

Abstract
To examine the function of ligand-gated ion channels in a defined membrane environment, we developed a robust sequential-mixing fluorescence-based stopped-flow assay. Channel activity is determined using a channel-permeable quencher (e.g., thallium, Tl+) of a water-soluble fluorophore (8-aminonaphthalene-1,3,6-trisulfonic acid) encapsulated in large unilamellar vesicles in which the channel of interest has been reconstituted, which allows for rapid solution changes. To validate the method, we explored the activation of wild-type KcsA channel, as well as it's noninactivating (E71A) KcsA mutant, by extravesicular protons (H+). For both channel types, the day-to-day variability in the reconstitution yield (as judged from the time course of fluorescence quenching) is <10%. The activation curve for E71A KcsA is similar to that obtained previously using single-channel electrophysiology, and the activation curves for wild-type and E71A KcsA are indistinguishable, indicating that channel activation and inactivation are separate processes. We then investigated the regulation of KcsA activation by changes in lipid bilayer composition. Increasing the acyl chain length (from C18:1 to C22:1 in diacylphosphatidylcholine), but not the mole fraction of POPG (>0.25) in the bilayer-forming phospholipid mixture, alters KcsA H+ gating. The bilayer-thickness-dependent shift in the activation curve is suggestive of a decrease in an apparent H+ affinity and cooperativity. The control over bilayer environment and time resolution makes this method a powerful assay for exploring ligand activation and inactivation of ion channels, and how channel gating varies with changes in the channels’ lipid bilayer environment or other regulatory processes.
Introduction
Membrane protein function is regulated by changes in lipid bilayer composition (1–6). This regulation of membrane protein function results from a combination of specific lipid-protein interactions (7–10), including interactions important for membrane protein structural integrity (7,11–13), and nonspecific (10) lipid bilayer-protein interactions, where membrane protein function varies with changes in lipid bilayer physical properties (2).
The lipid bilayer regulation of membrane protein function arises because the proteins are coupled to their host bilayer through hydrophobic interactions, such that protein conformational changes produce local bilayer deformations. Membrane protein's conformational equilibrium thus is coupled energetically to the bilayer because the total free-energy difference between two protein conformations, I and II , is the sum of contributions from the protein conformational change and the associated bilayer deformation energy (4,14–16):
| (1) |
The generality of this bilayer regulation of membrane protein function is evident from the ability to predict how changes in lipid bilayer properties alter the function of voltage-dependent calcium and sodium channels and ligand-activated Cys-loop channels based on their effects on gramicidin channels (17–19). To gain further insight into the bilayer regulation of complex membrane proteins, we have developed a method that allows us to alter the lipid bilayer composition and monitor how the associated changes in lipid bilayer properties alter the function of an ion channel, which we illustrate with the prototypical potassium channel, KcsA.
KcsA is a bacterial H+-activated potassium channel (20–22) that has been extensively characterized using single-channel and macroscopic recording methods (23–28). High-resolution structural data show that KcsA can exist in multiple conformations (29–32) (Fig. S1 in the Supporting Material), making it an ideal model for exploring the bilayer regulation of membrane protein function. Wild-type (WT) KcsA is closed at pH 7 and activates at lower pH values where it inactivates on the seconds timescale, resulting in low steady-state open probability (24,33) and thereby complicating studies of WT KcsA behavior using single-channel electrophysiology. Mutation of an amino acid adjacent to the KcsA selectivity filter (E71A) abolishes inactivation and increases the steady-state open probability (at pH <4) to ∼1 (25). Whereas the E71A mutation allows for characterization of the H+ dependence of steady-state open probability in the absence of inactivation (28), it is difficult to obtain this information for WT KcsA. It therefore is not known how the activation of WT KcsA (gauged by the apparent H+ affinity and cooperativity) compares to that of the E71A mutant. Indeed, WT KcsA affinity and cooperativity obtained with macroscopic current recording (26) and radioisotope flux assays (34,35) yield divergent results, which furthermore differ from those obtained with E71A KcsA (28).
To assay for KcsA activity as a function of lipid bilayer composition, we built on a fluorescence-based gramicidin assay (36) that takes advantage of an ion channel’s permeability to a quencher of an intravesicular fluorophore, in this case Tl+ and 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (ANTS) (37–39). The activity of KcsA reconstituted into large unilamellar vesicles (LUVs) thus is assayed by the rate of Tl+ influx into the ANTS-loaded LUVs. It is important to note that this method allows for examination of both specific ligand-binding-mediated and bilayer-mediated regulation of ion channel function.
