

Abstract
Cyanovirin-N (CVN), a cyanobacterial lectin, exemplifies a class of antiviral agents that inhibit HIV by binding to the highly glycosylated envelope protein gp120. Here, we investigate the energetics of glycan recognition using a computationally inexpensive flexible docking approach, backbone perturbation docking (BP-Dock). We benchmarked our method using two mutants of CVN: P51G-m4-CVN, which binds dimannose with high affinity through domain B, and CVN(mutDB), in which binding to domain B has been abolished through mutation of five polar residues to small nonpolar side chains. We investigated the energetic contribution of these polar residues along with the additional position 53 by docking dimannose to single-point CVN mutant models. Analysis of the docking simulations indicated that the E41A/G and T57A mutations led to a significant decrease in binding energy scores due to rearrangements of the hydrogen-bond network that reverberated throughout the binding cavity. N42A decreased the binding score to a level comparable to that of CVN(mutDB) by affecting the integrity of the local protein structure. In contrast, N53S resulted in a high binding energy score, similar to P51G-m4-CVN. Experimental characterization of the five mutants by NMR spectroscopy confirmed the binding affinity pattern predicted by BP-Dock. Despite their mostly conserved fold and stability, E41A, E41G, and T57A displayed dissociation constants in the millimolar range. N53S showed a binding constant in the low micromolar range, similar to that observed for P51G-m4-CVN. No binding was observed for N42A. Our results show that BP-Dock is a useful tool for rapidly screening the relative binding affinity pattern of in silico-designed mutants compared with wild-type, supporting its use to design novel mutants with enhanced binding properties.
Introduction
Cyanovirin-N (CVN), a 101 aa lectin isolated from Nostoc ellipsosporum, exemplifies a novel class of therapeutic agents that target the surface glycans of HIV and other enveloped viruses (1–7). Its potent activity against HIV is mediated by high-affinity binding to the mannose-rich glycans that decorate the surface of gp120, and requires multivalent interactions through two binding domains within the protein (8–12). CVN is unique among lectins in recognizing a glycan unit, Manα(1,2)Manα, with high affinity and specificity: glycan array analysis reveals that only mannose-rich structures containing the specific glycosidic linkage α-Man-(1,2)-α-Man are recognized (13,14). For these reasons, the protein is emerging as a convenient model system for investigating glycan-protein interactions and testing computational approaches to glycan recognition.
The structure of CVN reveals a novel (to our knowledge) β-sheet fold that comprises two quasi-symmetric domains, defined as A (residues 1–38/90–101) and B (residues 39–89), connected on each side by a short 310 helix. Despite a backbone root mean-square standard deviation (RMSD) of 0.76 Å, the two domains are ∼25% identical in sequence, resulting in slightly different glycan selectivity and affinity. Experimental and computational simulation data show a preference for linear trimannose in domain A, and a preference for dimannose (Manα(1,2)Manα) in domain B (1–7,9,15–21). Domain B is generally considered the high-affinity binding site: the reported binding constants are in good agreement and show low-micromolar affinity for Manα(1,2)Manα (8–12,15,16,22,23). Structural data for CVN and mutants containing an intact domain B in the apo and dimannose-bound forms (8,9,13–15,18,19,23), complemented by binding studies using oligomannose derivatives (24), contributed to mapping the key interactions in the glycan recognition event on both the protein and the glycan. The origins of the unique specificity and high affinity of WT CVN for dimannose are poorly understood (25). In particular, the energetic contribution of backbone versus side-chain interactions, and the role of water-mediated hydrogen bonds in determining binding affinity are still under debate.
Here, we analyzed glycan binding by CVN and its mutants by applying a flexible docking approach called backbone perturbation docking (BP-Dock), which is based on perturbation response scanning (PRS), a method that combines elastic network models with linear response theory (26–29). This approach accurately predicted binding affinity patterns of the PDZ domain of PICK1 with various PDZ motifs (29). BP-Dock enabled us to integrate backbone and side-chain conformational changes of the protein, as well as the flexibility of the glycan ligand, in the docking analysis, which is crucial for accurate prediction of binding affinity (29). Compared with computationally expensive traditional molecular-dynamics (MD) calculations performed on CVN (20,30–32), this method allows for rapid in silico screening of a series of mutants.
For simplicity, we limited our analysis to the high-affinity site, domain B. We benchmarked our method by comparing two well-characterized mutants of CVN (Fig. 1): one that comprises an intact domain B (Fig. 1 B) and one in which binding to domain B has been abolished (Fig. 1 C). The computed binding affinities are consistent with high-affinity binding for one, and with no binding for the other. Extending our docking analysis to single mutants of P51G-m4-CVN, we observed that some mutations led to less favorable binding-energy scores, whereas others did not. Based on these predictions, we experimentally characterized five mutants and obtained binding affinities in agreement with the predictions.
Figure 1.
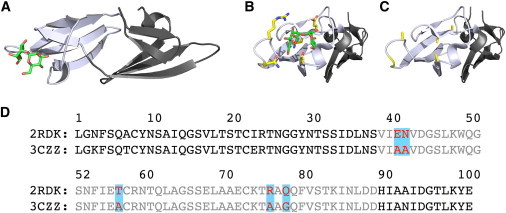
(A) Crystal structure of P51G-m4-CVN (PDB ID: 2RDK) with dimannose bound. (B) Domain B of P51G-m4-CVN, with residues focused on in this study highlighted in yellow (blue, nitrogen atoms; red, oxygen atoms). (C) Domain B of CVN(mutDB) (PDB ID: 3CZZ), with residues that differ from those of P51G-m4-CVN highlighted in yellow. Domain B of CVN(mutDB) does not bind dimannose. (D) The sequences of the two mutants are shown aligned with domain A (black) and domain B (gray). The differences between P51G-m4-CVN and CVN(mutDB) in domain B are highlighted. To see this figure in color, go online.
Our work demonstrates that rapid docking can be an efficient tool for predicting binding-affinity patterns of various mutants in glycan recognition. However, the method relies on accurate prediction of the initial unbound mutant structure. With such tools, it should be possible to predict mutations that change affinity and specificity for given glycan targets.
Materials and Methods
Benchmark
We analyzed bound mutant structures of P51G-m4-CVN (8,9) and CVN(mutDB) (16) in this study. The crystal structures for P51G-m4-CVN (PDB code: 2RDK) and CVN(mutDB) (PDB code: 3CZZ) were retrieved from the Protein Data Bank (http://www.rcsb.org/pdb) (33). These structures were used as templates for binding affinities to dimannose sugar in our flexible docking simulations.
