16.018495. [DOI] [PubMed] [Google Scholar]
- 23.Fiolka R., Belyaev Y., Stemmer A. Even illumination in total internal reflection fluorescence microscopy using laser light. Microsc. Res. Tech. 2008;71:45–50. doi: 10.1002/jemt.20527. [DOI] [PubMed] [Google Scholar]
- 24.Mattheyses A.L., Simon S.M., Rappoport J.Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 2010;123:3621–3628. doi: 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson D.S., Jaiswal J.K., Simon S.M. Total internal reflection fluorescence (TIRF) microscopy illuminator for improved imaging of cell surface events. Curr. Protoc. Cytom. 2012;61:12.29.1–12.29.19. doi: 10.1002/0471142956.cy1229s61. [DOI] [PubMed] [Google Scholar]
- 27.Bereiter-Hahn J., Fox C.H., Thorell B. Quantitative reflection contrast microscopy of living cells. J. Cell Biol. 1979;82:767–779. doi: 10.1083/jcb.82.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curl C.L., Bellair C.J., Delbridge L.M. Refractive index measurement in viable cells using quantitative phase-amplitude microscopy and confocal microscopy. Cytometry A. 2005;65:88–92. doi: 10.1002/cyto.a.20134. [DOI] [PubMed] [Google Scholar]
- 29.Liang X.J., Liu A.Q., Yap P.H. Determining refractive index of single living cell using an integrated microchip. Sens. Actuators A Phys. 2007;133:349–354. [Google Scholar]
- 30.Tokunaga M., Imamoto N., Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- 31.Konopka C.A., Bednarek S.Y. Variable-angle epifluorescence microscopy: a new way to look at protein dynamics in the plant cell cortex. Plant J. 2008;53:186–196. doi: 10.1111/j.1365-313X.2007.03306.x. [DOI] [PubMed] [Google Scholar]
- 32.Vaughan J.C., Jia S., Zhuang X. Ultrabright photoactivatable fluorophores created by reductive caging. Nat. Methods. 2012;9:1181–1184. doi: 10.1038/nmeth.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merrifield C.J., Feldman M.E., Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- 34.Mattheyses A.L., Atkinson C.E., Simon S.M. Imaging single endocytic events reveals diversity in clathrin, dynamin and vesicle dynamics. Traffic. 2011;12:1394–1406. doi: 10.1111/j.1600-0854.2011.01235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smalyukh I.I. Confocal microscopy of director structures in strongly confined and composite systems. Mol. Cryst. Liq. Cryst. 2007;477:23–41. [Google Scholar]
- 36.La Porta A., Wang M.D. Optical torque wrench: angular trapping, rotation, and torque detection of quartz microparticles. Phys. Rev. Lett. 2004;92:190801-1–190801-4. doi: 10.1103/PhysRevLett.92.190801. [DOI] [PubMed] [Google Scholar]
- 37.Oroszi L., Galajda P., Ormos P. Direct measurement of torque in an optical trap and its application to double-strand DNA. Phys. Rev. Lett. 2006;97:058301-1–058301-4. doi: 10.1103/PhysRevLett.97.058301. [DOI] [PubMed] [Google Scholar]
- 38.Gutiérrez-Medina B., Andreasson J.O., Block S.M. An optical apparatus for rotation and trapping. Methods Enzymol. 2010;475:377–404. doi: 10.1016/S0076-6879(10)75015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Presley J.F., Cole N.B., Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 40.Hirschberg K., Miller C.M., Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of golgi to plasma membrane transport intermediates in living cells. J. Cell Biol. 1998;143:1485–1503. doi: 10.1083/jcb.143.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmoranzer J., Goulian M., Simon S.M. Imaging constitutive exocytosis with total internal reflection fluorescence microscopy. J. Cell Biol. 2000;149:23–32. doi: 10.1083/jcb.149.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosell F.I., Boxer S.G. Polarized absorption spectra of green fluorescent protein single crystals: transition dipole moment directions. Biochemistry. 2003;42:177–183. doi: 10.1021/bi0266535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.