We show that LUV-reconstituted KcsA channels examined using our stopped-flow fluorescence-based ion flux assay recapitulate key aspects of KcsA channel function as determined in electrophysiological studies, including its requirement for anionic phospholipids (23), its activation by H+ (21,26–28), and the time course of activation and inactivation (24,26,33). We further show that KcsA function is altered by changes in phospholipid acyl chain length, and that this regulation is due primarily to changes in bilayer thickness rather than changes in the local lipid composition in the vicinity of the channels.
Materials and Methods
Materials
Synthetic lipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DC18:1PC), 1,2-dierucoyl-sn-glycero-3-phosphocholine (DC22:1PC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) were acquired from Avanti Polar Lipids (Alabaster, AL), 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate hydrate (CHAPS) (>98% purity) from Sigma-Aldrich (St. Louis, MO), and 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (ANTS) from Life Technologies (Carlsbad, CA). Bio-Beads SM-2 adsorbents were from Bio-Rad Laboratories (Hercules, CA) and TlNO3, KNO3, HEPES, and succinic acid, all >98% pure, were from Sigma-Aldrich.
KcsA expression and purification
KcsA protein was expressed and purified according to the protocol in Thompson et al. (28) with minor modifications. For detailed description see the Supporting Material.
KcsA reconstitution into ANTS-loaded LUVs
POPG and DC22:1PC or DC18:1PC phospholipids in chloroform were mixed at the desired mole fraction and dried under nitrogen gas. To remove any remaining chloroform, the dried lipid mixture was kept in a desiccator under vacuum overnight and then hydrated by vortexing with filling buffer (100 mM KNO3, 10 mM HEPES, 10 mM succinic acid, and 25 mM ANTS•2Na, pH 7.0). The lipid mixture was solubilized with 35 mM CHAPS and sonicated to clarity with an Avanti Sonicator (Avanti Polar Lipids). KcsA was added to the solubilized lipids at a 1:10,000 protein (KcsA tetramer)/lipid molar ratio and incubated for 30 min. CHAPS was removed by incubating the protein/lipid mixture with BioBeads SM-2, at a final concentration of 1 g of BioBeads to 35 mg of CHAPS, on a rotator (LabQuake, Barnstead/Thermolyne, Thermo Scientific, Waltham, MA) for 2 h at room temperature. To form LUVs, the vesicles formed after detergent removal were sonicated for 20 s on a Branson bath sonicator (Branson, Danbury, CT) and extruded through a 100-nm-pore-diameter polycarbonate filter using an Avanti Mini-Extruder (Avanti Polar Lipids). ANTS was removed from the extravesicular solution by running the LUVs through a desalting column PD-10 (GE Healthcare Life Sciences, Piscataway, NJ) and eluting with a Recording Buffer (140 mM KNO3, 10 mM HEPES, and 10 mM succinic acid, pH 7.0). Based on the average diameter of the LUVs, ∼150 nm (36), we estimate that each LUV contains ∼20 channels.
Stopped-flow ion flux assay
ANTS fluorescence was measured using an SX-20 stopped-flow spectrofluorometer (Applied Photophysics, Leatherhead, UK) (instrumental dead time ∼1.5 ms) as previously described (36), with the following modifications. In the single-mixing mode (Fig. 1 A, left) the LUVs in injector 1 were mixed with quench buffer (50 mM TlNO3, 94 mM KNO3, 10 mM HEPES, and 10 mM succinic acid) in injector 2, and the time course of fluorescence quenching was recorded. The pH of the quench buffer was adjusted such that, upon mixing 1:1 with the LUVs in recording buffer (pH 7), the pH in the optical cell had the desired pH. In the sequential-mixing mode (Fig. 1 A, right; see Results) the LUVs in injector 1 first are mixed with the activating solution (140 mM KNO3, 10 mM HEPES, and 10 mM succinic acid) in injector 2, with the pH adjusted such that upon mixing 1:1 with the LUVs we achieve the desired pH in the premix loop, where channel activation occurs. Except in the case of the KcsA activation and inactivation experiments (see Fig. 3), the incubation in the premix loop was 100 ms. After the incubation in the premix loop, the LUVs were mixed with the quench buffer (at the desired pH) in injector 3, and the time course of fluorescence quenching was recorded. The pH dependence of KcsA activation (Tl+ influx) is obtained by varying the pH in the quench (single-mixing) or activating and quench (sequential-mixing) solutions. The time course of KcsA activation and inactivation is determined by measuring the rate of Tl+-induced ANTS fluorescence quenching as a function of premix duration. (Because KcsA is more permeable to Tl+ than to K+ (PTl/PK = 3.2 (40)), the membrane potential will differ from 0 right after mixing the LUVs with the quench solution. The initial potential difference will be ∼17 mV (lumen positive); it will decay to zero as Tl+ equilibrates between the extravesicular and luminal solutions. Given the modest change in potential, we do not consider it in the data analysis.)