Modeling of single point mutant structures
We first checked the possible impact of single point mutations on the local structure, or the overall stability of the protein. By analyzing the hydrogen-bond network of each individual site within the binding pocket, we found that the side chains of some residues, such as Asn-42 and Thr-57, made critical hydrogen bonds with the backbone or side chain of other positions of CVN (Table S1 in the Supporting Material). Thus, mutating these positions to Ala could alter the stability or lead to local structural change. To capture those effects, we introduced the point mutations into the x-ray structure via Swiss PDB Viewer (34), and performed short (3 ns) replica-exchange MD (REMD) simulations to obtain well-equilibrated mutant models. The initial point mutant structure was seeded into the REMD (35) simulation, and the temperature of the replicas ranged from 270 K to 450 K. Replica spacing was set such that the swap likelihood between replicas approached 0.45. The AMBER force field (ff99SB) (36) with a generalized Born implicit solvent (37) model and a solvent-accessible penalty term of 5 cal/mol Å was used. An α-carbon covariance matrix was calculated from a 1 ns window of the trajectory, and the convergence was checked by the observed high correlation between the slowest fluctuation profiles of the two successive windows. After clustering the snapshots of the lowest replica, we used the most dominant structure (i.e., the lowest free-energy state) as the predicted mutant model structure.
PRS
When a ligand (i.e., dimannose sugar) approaches a receptor (CVN mutants), it exerts force on the binding site, inducing conformational changes. In this work, as a first-order approximation, we mimicked this by applying small random forces on the protein and then computing the displacement of each residue through PRS (26,38). As a matter of fact, the most intriguing aspect of the BP-Dock approach is that it generates a wide range of binding-induced conformations realized in nature by exerting random unit forces on each residue in the sequence without the presence of ligand. This enabled us to integrate the conformational changes of the receptor (CVN mutants) in the docking analysis, which in turn increased the accuracy of the binding predictions. Thus, ensembles for P51G-m4-CVN, CVN(mutDB), and the individual domain B mutant models were obtained by PRS. PRS is a coarse-grained approach that couples elastic network models with linear response theory (26,28,39). Previously, we have shown that PRS can 1), capture conformational changes upon binding (26); 2), identify key residues that mediate long-range communication and find allosteric pathways (28); and 3), discriminate disease-associated mutants from neutral ones (40). We compared the dynamics of PRS, which is based on a coarse-grained approach, with those obtained from all-atom MD simulations, and these two estimates showed high correlations (28,40). However, PRS requires four orders of magnitude less CPU time than MD simulations. Furthermore, although convergence is a bottleneck for MD simulations of larger proteins, it is not an issue in the case of PRS. Therefore, PRS is a fast and efficient approach to incorporate backbone flexibility (27–29), which most of the conventional docking protocols cannot do because it is a computationally expensive and difficult procedure.
In the model, the protein structure is viewed as a 3D elastic network in which the Cα atom acts as the nodes and identical springs connect the interacting α-carbons. We applied a random unit force, F, in a random direction to the α-carbon atom of each residue one at a time (i.e., perturbation of a single residue in the chain with a random Brownian kick) sequentially along the chain. The perturbation cascaded through the residue interaction network of the protein and caused other amino acid positions to respond. By using the PRS method, we computed this fluctuation response as a profile, in both direction and magnitude. Thus, the response vector, ΔR, gave the deviation of each residue from its mean position in the x, y, and z directions upon perturbation. This procedure indeed mimicked the natural process of protein binding interactions in a cell as a first approximation, since an approaching ligand applies forces on the receptor protein, inducing conformational change. To minimize the effects of randomness, we performed this procedure 10 times to ensure that the force applied was isotropic and the response vector was averaged. These response fluctuation profiles were used to generate low-resolution multiple receptor conformations (MRCs) based on Cα atoms. If ΔF is a 3N×1 vector that contains the components of the externally applied force on the selected residues, and H is the Hessian, a 3N × 3N matrix composed of the second derivatives of the potential with respect to the components of the position vectors, we can calculate the response of the elastic network of residues using Eq. 1:
| (1) |
where ΔR is the 3N-dimensional vector of the response fluctuations of all the residues.
To derive the perturbed structure, we calculated the final perturbed coordinates for each residue as follows:
| (2) |
where R0 is the vector showing initial coordinates of the residues before perturbation, and α is a scaling factor with a value of 50. The arbitrary value of 50 was chosen such that it could yield structures in a range of 0.5–2.0 Å RMSD from the original (unperturbed) crystal structure (28,41).
The perturbed structures were then clustered to discard similar conformations generated from the perturbations on different residues in the protein. The perturbed structures generated from PRS varied from each other and had a backbone RMSD of 0.58–2.15 Å relative to the initial structure. Once the backbone was perturbed, coarse-grained conformations were obtained by integrating the response fluctuation profile into the original Cartesian coordinates; the side chains were derived from the original unperturbed structure. An all-atom minimization of these structures was done using the AMBER package (42,43) to account for the fluctuation in the side chains upon perturbation on the backbone of the protein and to relieve strain in the system. Another way to couple the side-chains to the perturbed backbone conformations is to update the side-chain positions using the internal coordinates of the initial structure and then follow up by energy minimization. We tested this approach for 2RDK and the E41A and E41G mutants. However, the RMSD between the conformations obtained after minimization with and without updated internal coordinates were in the range of 0.26–0.49 Å for 2RDK, 0.03–0.2 Å for E41A, and 0.27–0.43 Å for E41G. We also observed that the docking results did not show any significant difference compared with results from the minimization approaches discussed above (Table S2). This could be because side-chain conformations are again repacked using either rotamer trials or a full combinatorial search in RosettaLigand (44,45).
Docking with RosettaLigand
An ensemble docking using PRS-generated conformations was performed with RosettaLigand (44,45). The docking simulations for each structure in the ensemble were performed using the RosettaLigand protocol in the ROSETTA package. RosettaLigand is a method that was specifically developed for docking ligands into protein-binding sites. This method incorporates ligand flexibility by changing torsional angles and the backbone of the ligand, whereas the whole protein is held fixed throughout the docking simulation. In each case, the coordinates of the dimannose sugar were taken from the crystallographic complex of 2RDK and were treated as a single residue. The ligand was represented as a set of discrete conformations and the docking procedure, ensuring that the ensembles spun a maximal range of the conformational space (45). In this study, we perturbed the ligand position and orientation randomly with the translation of mean 0.1 Å and rotations of mean 3°, respectively. The rigid-body orientation and side-chain χ angles of the ligand were optimized using the gradient-based Davidson-Fletcher-Powell algorithm (44,45). Each docking trajectory consisted of 50 of the Monte Carlo minimization cycles, and we computed 10,000 trajectories to generate a comprehensive ensemble of conformations of the receptor-glycan complex for each protein. The formation of a distinct binding funnel in energy score/RMSD plots was considered as an indication of successful docking, and the final docked conformations were selected based on the lowest free-energy pose in the protein-binding site. The lowest free-energy pose had the lowest Ligsum score among all other docking poses (Ligsum is a RosettaLigand scoring function composed of five energy terms: attractive, repulsive, solvation, hydrogen bonding, and coulombic (44,45)). The overall flexible docking approach is outlined in Fig. 2. The binding energies of the bound complexes with the lowest Ligsum score were evaluated with X-Score. X-Score is an empirical scoring function that was developed to rerank the protein-ligand complex and give a more accurate estimate of the binding free energies, which have been found to correlate with the experimental binding constants (46).
Figure 2.
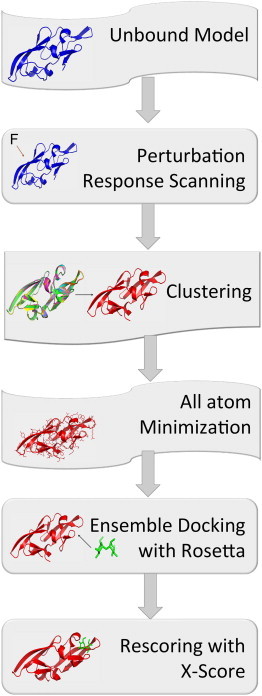
Flow chart of the flexible docking method. In our flexible docking approach using the PRS model, we generate an ensemble of receptor conformations through several steps: (i) sequentially exerting random external force on each single-residue of the CVN mutants, (ii) calculating the response fluctuation vector using the PRS method, (iii) constructing the low-resolution deformed structures (i.e., backbone) using the response vectors after each single residue perturbation, (iv) clustering the perturbed conformations using the k-clustering method, and (v) all-atom minimization of each clustered conformation. Once the MRCs ensemble is completed, we perform a docking simulation using the RosettaLigand option in the ROSETTA package for each minimized structure in the ensemble. (vi) Lastly, the binding energies of the CVN-dimannose complexes with the lowest Ligsum score are evaluated using X-Score. To see this figure in color, go online.
Protein cloning, expression, and purification
Mutagenic primers were used to introduce the E41A, E41G, T57A, N42A, and N53S single mutations into a P51G-m4-CVN sequence in a pET26b(+) vector and then transformed into BL21(DE3) cells (Novagen, Madison, WI). Proteins were expressed as previously described (9). Briefly, 100 mL cell cultures were incubated overnight at 37°C, and then 10 mL of this culture was added to a 1 L of lysogeny broth (LB). For NMR experiments, a modified M9 minimal media containing 15NH4Cl (Cambridge Isotope Laboratories, Andover, MA) as the sole nitrogen source was used in place of LB. Cells were induced with addition of 1 mM isopropyl β-D-1-thiogalactopyranoside when the OD600 reached 0.8 and were grown overnight at 37°C, and harvested via centrifugation. The cell pellet was lysed by sonication and the insoluble fraction was solubilized using an 8 M urea buffer. The 6× histidine-tagged proteins were purified on a GE HisTrap HP column (GE Healthcare Bio-Sciences, Piscataway, NJ) using a Bio-Rad EconoPump (Bio-Rad, Richmond, CA) under denaturing conditions. Proteins were refolded by dialysis into a 2 M urea buffer, pH 8.0, at room temperature for 12 h, and then overnight into 10 mM Tris, 100 mM NaCl, pH 8.0, replacing the Tris buffer once. The protein secondary structure and stability were verified by circular dichroism (CD) on a JASCO J-815 CD spectrometer (JASCO, Easton, MD). Temperature denaturation experiments were performed by following the molar ellipticity at 202 nm of samples containing 20 μM protein in 15 mM potassium phosphate; samples were heated over a range of 4–90°C at 1°C/min with a data pitch of 0.5°C.
NMR titrations
Purified 15N-labeled protein was buffer exchanged by gel filtration with a GE HiLoad 16/600 Superdex 75 pg column (GE Healthcare Bio-Sciences) on an Agilent 1260 BIO-inert HPLC system (Agilent Technologies, Santa Clara, CA) into 20 mM sodium phosphate buffer, pH 6.0. Fractions containing the monomer of the protein were pooled and concentrated using Amicon Ultracel-3K centrifugal filters (Millipore, Billerica, MA) to 300 μM protein.
Aliquots from a stock solution of 2α-mannobiose (Sigma Aldrich, St. Louis, MO) were titrated into the protein. 1H-15N heteronuclear single quantum coherence (HSQC) spectra were obtained on a Varian Innova 500 MHz spectrometer equipped with a room-temperature triple-resonance probe at gradually increasing molar ratios of ligand to protein. Chemical shifts were observed and the Δδ were determined according to Eq. 3 (47,48):
| (3) |
The chemical shifts were plotted against the molar ratio ([ligand]/[protein]) and fitted to Eq. 4:
| (4) |
Results
Our analysis focused on domain B, which has higher affinity and specificity for dimannose (Manα(1,2)Manα) in WT CVN, and for which extensive mutational analysis is available. We used two engineered forms of CVN, P51G-m4-CVN and CVN(mutDB), which present the two extremes of the binding affinity range: P51G-m4-CVN contains a wild-type (WT) domain B and thus binds dimannose with high affinity (8,9), whereas CVN(mutDB) contains an impaired domain B and thus does not bind dimannose at concentrations as high as millimolar (49). P51G-m4-CVN and CVN(mutDB) each contain a P51G mutation in the hinge region that stabilizes the monomeric form relative to the domain-swapped form of CVN (50). The two mutants have been thoroughly characterized, with crystal structures available for CVN(mutDB) (16) and for P51G-m4-CVN in its apo and bound forms (8,9).
Using the crystallographic coordinates as a starting point, we generated ensembles of binding-induced conformations by subjecting each protein to PRS. Specifically, we first obtained backbone conformational changes upon perturbing different positions with a random force. We generated the full-atom models by adding the side chains and performing an all-atom minimization for each conformation in the ensemble. We used RosettaLigand to dock the dimannose ligand into the cavity or binding pocket for each structure in the ensemble, and scored the lowest-energy poses as described in Materials and Methods. The binding energy of dimannose to domain B of P51G-m4-CVN calculated by X-Score is −7.58 kcal/mol and the RMSD of the ligand obtained from the lowest-energy docked pose is only 0.19 Å. In contrast, analysis of CVN(mutDB) shows a drastic change in the binding energy making it less favorable (−4.68 kcal/mol), corresponding to a change in dissociation constant of four orders of magnitude, and a ligand RMSD of 0.38 Å obtained from the lowest-energy docked pose. Fig. 3 compares the binding profiles of P51G-m4-CVN and CVN(mutDB) for dimannose, and the docking profile observed for P51G-m4-CVN indicates a well-converged distinct binding funnel. In contrast, CVN(mutDB) shows a poor docking profile in which energetically degenerate structures have high RMSDs.