Figure 1.
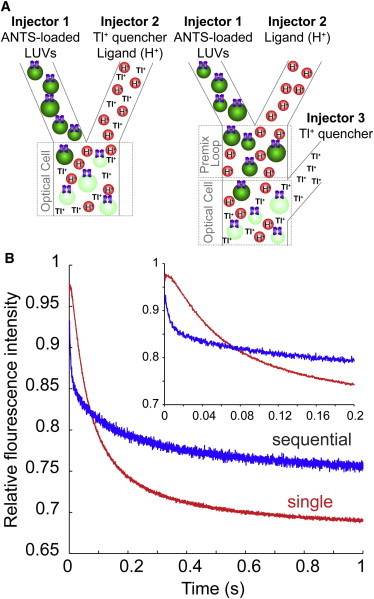
Single- and sequential-mixing stopped-flow experiments. (A) In the single-mixing mode (left), KcsA reconstituted into ANTS-loaded LUVs (in injector 1) is mixed with a Tl+- and H+-containing buffer solution (in injector 2) in the optical cell where the ANTS fluorescence signal is measured. Exposure of KcsA to H+ in the presence of Tl+ initiates KcsA activation and Tl+ influx. In the sequential-mixing mode (right), the LUV-reconstituted KcsA (in injector 1) is sequentially mixed with an activating H+-containing buffer (in injector 2) and followed by a Tl+-containing quench buffer (in injector 3): in the first, premix (or activation) step, KcsA is incubated with H+ for a sufficient time to reach peak activation (in this case, 100 ms); in the second, test step, the activated KcsA-LUVs are mixed with the quench buffer in the optical cell. (B) Tl+ influx into ANTS-loaded LUVs quenches the ANTS fluorescence. The time course of ANTS quenching in single-mixing mode is slower than that in sequential-mixing mode, mixing mode corresponding to each trace is noted on the figure. E71A KcsA was incorporated into DC22:1PC/POPG 3:1 LUVs, and channel activation was tested at pH 5. (Inset) Initial 200 ms of the fluorescence-quench time course for either mixing mode. To see this figure in color, go online.
Figure 3.
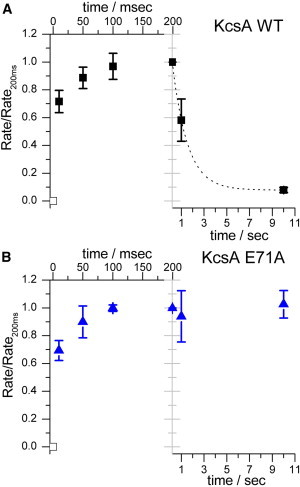
Time course of KcsA activation and inactivation. (A and B) In separate experiments, WT KcsA (A) and E71A KcsA (B) were premixed at 100 μM H+ (pH 4) for times varying between 10 ms and 10 s. The first 200 ms (upper abscissa) shows the time course of activation, and the latter 9.8 s shows the time course of inactivation (for WT KcsA); note the difference in timescale between the two time courses. The quench rate at each time point was normalized to the rate at 200 ms. The activation time course was very fast, with 70% activation at the first time point, 10 ms, and the results could not be fit by a single-exponential function; the time course of WT KcsA inactivation between 200 ms and 10 s exhibited 50% decrease in activity at 1 s, E71A maintained 100% activity over 10 s; n = 2, mean ± range/2. The LUVs were formed from a DC18:1PC/POPG 3:1 phospholipid mixture. To see this figure in color, go online.