Figure 3.
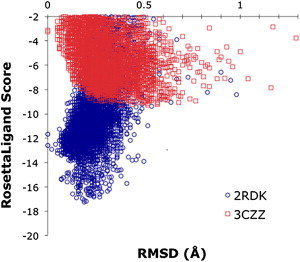
RosettaLigand energy scores (in kcal/mol) versus the RMSD of the docked complex for CVN mutants and dimannose sugar. P51G-m4-CVN (2RDK) is shown in blue circles and CVN(mutDB) (3CZZ) is shown in red squares. The funnel shape of the graph indicates good docking for P51G-m4-CVN. In agreement with experiments, P51G-m4-CVN-dimannose has a significantly low binding-energy score (−17.25 kcal/mol), whereas the complex of CVN(mutDB) (−9.28 kcal/mol) does not. To see this figure in color, go online.
Next, we analyzed each residue’s energetic contribution to binding by systematically mutating each position in P51G-m4-CVN to the corresponding amino acid in CVN(mutDB): Glu-41, Asn-42, Thr-57, Arg-76 were changed to Ala, and Gln-78 was changed to Gly. We also mutated residue 41 to glycine, which is the most common substitution at that position in the CVN-homologous family of proteins, CVNH (13,51). In addition to the positions mutated in CVN(mutDB), we mutated N53, which lines the binding pocket and interacts with the glycan, to serine, because serine is the most common substitution at this position in CVNH (13,51).
In the absence of crystal structures for the mutants, we generated starting models by introducing the individual point mutations into the coordinates of P51G-m4-CVN and performing a short REMD run (3 ns). Starting from the most dominant structure of the lowest-temperature REMD run (i.e., the structure with the lowest free energy), we applied our flexible docking approach to each mutant (see Materials and Methods for details). The mutations at positions 41, 42, and 57 resulted in decreased affinity compared with that of P51G-m4-CVN (Table 1). Position 42 accounts for most of the loss of binding energy between the intact domain B in P51G-m4-CVN and the defective domain B in CVN(mutDB): the X-Score binding energy of N42A (−5.37 kcal/mol) is in close proximity to the nonbinder CVN(mutDB) (i.e., −4.68 kcal/mol). This drastic loss in binding energy score may indicate the critical role of N42 in stabilizing the structure of the binding pocket. The side chain of N42 forms hydrogen bonds to the backbone of neighboring residues (Table S1); further, this position is very well conserved in the CVNH family (13,46). T57A, E41A, and E41G also showed a significant decrease in binding affinity, albeit not as drastic as that observed for N42A. In contrast, the binding energy score of N53S mutant (−7.06 kcal/mol) is comparable to that of P51G-m4-CVN (−7.58 kcal/mol). To verify the accuracy of our predictions, we recombinantly expressed E41A, E41G, T57A, N42A, and N53S on the background of P51G-m4-CVN (8,9), and subjected them to experimental characterization.
Table 1.
Binding energy scores (Ebind) evaluated from X-Score along with corresponding RMSDs of the ligand (RMSDligand) and protein side chain positions in the binding site (RMSDsidechain) of the lowest energy docked pose for various CVN mutants are shown. Higher negative values indicate a higher binding affinity prediction.
| Protein | RMSDligand (Å) | RMSDside chain (Å) | X-Score Ebind (kcal/mol) | Experimental Kd |
|---|---|---|---|---|
| P51Gm4CVN | 0.19 | 0.189 | −7.58 | low μM |
| CVN(mutDB) | 0.38 | 1.866 | −4.68 | no binding (49) |
| E41A | 0.74 | 1.155 | −6.7 | 541 ± 118 μM |
| E41G | 0.82 | 1.261 | −6.82 | 389 ± 73 μM |
| N42A | 1.08 | 1.808 | −5.37 | no binding |
| T57A | 0.99 | 1.08 | −6.46 | low mM |
| R76A | 1.06 | 1.036 | −6.9 | NA |
| Q78G | 0.98 | 0.845 | −6.85 | NA |
| N53S | 0.88 | 1.365 | −7.06 | low μM |
Effect of single point mutations on fold and stability of CVN
We confirmed that the mutants are monomers, similar to the parent construct, using size exclusion chromatography. The elution profiles of P51G-m4-CVN and the mutants each exhibit a main peak at 89 mL, which accounts for over 80% of the total protein; small peaks at 70 mL correspond to dimer (Fig. S1). We assigned monomer and dimer peaks by comparing the elution profiles with those of recombinant WT CVN, in which the ratio of domain-swapped dimer to monomer is ∼40:60 (9,50).
The mutations do not affect the overall secondary and tertiary structure of P51G-m4-CVN, as assessed by CD spectroscopy and 2D-NMR. The CD spectra of the mutants are identical to that of P51G-m4-CVN, indicating mainly β structure, with a characteristic minimum at 212 nm and maximum at 190 nm (Fig. S2). The 1H-15N HSQC correlation spectra of isotopically labeled mutants show that the backbone amide resonances are well dispersed in both dimensions and resemble the spectrum of P51G-m4-CVN (Fig. 4, P51G-m4-CVN; Fig. 5, E41A and E41G, red spectra; Figs. S3–S5 for N42A, T57A, and N53S, respectively). Taken together, the spectroscopic results demonstrate that the mutants display the hallmarks of native proteins, and are consistent with the conservation of the CVN fold.
Figure 4.
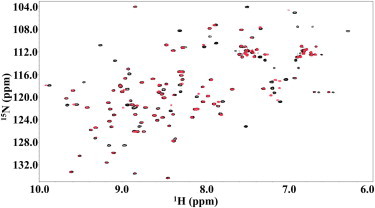
1H-15N HSQC spectra of P51G-m4-CVN without dimannose (red) and with 12.5 molar equivalents of dimannose (black). To see this figure in color, go online.
Figure 5.