Data analysis
KcsA activation was determined from the ANTS fluorescence quench rates (Tl+ influx), which were evaluated as previously described (36) using MATLAB (The MathWorks, Natick, MA). Briefly, for each [H+] and premix duration, the time course of fluorescence quenching was determined in several (single- or sequential-mixing) repeats. Reflecting the dispersion in LUV sizes, and the variation in the number of KcsA channels in each LUV, the fluorescence-quench time courses could not be fit with either a single- or a double-exponential decay. Instead the time courses were fit with a stretched exponential (Fig. 2 A), which is a computationally efficient way to represent sums of exponentials with a distribution of time constants (41,42):
| (2) |
where F(t) is the fluorescence intensity at time t, τ0 is a parameter with units of time, and β is a measure of sample dispersity (0 < β ≤ 1). The quench rate at 2 ms then was determined as (42)
| (3) |
The quench rates from the individual repeats at a given pH were averaged, and subsequent analysis was performed using Origin 6.1 (OriginLab, Northampton, MA). To obtain the [H+] activation curve for KcsA, the averaged rates at each pH were normalized to the rate at pH 3.5 from the same experiment, except for the plot comparing the results obtained using sequential mixing to those obtained using single mixing, where rates were normalized to the rate at pH 3. The resulting normalized rates were plotted as a function of [H+], and fitted with the Hill equation,
| (4) |
to determine the midpoint of the activation curve (pH0.5) and the Hill coefficient (nH).
Figure 2.
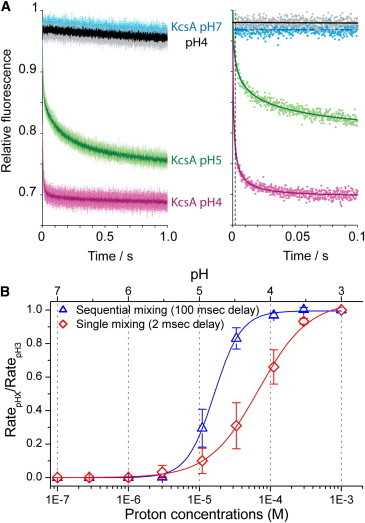
Tl+ influx rate into LUVs with reconstituted E71A KcsA as a function of extravesicular pH in the single- and sequential-mixing modes. (A, left) Normalized fluorescence quench rates from several sequential-mixing repeats (individual points) and their average (solid trace). The indicated pH is the pH in the premix loop. Fluorescence-quench time course from KcsA-free LUVs at pH 4 (upper black trace) overlaps that from KcsA-containing LUVs at pH 7. (A, right) An individual-mixing repeat (scattered points) corresponding to the experiments in the sequential-mixing mode (left), fit with a stretched exponential (solid line). (B) Plot of fluorescence quench rates, obtained from the stretched exponential fit, as a function of [H+] and the fits of the Hill equation to the data (solid lines). All quench rates are normalized to the rate at 1 mM [H+] (pH 3). H+ activation curves for KcsA in single-mixing mode (open diamonds) and sequential-mixing mode (open triangles). The LUVs were prepared from a DC22:1PC/POPG 3:1 mixture. Symbols represent the mean ± SD, n = 3–4. In the single-mixing mode, pH0.5 = 4.2 ± 0.2 and nH = 1.3 ± 0.2; in the sequential-mixing mode, pH0.5 = 4.8 ± 0.1 and nH = 2.4 ± 0.3. To see this figure in color, go online.
Statistical analysis
The nH and pH0.5 from each experiment were grouped by experimental condition and averaged. In most cases, the final results are based on three experiments and reported as the mean ± SD, and pairwise comparisons were made by two-tailed, independent Student’s t-test; p ≤ 0.05 is statistically significant. If the results are based on only two independent measurements, they are reported as the mean ± range/2.
Results
Fluorescence-based ion flux assay: single and sequential mixing
The fluorescence-based ion flux assay takes advantage of KcsA’s permeability to Tl+, which quenches the ANTS fluorescence (43) as it enters the LUV lumen through open KcsA channels. The rate of fluorescence quenching thus reports on the number of activated KcsA channels in the LUV membrane (given that the activation state of the channels doesn't vary on the same timescale as the influx measurement, i.e., in the sequential-mixing but not the single-mixing experiments).
Fig. 1 shows a schematic of the stopped-flow system (Fig. 1 A) and the time course of fluorescence quenching of ANTS-loaded DC22:1PC/POPG 3:1 LUVs containing the noninactivating E71A KcsA mutant (25) (Fig. 1 B). When the LUV-reconstituted E71A KcsA is activated by H+ and exposed to Tl+ in the extravesicular solution, Tl+ influx through the open E71A KcsA channels quenches the ANTS fluorescence. The stopped-flow spectrofluorometric assay can be performed using two different mixing modes, single and sequential. In the single-mixing mode (Fig. 1 A, left), the LUVs in one injector are combined with the Tl+- and H+-containing solution in the second injector, such that the H+-dependent KcsA activation occurs concurrently with the influx of Tl+ (and quenching of ANTS).