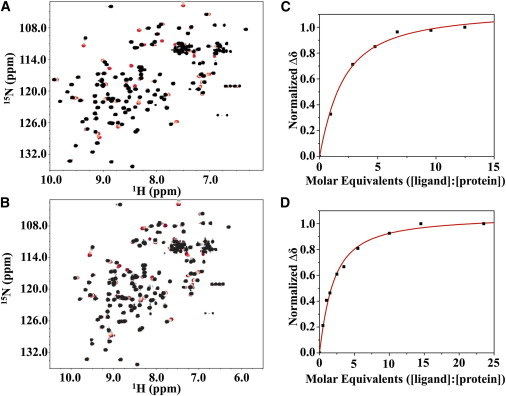
1H-15N HSQC spectra of (A) E41A and (B) E41G from titrations with increasing amounts of dimannose (red to black). The resonances are well dispersed and resemble that of the P51G-m4-CVN spectrum, indicating the two mutants are well folded. Example plots and fitting of molar equivalents versus normalized peak shift are shown for a single resonance (E41A in C; E41G in D). The average Kd values from the fits for E41A and E41G are 541 μM and 389 μM, respectively. To see this figure in color, go online.
We assessed the thermodynamic stability of each mutant by thermal denaturation, monitoring the loss of secondary structure at increasing temperatures from 4°C to 90°C by CD at 202 nm, the wavelength at which the largest change is observed between folded and unfolded spectra. The thermal denaturation of WT CVN and its mutants is not reversible, which precludes a rigorous analysis of the folding thermodynamics. It is possible, however, to compare the thermal denaturation profiles of a series of mutants and calculate the apparent midpoint of the melting transitions (Tm). The melting curves of the mutants (Fig. 6) show that, with the exception of T57A and N42A, the single point mutations do not affect protein stability significantly. The apparent melting points Tm are shown in Table 2.
Figure 6.
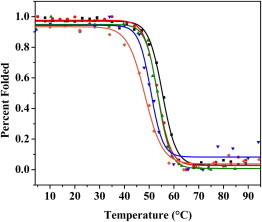
Thermal denaturation profiles of E41A (red circles), E41G (black squares), N53S (green triangles), T57A (blue upside-down triangles), and N42A (orange diamonds). The unfolding transitions were monitored at 202 nm from 4°C to 90°C using 20 μM protein in 15 mM potassium phosphate. The melting points are 53°C, 55°C, 53°C, 50.6°C, and 48°C, respectively. To see this figure in color, go online.
Table 2.
Melting temperatures of the proteins used in this study
| Protein | Tm (°C) |
|---|---|
| P51G-m4-CVN | 59 |
| E41A | 53.4 |
| E41G | 55.3 |
| N53S | 53.6 |
| T57A | 50.6 |
| N42A | 48.2 |
The proteins unfold irreversibly, preventing a rigorous analysis of folding thermodynamics.
Binding to dimannose (Man2)
We used 2D-NMR spectroscopy to quantify the binding of dimannose to P51G-m4-CVN and each of the mutants, by monitoring the changes in chemical shift of the backbone amide signals in uniformly labeled proteins upon addition of ligand through 1H-15N-correlation spectroscopy (HSQC). This technique is particularly well suited for measuring dissociation constants in the mid- to high-micromolar range (52). Titrations were carried out by adding dimannose from a stock solution up to at least a 15 molar equivalent in 300 μM protein. A comparison of the 1H-15N HSQC spectra of the P51G-m4-CVN before (red) and after (black) addition of 12.5 molar equivalents of dimannose is reported in Fig. 4. The 1H-15N HSQC spectra resulting from the titration of increasing amounts of dimannose (red to black color) into E41A and E41G are shown in Fig. 5, A and B, respectively. The binding isotherms derived by plotting changes in normalized chemical shifts (Δδ, ppm) of selected resonances as a function of added dimannose are shown in Fig. 5, C and D. As expected, P51G-m4-CVN bound tightly to dimannose, and the changes in chemical-shift signals saturated upon addition of slightly more than one equivalent. In contrast, addition of dimannose to E41A and E41G resulted in much weaker binding. The monitored chemical-shift changes reached saturation only upon addition of several excess equivalents of dimannose (5.5 molar equivalents). We calculated the equilibrium dissociation constants (Kd) for E41A and E41G by analyzing the changes in chemical shifts versus ligand concentration for several resonances, as described in Materials and Methods. This analysis yielded Kd values of 541 ± 118 μM for E41A, and 389 ± 74 μM for E41G. For T57A, the chemical-shift changes were initially small (<2 molar equivalents) and did not reach saturation even after the addition of 15 molar equivalents (Fig. S4). This behavior suggests a Kd in the low millimolar range. N42A did not show any binding to dimannose, as no change in chemical shift was observed in molar equivalents up to 20 (Fig. S3). In contrast, binding of dimannose to P51G-m4-CVN displayed the hallmark of tight binding and occurred in fast-to-intermediate exchange mode in the NMR timescale. This behavior prevented an accurate determination of Kd by NMR. Consistent with the computational predictions, N53S behaved similarly to P51G-m4-CVN, and binding was in intermediate exchange (Fig. S5). In both cases, analysis of the spectra revealed that the protein was saturated upon addition of one equivalent of dimannose, and further addition of ligand did not result in changes in the signals.
Discussion
Computationally intensive explicit-solvent MD simulations along with Poisson-Boltzmann-based calculations have been used to evaluate binding free energies and analyze binding interaction networks, using dimannose and trimannose as ligands. Most computational studies have focused on WT CVN (17,20,53) and P51G-m4-CVN (21,54). The calculated binding energies, in general, reproduced experimental values reported in the literature. Explicit-solvent MD simulations indicated the critical role of water and the flexibility of the binding pocket in the binding free energy calculations (20), which were later confirmed experimentally (8,9). These computational studies showed that several hydrogen-bond interactions originate from the backbone amides and carbonyl moieties (21,54); however, some side-chain hydrogen-bond interactions were also observed, especially in α-Man-(1,2)-α-Man-(1,2)-α-Man (trimannose) recognition (53).
Despite the wealth of structural and computational data available for WT CVN and its mutants, the role of main-chain interactions versus side-chain interactions in stabilizing CVN-dimannose complexes remained unclear. Here, we extended our flexible docking protocol to the analysis of dimannose recognition in CVN, using two engineered variants (P51G-m4 and CVNmutDB) as benchmarks. We generated backbone-perturbed conformations of CVN mutants by applying random unit forces to individual residues in a sequential manner. We then performed an all-atom minimization after adding the side chains to the backbone-perturbed structures to account for any rotamer changes of the side chains and to relieve any strain in the structure, as perturbations on the backbone can lead to significant changes in the orientation of the side chains. To understand the drastic decrease in binding affinity of CVN(mutDB) relative to P51G-m4-CVN, we analyzed the hydrogen-bond network in the lowest-energy docked poses of P51G-m4-CVN-dimannose and CVN(mutDB)-dimannose using Chimera with a cutoff of 4.0 Å. This approach is commonly used in docking studies to roughly estimate critical ligand-receptor interactions (55). The network for the P51G-m4-CVN-dimannose complex comprises of main-chain nitrogen atoms of Asn-42 and Asp-44 as donors; main-chain oxygen atoms of Asn-42, Ser-52, Asn-53, and Lys-74 as acceptors; and side-chain Oγ of Thr-57 as both acceptor and donor (Fig. 7 A). This hydrogen-bond network is similar to the hydrogen-bond network identified in previous studies (54,56). In addition, we observed side-chain hydrogen-bonding interactions of OE2 of Glu-41 as acceptor, and NE of Arg-76, NH2 of Arg-76, and NE2 of Gln-78 as donor with the dimannose (Fig. 7 A), which was previously seen only in the case of trimannose binding (53). CVN(mutDB) retained all of the main-chain hydrogen bonds at Ala-42, Asp-44, Ser-52, Asn-53, and Lys-74; in contrast, all side-chain hydrogen-bonding interactions observed in P51G-m4-CVN were lost. Only one side-chain hydrogen bond, with OE1 of Glu-56 as acceptor, was observed for CVN(mutDB). These results emphasize the critical role played by side-chain interactions of residues 41, 57, 76, and 78 in dimannose.