KcsA activation by H+ is not instantaneous, however; the time course of activation is on the order of 100 ms, varying with pH (24,26). Therefore, the rate of fluorescence quenching immediately after exposure to H+ in the single-mixing mode does not reflect peak channel activation. This is evidenced from the time course of quenching, where the initial shoulder in the quenching time course (Fig. 1 B, inset) suggests that Tl+ influx obtained in the single-mix mode occurs over the time course of E71A KcsA activation. To probe the influx rates at peak E71A KcsA activation, we therefore used the sequential-mixing mode (Fig. 1 A, right). In this mode, LUVs reconstituted with E71A KcsA were incubated at the desired pH for a given time, in this case 100 ms, which allows the channels to be activated before we test for their function. Subsequently the E71A KcsA-containing LUVs were mixed with the Tl+-containing quench solution and the time course of fluorescence quenching was measured.
Comparing the fluorescence time courses recorded in the single- and sequential-mixing modes reveals that the time course recorded in the sequential-mixing mode is faster than that recorded in the single-mixing mode (cf. the two traces in Fig. 1 B). Moreover, the initial shoulder in the quench curve obtained with the single- but not the sequential-mixing mode (cf. the two traces in Fig. 1 B, inset) demonstrates that another process, presumably channel activation, is taking place concurrently with the Tl+ influx in the single-mixing mode. To determine the H+ activation curve, LUVs reconstituted with E71A KcsA were exposed to extravesicular pH values varying between pH 7, where the channel is fully closed, and pH 3, where it is fully active (21,28). As expected, increasing pH from 4 to 5 results in a slowing of the ANTS fluorescence-quench time course, and at pH 7 the quench time course was equivalent to KcsA-free LUVs (Fig. 2 A). These results moreover indicate that the previously reported orientation asymmetry of KcsA incorporation into lipid bilayers, with most channels oriented such that their proton sensor on the cytoplasmic domain faces the intravesicular solution (21,22), does not limit our ability to study H+-dependent KcsA activation—meaning that a population of KcsA is incorporated such that the proton sensor is facing the extravesicular solution. The downward drift in fluorescence is due to slow permeation of Tl+ across the lipid bilayer, presumably as an ion pair (44,45).
Quench rates were obtained by fitting individual time courses with a stretched exponential (Fig. 2 A, right), and channel activation curves were obtained by plotting the relative changes in the quench rate as a function of [H+] (Fig. 2 B). The activation curve obtained using the single-mixing mode (with the midpoint of the activation curve, pH0.5 = 4.2 ± 0.2, and a Hill coefficient of nH = 1.3 ± 0.2) is right-shifted and shallower than the activation curve obtained in the sequential-mixing mode (pH0.5 = 4.8 ± 0.1 and nH = 2.4 ± 0.3). These results are consistent with the notion that in the single-mixing mode, only a fraction of KcsA channels are active just after the initial exposure to the test solution; therefore, the resulting quench rates underestimate the peak activation (at the given pH) and consequently the sensitivity and cooperativity of H+-activation. In the sequential-mixing mode, the time dependence of activation does not contribute to the measurement, resulting in a more accurate reflection of KcsA H+ activation. All subsequent experiments are performed using the sequential-mixing mode.
Characterizing KcsA ligand gating using sequential-mixing mode stopped flow
The activation curve obtained in the sequential-mixing mode (Fig. 2 B) shows that the noninactivating E71A KcsA mutant is activated at acidic pH with a steep concentration dependence, as indicated by the 1.5 pH unit span between the fully inactive and fully active states. The pH0.5 and Hill coefficient obtained with our assay are comparable to those previously determined using single-channel and macroscopic recording (Table 1). Moreover, the preparation-to-preparation variability in the reconstitution yield, as judged from the time course of fluorescence quenching, is <10%.
Table 1.
Effects of the lipid bilayer composition on KcsA ligand gating
| Protein | Lipid | pH0.5 ± SD | nH ± SD | Rate ± SD |
|---|---|---|---|---|
| WT KcsA | DC18:1PC/POPG 3:1 | 5.00 ± 0.04 | 2.9 ± 0.2 | 203 ± 14 |
| WT KcsA | DC18:1PC/POPG 1:1 | 5.0 ± 0.2 | 3.2 ± 0.3 | 213 ± 49 |
| E71A KcsA | DC18:1PC/POPG 3:1 | 5.00 ± 0.04 | 3.3 ± 0.2 | 247 ± 5 |
| WT KcsAa | DC22:1PC/POPG 3:1 | 4.60 ± 0.05 | 2.1 ± 0.6 | 206 ± 34 |
| E71A KcsA | DC22:1PC/POPG 3:1 | 4.8 ± 0.1 | 2.4 ± 0.3 | 305 ± 9 |
| E71A KcsAb | DC22:1PC/POPG 3:1 | 4.2 ± 0.2 | 1.3 ± 0.2 | 344 ± 7 |
| E71A KcsAc | POPE/POPG 3:1 | 5.30 ± 0.01 | 4.4 ± 0.1 |
Values are given as the mean ± range/2, n = 2.