Figure 7.
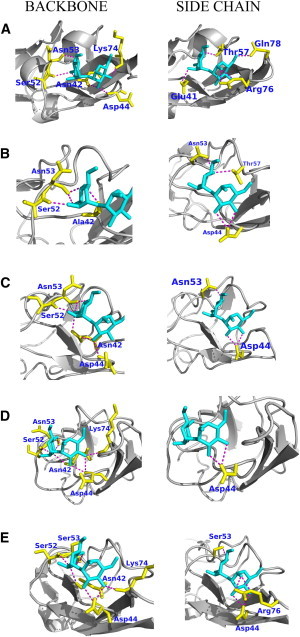
Main-chain and side-chain hydrogen-bond networks for the lowest-energy docked pose of (A) P51G-m4-CVN, (B) N42A, (C) E41A, (D) T57A, and (E) N53S. The dimannose sugar is shown in cyan, the residues that form a hydrogen bond with the sugar are shown in yellow, and the hydrogen bond is shown in magenta. To see this figure in color, go online.
A comparison of the hydrogen-bond networks of the E41A, N42A, and T57A mutants with that of the P51G-m4-CVN-dimannose complex showed rearrangements in main-chain and side-chain networks. Specifically, in N42A-dimannose, the main-chain hydrogen bond interactions of the nitrogen of Asn-42 and Asp-44 as donors, and oxygen of Lys-74 as acceptor were lost. The side-chain interactions of OE2 Glu-41, NE and NH2 of Arg-76, and NE2 Gln-78 were lost; however, the side-chain interactions of OD1 Asp-44 and OD1 Asn-53 were gained, preserving the interaction with OG1 of Thr-57 (Fig. 7 B). In E41A-dimannose, the main-chain carbonyl oxygen of Lys-74 as acceptor was lost and another oxygen of Ser-52 and Asn-53 were gained as acceptors. The side-chain interactions of OE2 Glu-41, NE and NH2 of Arg-76, NE2 Gln-78, and OG1 of Thr-57 were lost, and OD1 Asp-44 and OD1 Asn-53 were gained (Fig. 7 C). The T57A mutant retained all of the main-chain hydrogen-bond interactions with dimannose; however, it lost all of the side-chain interactions except OD1 of Asp-44, which explains the slightly higher decrease in binding affinity as compared with the E41A mutant (Fig. 7 D). E41G displayed a similar hydrogen-bond network (Fig. S6). The analysis shows that compared with the hydrogen-bond network of the P51G-m4-CVN-dimannose complex, N42A lost the highest number of hydrogen bonds (most of which were backbone hydrogen bonds) to dimannose. The loss of hydrogen bonds in T57A and E41A corresponded mostly to side-chain hydrogen bonds. These findings suggest that the identity of the side chains at positions 41 and 57 plays a critical role in dimannose binding. Conversely, our results suggest that N42 defines the integrity (i.e., the local stability) of the binding pocket by making critical side-chain-to-backbone hydrogen bonds to the neighboring β-sheet. Although polar-to-Ala mutations have disruptive effects on binding, we observe that conservative Asn to Ser mutation at position 53 preserves a hydrogen-bond pattern conducive to binding. The interactions of main-chain N of Asn-42, OE2 of Glu-41, OG1 of Thr-57, and NE2 of Gln-78 were lost, but additional side-chain interactions of N Asp-44, O of Ser-53, OD1 of Asp-44, and OG of Ser-53 were gained compared with P51G-m4-CVN-dimannose (Fig. 7 E). Details of the hydrogen-bond analysis are provided in Table S3.
One common approach in ensemble docking is to use snapshots of the MD trajectories (57,58). However, the success of this approach depends on whether the simulation covers the wide range of conformations sampled during binding. To gain a better idea of whether BP-Dock can help improve the binding affinity prediction beyond MD, we also performed ensemble docking using clustered snapshots of the REMD trajectories, and compared the results with those obtained by our flexible BP-Dock approach, which is based on a single mutant model. The binding affinities obtained from ensemble MD docking (Table S4) were all in the same range and could not really differentiate between a binder (i.e., N53S) and a nonbinder (N42A). Moreover, it did not capture the relative order of binding affinity (with N53S being the strongest, the binding score decreased in order from E41G to E41A and finally T57A, the weakest binder). However, BP-Dock approach captured the relative order of binding affinities observed experimentally and was capable of differentiating binders from nonbinders.
Our biophysical characterization by CD, thermal denaturation experiments, and 2D-NMR showed that the N42A and T57A mutations slightly affected the stability of the protein. As observed in P51G-m4-CVN, N53S bound dimannose with dissociation constants in the low micromolar range and in intermediate exchange conditions, indicative of tight binding. On the other hand, a single point mutation of E41A, E41G, and T57A resulted in much weaker dissociation constants (in the low millimolar range) and in fast exchange on the NMR timescale. However, mutation N42A abolished binding to dimannose.
Overall, our experimental characterizations of these five mutants show that BP-Dock can capture the binding-affinity pattern. The X-Score binding energy scores suggest that the mutations of E41A, E41G, and T57A resulted in a lower binding affinity for dimannose compared with P51G-m4-CVN, indicating the critical role of these side chains in glycan recognition. Mutation N42A was more disruptive than the other mutations, with a binding score closer to that of the nonbinder CVN(mutDB).