Single-mixing mode.
Data taken from Thompson et al. (28).
For WT KcsA, channel inactivation results in low steady-state open probability (24,33). Moreover, radioisotope flux assays (35) and macroscopic current measurements (26) of the H+ activation of WT KcsA yield divergent results. Unlike in single-channel recordings, however, the sequential-mixing stopped-flow mode allows us to deconvolute activation and inactivation (see below and Fig. 3), and thus determine the H+ dependence of WT KcsA activation and compare it to that of E71A KcsA under the same experimental conditions. We therefore reconstituted WT KcsA into LUVs, determined the H+-activation curve as described above for E71A KcsA (Fig. 4), and found that pH0.5 = 5.0 ± 0.04 and nH = 2.9 ± 0.2, which are statistically indistinguishable from those obtained for E71A KcsA (Table 1).
Figure 4.
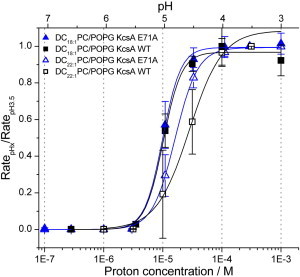
The H+ activation of KcsA varies with lipid bilayer composition. H+ activation curves for WT (squares) and E71A (triangles) KcsA were determined in LUVs prepared from DC18:1PC/POPG 3:1 (solid symbols) and DC22:1PC/POPG (open symbols) 3:1 mixtures. The quench rates plotted as a function of [H+] are normalized to the rate at pH 3.5 and fit by a Hill equation (solid lines). Except for the results with WT KcsA in the DC22:1PC/POPG membranes, where n = 2, the results are based on n ≥ 3; there was no overlap between the pH0.5 and nH values for the individual WT KcsA experiments in DC18:1PC/POPG (pH0.5 range, 5.0–5.1; nH range, 2.7–3.1) and DC22:1PC/POPG (pH0.5 range, 4.5–4.7, nH range, 1.5–2.6). To see this figure in color, go online.
The maximum unnormalized quench rates for WT KcsA and the E71A mutant differ, however: the WT rate is 203 ± 14 s−1 (mean ± S.D.) and the E71A rate is 247 ± 5 s−1 in DOPC/POPG 3:1 (Table 1). Because the channels were reconstituted using identical protocols (including the KcsA:lipid ratio; see Materials and Methods), the smaller maximal quench rate observed with WT KcsA may reflect contributions from inactivation (Table 1).
To further explore the activation and inactivation of KcsA, we used the sequential-mixing protocol where the interval between the first and the second push varied between 10 ms and 10 s, which allows for mapping the time course of activation and inactivation (Fig. 3). The resolution of the stopped flow did not allow us to determine the time course of activation at pH 4, which for both WT and E71A KcsA was 70% complete within 10 ms. The time course of activation is pH-dependent (26), an issue we did not explore here, and we may have underestimated the peak rates at pH >4.5, which would tend to decrease the apparent cooperativity of H+ activation and, to a lesser extent, underestimate pH0.5. In the case of the WT channels, the fluorescence quench rates show evidence of inactivation at longer incubation times, and at pH 4, ∼50% of the channels were inactivated after 1 s, similar to what was observed using macroscopic current recordings (24,26,46). Based on these results, we conclude that the stopped-flow-based assay provides for robust and accurate monitoring of KcsA function.
Bilayer phospholipid composition alters KcsA function
Native KcsA requires anionic phospholipids to be activated by H+ ((7,9,23), but see Alvis et al. (9), Iwamoto and Oiki (47), and Marius et al. (48). Phosphatidylglycerol (PG) has been observed in a so-called nonannular binding site at the extracellular lipid bilayer leaflet (7), but there is little selectivity among different anionic phospholipid species (48,49). In addition to this anionic phospholipid requirement, the KcsA conformational preference varies with changes in lipid bilayer thickness (imposed by increasing the acyl chain of monounsaturated, symmetric diacylphosphatidylcholines) (9,50). However, it remains unclear whether the closed/open equilibrium is altered by changes in bilayer thickness; in addition, the lipid regulation of KcsA structure (and function) may occur via several mechanisms, one of which appears to be nonspecific and bilayer-mediated.
Consistent with the above, we found that E71A KcsA does not mediate detectable Tl+ influx when reconstituted in DC22:1PC LUVs (Fig. S2), indicating a marked reduction in channel activity relative to that observed when the channels were reconstituted into 3:1 DC22:1PC/POPG bilayers (Figs. 1 and 2). To further explore how bilayer composition modulates KcsA function, we reconstituted WT and E71A KcsA into DC22:1PC/POPG 3:1 or DC18:1PC/POPG 3:1 bilayers and compared the H+-activation curves obtained for channels in these membrane environments (Fig. 4).
When reconstituted in LUVs composed of DC18:1PC/POPG 3:1, the activation curves of both WT and E71A KcsA are shifted to the left and exhibit a steeper [H+] dependence relative to DC22:1PC/POPG 3:1-reconstituted KcsA (Table 1). The change in the average acyl chain length alters KcsA function even though the PC/PG ratio does not change.
The shift in the activation curve could result simply from the changes in membrane thickness. However, another possibility is that the radial distribution of POPG adjacent to the channel changes when the phosphatidylcholine acyl chain length is changed (51–53). Thus, the acyl chain length mismatch between DC22:1PC and POPG may alter the POPG distribution, which in turn may result in local enrichment or depletion of POPG in the vicinity of KcsA, thereby altering the local mole-fraction of POPG in the first shell around the channel. Increasing the mole-fraction of POPG upregulates KcsA activity by increasing its open probability and current amplitude (47,48). Therefore, the effect of increasing the bilayer thickness could also be due to a local redistribution (in this case depletion) of POPG, which could result in a shift of the H+-activation curve.
To determine whether the differences in KcsA activation observed above reflect changes in the mole fraction of POPG adjacent to the protein, we compared the WT KcsA activation curves in LUVs formed from DC18:1PC/POPG 3:1 and DC18:1PC/POPG 1:1 (Fig. 5). Neither pH0.5 (5.0 ± 0.2) nor nH (3.2 ± 0.3) in DC18:1PC/POPG 1:1 LUVs differ from the corresponding values in DC18:1PC/POPG 3:1 LUVs (see Table 1). KcsA H+ activation is thus similar in these PC/PG mixtures. These results show that the right shift in the activation curve and the decrease in nH in DC22:1PC/POPG 3:1 LUVs were not due to changes in POPG distribution in the vicinity of KcsA; we conclude that the shift reflects changes in the average bilayer thickness.
Figure 5.
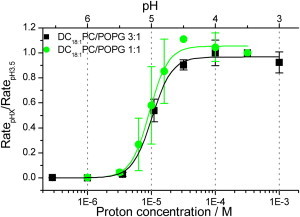
Increasing the POPG mole fraction to >0.25 does not alter the apparent H+ affinity of KcsA. H+ activation curves for WT KcsA reconstituted into LUVs prepared from DC18:1PC/POPG 3:1 (squares) and DC18:1PC/POPG 1:1 mixtures (circles). The quench rates are normalized to the rate at pH 3.5 and fit with a Hill equation. To see this figure in color, go online.
Discussion
We have developed a robust assay for ion channel function using a simple and efficient fluorescence-based ion flux method. Using the KcsA potassium channel as a prototype, we show that the LUV-reconstituted KcsA recapitulates ion channel H+ gating as observed using single-channel electrophysiology (27), and that the activation of the WT KcsA is very similar to that of the noninactivating E71A KcsA (28). We further find that KcsA function is sensitive to the lipid bilayer composition, as would be expected for a membrane protein that undergoes conformational transitions that involve the protein/bilayer boundary, and that changes in channel function most likely reflect changes in lipid bilayer thickness.
Validating the fluorescence-based ion flux assay
The incorporation of KcsA into phospholipid vesicles is asymmetric in the sense that most channels are oriented such that their cytoplasmic surface, with the proton sensor, faces the intravesicular milieu (21,22). This asymmetry is unlikely to be absolute, and our results show that the asymmetry does not limit our ability to study H+-dependent KcsA activation, suggesting that a small population of the reconstituted KcsA has the proton sensor facing the extravesicular solution. In this context, it is important that the rapid decrease in fluorescence observed after H+ activation constitutes only ∼70% of the initial value, in contrast to the 40% that would be expected if all ANTS was accessible to Tl+ (36)—and if all the LUVs had at least one KcsA that could be activated by brief increases in extravesicular H+. Given the KcsA/phospholipid ratio in the reconstitution mixture (1:10,000) and the number of lipids per LUV (200,000), there may be 20 KcsAs per LUV, and fewer than one of these has its proton sensor facing the extravesicular solution. Our results are thus consistent with the reported asymmetry of incorporation.