Conclusions
This work highlights the role of side-chain interactions in stabilizing the complex of dimannose and P51G-m4-CVN, with N42, T57, and E41 emerging as key factors for strong binding to dimannose. As shown here, N42 also appears to stabilize the binding pocket; its absence alters the hydrogen bond network within the protein and the ligand, thus abolishing binding. These effects add to other previously identified key interactions in CVN, such as the hydrogen bond networks with the side chain of Arg-76 (20,53) and the main-chain (54), suggesting that a complex interplay between hydrogen bonds contributes to the remarkable affinity and specificity of CVN for α(1,2)-linked mannose.
The experimental verification of flexible BP-Dock suggests that our approach could be utilized as an in silico screening tool to assess the effect of mutations on the binding affinity and specificity of CVN-based lectins as compared with the WT.
Acknowledgments
The authors thank the Fulton High Performance Computing Initiative for computer time and anonymous authors for their useful feedback.
This work was supported by grant 1121276 from the Division of Molecular and Cellular Biosciences, National Science Foundation.
Footnotes
A. Bolia and B.W. Woodrum contributed equally to this work
Contributor Information
S. Banu Ozkan, Email: banu.ozkan@asu.edu.
Giovanna Ghirlanda, Email: Giovanna.Ghirlanda@asu.edu.
Supporting Material
References
- 1.Mori T., Boyd M.R. Cyanovirin-N, a potent human immunodeficiency virus-inactivating protein, blocks both CD4-dependent and CD4-independent binding of soluble gp120 (sgp120) to target cells, inhibits sCD4-induced binding of sgp120 to cell-associated CXCR4, and dissociates bound sgp120 from target cells. Antimicrob. Agents Chemother. 2001;45:664–672. doi: 10.1128/AAC.45.3.664-672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esser M.T., Mori T., Lifson J.D. Cyanovirin-N binds to gp120 to interfere with CD4-dependent human immunodeficiency virus type 1 virion binding, fusion, and infectivity but does not affect the CD4 binding site on gp120 or soluble CD4-induced conformational changes in gp120. J. Virol. 1999;73:4360–4371. doi: 10.1128/jvi.73.5.4360-4371.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Botos I., Wlodawer A. Cyanovirin-N: a sugar-binding antiviral protein with a new twist. Cell. Mol. Life Sci. 2003;60:277–287. doi: 10.1007/s000180300023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrientos L.G., O’Keefe B.R., Boyd M.R. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antiviral Res. 2003;58:47–56. doi: 10.1016/s0166-3542(02)00183-3. [DOI] [PubMed] [Google Scholar]
- 5.Bolmstedt A.J., O’Keefe B.R., Boyd M.R. Cyanovirin-N defines a new class of antiviral agent targeting N-linked, high-mannose glycans in an oligosaccharide-specific manner. Mol. Pharmacol. 2001;59:949–954. doi: 10.1124/mol.59.5.949. [DOI] [PubMed] [Google Scholar]
- 6.Balzarini J. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat. Rev. Microbiol. 2007;5:583–597. doi: 10.1038/nrmicro1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scanlan C.N., Offer J., Dwek R.A. Exploiting the defensive sugars of HIV-1 for drug and vaccine design. Nature. 2007;446:1038–1045. doi: 10.1038/nature05818. [DOI] [PubMed] [Google Scholar]
- 8.Fromme R., Katiliene Z., Ghirlanda G. Conformational gating of dimannose binding to the antiviral protein cyanovirin revealed from the crystal structure at 1.35 A resolution. Protein Sci. 2008;17:939–944. doi: 10.1110/ps.083472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fromme R., Katiliene Z., Ghirlanda G. A monovalent mutant of cyanovirin-N provides insight into the role of multiple interactions with gp120 for antiviral activity. Biochemistry. 2007;46:9199–9207. doi: 10.1021/bi700666m. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y., Carroll J.R., Ghirlanda G. Multivalent interactions with gp120 are required for the anti-HIV activity of Cyanovirin. Biopolymers. 2009;92:194–200. doi: 10.1002/bip.21173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matei E., Zheng A., Gronenborn A.M. Anti-HIV activity of defective cyanovirin-N mutants is restored by dimerization. J. Biol. Chem. 2010;285:13057–13065. doi: 10.1074/jbc.M109.094938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keeffe J.R., Gnanapragasam P.N.P., Mayo S.L. Designed oligomers of cyanovirin-N show enhanced HIV neutralization. Proc. Natl. Acad. Sci. USA. 2011;108:14079–14084. doi: 10.1073/pnas.1108777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patsalo V., Raleigh D.P., Green D.F. Rational and computational design of stabilized variants of cyanovirin-N that retain affinity and specificity for glycan ligands. Biochemistry. 2011;50:10698–10712. doi: 10.1021/bi201411c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koharudin L.M.I., Viscomi A.R., Gronenborn A.M. The evolutionarily conserved family of cyanovirin-N homologs: structures and carbohydrate specificity. Structure. 2008;16:570–584. doi: 10.1016/j.str.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Bewley C.A., Otero-Quintero S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. J. Am. Chem. Soc. 2001;123:3892–3902. doi: 10.1021/ja004040e. [DOI] [PubMed] [Google Scholar]
- 16.Matei E., Furey W., Gronenborn A.M. Solution and crystal structures of a sugar binding site mutant of cyanovirin-N: no evidence of domain swapping. Structure. 2008;16:1183–1194. doi: 10.1016/j.str.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto Y.K., Terbush R.N., Green D.F. Computational models explain the oligosaccharide specificity of cyanovirin-N. Protein Sci. 2008;17:2008–2014. doi: 10.1110/ps.034637.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bewley C.A. Solution structure of a cyanovirin-N:Man α1-2Man α complex: structural basis for high-affinity carbohydrate-mediated binding to gp120. Structure. 2001;9:931–940. doi: 10.1016/s0969-2126(01)00653-0. [DOI] [PubMed] [Google Scholar]
- 19.Botos I., O’Keefe B.R., Wlodawer A. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. J. Biol. Chem. 2002;277:34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 20.Margulis C.J. Computational study of the dynamics of mannose disaccharides free in solution and bound to the potent anti-HIV virucidal protein cyanovirin. J. Phys. Chem. B. 2005;109:3639–3647. doi: 10.1021/jp0406971. [DOI] [PubMed] [Google Scholar]
- 21.Vorontsov I.I., Miyashita O. Solution and crystal molecular dynamics simulation study of m4-cyanovirin-N mutants complexed with di-mannose. Biophys. J. 2009;97:2532–2540. doi: 10.1016/j.bpj.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bewley C.A., Kiyonaka S., Hamachi I. Site-specific discrimination by cyanovirin-N for α-linked trisaccharides comprising the three arms of Man(8) and Man(9) J. Mol. Biol. 2002;322:881–889. doi: 10.1016/s0022-2836(02)00842-2. [DOI] [PubMed] [Google Scholar]
- 23.Shenoy S.R., Barrientos L.G., Boyd M.R. Multisite and multivalent binding between cyanovirin-N and branched oligomannosides: calorimetric and NMR characterization. Chem. Biol. 2002;9:1109–1118. doi: 10.1016/s1074-5521(02)00237-5. [DOI] [PubMed] [Google Scholar]
- 24.Sandström C., Berteau O., Gronenborn A.M. Atomic mapping of the interactions between the antiviral agent cyanovirin-N and oligomannosides by saturation-transfer difference NMR. Biochemistry. 2004;43:13926–13931. doi: 10.1021/bi048676k. [DOI] [PubMed] [Google Scholar]
- 25.Woodrum B.W., Maxwell J.D., Ghirlanda G. The antiviral lectin cyanovirin-N: probing multivalency and glycan recognition through experimental and computational approaches. Biochem. Soc. Trans. 2013;41:1170–1176. doi: 10.1042/BST20130154. [DOI] [PubMed] [Google Scholar]
- 26.Atilgan C., Gerek Z.N., Atilgan A.R. Manipulation of conformational change in proteins by single-residue perturbations. Biophys. J. 2010;99:933–943. doi: 10.1016/j.bpj.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerek Z.N., Ozkan S.B. A flexible docking scheme to explore the binding selectivity of PDZ domains. Protein Sci. 2010;19:914–928. doi: 10.1002/pro.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerek Z.N., Ozkan S.B. Change in allosteric network affects binding affinities of PDZ domains: analysis through perturbation response scanning. PLOS Comput. Biol. 2011;7:e1002154. doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolia A., Gerek Z.N., Dev K.K. The binding affinities of proteins interacting with the PDZ domain of PICK1. Proteins. 2012;80:1393–1408. doi: 10.1002/prot.24034. [DOI] [PubMed] [Google Scholar]
- 30.Xia J., Margulis C.J. Computational study of the conformational structures of saccharides in solution based on J couplings and the “fast sugar structure prediction software”. Biomacromolecules. 2009;10:3081–3088. doi: 10.1021/bm900756q. [DOI] [PubMed] [Google Scholar]
- 31.Xia J., Margulis C.J., Case D.A. Searching and optimizing structure ensembles for complex flexible sugars. J. Am. Chem. Soc. 2011;133:15252–15255. doi: 10.1021/ja205251j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramadugu S.K., Chung Y.-H., Margulis C.J. When sugars get wet. A comprehensive study of the behavior of water on the surface of oligosaccharides. J. Phys. Chem. B. 2009;113:11003–11015. doi: 10.1021/jp904981v. [DOI] [PubMed] [Google Scholar]
- 33.Berman H.M., Westbrook J., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 35.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 36.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsui V., Case D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2000-2001;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 38.Atilgan C., Atilgan A.R. Perturbation-response scanning reveals ligand entry-exit mechanisms of ferric binding protein. PLOS Comput. Biol. 2009;5:e1000544. doi: 10.1371/journal.pcbi.1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikeguchi M., Ueno J., Kidera A. Protein structural change upon ligand binding: linear response theory. Phys. Rev. Lett. 2005;94:078102. doi: 10.1103/PhysRevLett.94.078102. [DOI] [PubMed] [Google Scholar]
- 40.Nevin Gerek Z., Kumar S., Banu Ozkan S. Structural dynamics flexibility informs function and evolution at a proteome scale. Evol. Appl. 2013;6:423–433. doi: 10.1111/eva.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bahar I., Lezon T.R., Eyal E. Global dynamics of proteins: bridging between structure and function. Annu. Rev. Biophys. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kollman P.A., Dixon R., Cornell W., Fox T., Chipot C. The development/application of a “minimalist”organic/biochemical molecular mechanic force field using a combination of ab initio calculations and experimental data. Comput. Simul. Biomol. Syst. 1997;3:83–96. [Google Scholar]
- 43.Tsui V., Case D.A. Molecular dynamics simulations of nucleic acids with a generalized Born solvation model. J. Am. Chem. Soc. 2000;122:2489–2498. [Google Scholar]
- 44.Davis I.W., Baker D. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 45.Meiler J., Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 46.Wang R., Lai L., Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 47.Farmer B.T., 2nd, Constantine K.L., Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat. Struct. Biol. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 48.Williamson M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Barrientos L.G., Matei E., Gronenborn A.M. Dissecting carbohydrate-Cyanovirin-N binding by structure-guided mutagenesis: functional implications for viral entry inhibition. Protein Eng. Des. Sel. 2006;19:525–535. doi: 10.1093/protein/gzl040. [DOI] [PubMed] [Google Scholar]
- 50.Barrientos L.G., Louis J.M., Gronenborn A.M. The domain-swapped dimer of cyanovirin-N is in a metastable folded state: reconciliation of X-ray and NMR structures. Structure. 2002;10:673–686. doi: 10.1016/s0969-2126(02)00758-x. [DOI] [PubMed] [Google Scholar]
- 51.Percudani R., Montanini B., Ottonello S. The anti-HIV cyanovirin-N domain is evolutionarily conserved and occurs as a protein module in eukaryotes. Proteins. 2005;60:670–678. doi: 10.1002/prot.20543. [DOI] [PubMed] [Google Scholar]
- 52.Bewley C.A., Shahzad-Ul-Hussan S. Characterizing carbohydrate-protein interactions by nuclear magnetic resonance spectroscopy. Biopolymers. 2013;99:796–806. doi: 10.1002/bip.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujimoto Y.K., Green D.F. Carbohydrate recognition by the antiviral lectin cyanovirin-N. J. Am. Chem. Soc. 2012;134:19639–19651. doi: 10.1021/ja305755b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vorontsov I.I., Miyashita O. Crystal molecular dynamics simulations to speed up MM/PB(GB)SA evaluation of binding free energies of di-mannose deoxy analogs with P51G-m4-Cyanovirin-N. J. Comput. Chem. 2011;32:1043–1053. doi: 10.1002/jcc.21683. [DOI] [PubMed] [Google Scholar]
- 55.Gray J.J., Moughon S.E., Baker D. Protein-protein docking predictions for the CAPRI experiment. Proteins. 2003;52:118–122. doi: 10.1002/prot.10384. [DOI] [PubMed] [Google Scholar]
- 56.Matei E., Louis J.M., Gronenborn A.M. NMR solution structure of a cyanovirin homolog from wheat head blight fungus. Proteins. 2011;79:1538–1549. doi: 10.1002/prot.22981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Totrov M., Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr. Opin. Struct. Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andrusier N., Mashiach E., Wolfson H.J. Principles of flexible protein-protein docking. Proteins. 2008;73:271–289. doi: 10.1002/prot.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.