When comparing the E71A KcsA activation curve obtained using the fluorescence-based assay with the curve obtained using single-channel electrophysiology (28), our nH and pH0.5 values were lower than those obtained by Thompson et al. (Table 1). This difference most likely reflects the difference in experimental approach, because the ensemble-based fluorescence assay reports on the behavior of the full population of channels with a distribution of gating properties, whereas the single-molecule assay focuses on channels that satisfy a defined set of criteria (and a more limited spectrum of gating properties). When comparing our WT KcsA activation curve (Fig. 4) to that obtained using single-channel current recordings, the curve obtained from the stopped-flow assay (Table 1) is right-shifted and steeper than the curve obtained in the single-channel studies (pH0.5 = 4.37 ± 0.02 and nH = 1.51 ± 0.06) (27), with the latter being more similar to what we observe using the single-mixing mode. This difference is most likely because the time resolution provided by the stopped-flow methodology reduces the impact of channel inactivation on the results, but we cannot exclude the possibility that the fixed 100 ms activation time in the sequential-mixing experiments may have led to underestimation of both pH0.5 and nH.
Comparison of the activation curves for the noninactivating E71A KcsA and the inactivating WT KcsA show that their pH0.5 and nH values are statistically indistinguishable (Table 1). This is in contrast to previous studies, which showed greater apparent H+ affinity and cooperativity for E71A (28) than for WT KcsA, even though the latter was studied using macroscopic recordings, which should be less affected by inactivation (26). Rb+ flux experiments with WT KcsA reconstituted into LUVs yielded similarly low nH values, although the H+ affinity was markedly greater than those obtained using electrophysiological techniques (35). Although we cannot rule out other explanations, these differences likely reflect the different lipid bilayer composition and aqueous electrolyte compositions (and the time course of the pH change) in the different studies. The stopped-flow method, with its rapid mixing capability, has the advantage of quickly changing the extravesicular pH, which minimizes the overlap between channel activation and inactivation and thus gives us confidence that WT and E71A KcsA indeed have very similar activation.
The similar activation curves for the inactivating WT KcsA and the noninactivating E71A mutant suggests that the H+-dependent channel activation and inactivation are separate processes, which is consistent with WT KcsA inactivation occurring at the selectivity filter (25,34,54), independently of H+ sensing (24) and activation at the helix-bundle crossing gate (28,55–57). The smaller unnormalized quench rates obtained with WT KcsA, as compared to the E71A mutant (Table 1), may reflect that a fraction of WT channels reside in an inactivated state. This small difference in the maximal quench rate is in contrast to the >10-fold difference in the steady-state open probability between WT KcsA and the E71A mutant (25), indicating that we probe WT KcsA function before the majority of the channels have inactivated.
KcsA function is regulated by the lipid bilayer composition
The impetus for this study was to explore whether KcsA function is regulated by the lipid bilayer. That is indeed the case, as increasing the acyl chain length of the PC-containing phospholipids results in decreased apparent H+ sensitivity and gating cooperativity (Fig. 4). Although we cannot exclude the possibility that some of the shift is due to a slower activation of channels embedded in the thicker DC22:1PC/POPG 3:1 bilayers, this result could arise for two different reasons. First, the increase in the average bilayer thickness may shift the closed↔open equilibrium toward the closed conformation, which by atomic force microscopy appears to have a greater hydrophobic length (58), such that greater [H+] is needed to open the channel, leading to a right shift in the activation curves and a decrease in nH (cf. Rubin and Changeux (59)). Second, the POPG distribution adjacent to the channel may be altered as a result of the length mismatch between the PC and PG components in the presence of the longer-acyl-chain PC (DC22:1PC) (cf. Beaven et al. (53) and Maer et al. (60)).
Given the acyl-chain-length mismatch between DC22:1PC and POPG, any such redistribution would most likely tend to increase the POPG mole fraction in the vicinity of the KcsA, which has been determined by single-channel electrophysiology to stimulate KcsA activity (48). However, the WT KscA activation curve at the higher POPG mole fraction (DC18:1PC/POPG 1:1) was indistinguishable from that for DC18:1PC/POPG 3:1. This suggests that the changes in channel function between KcsA reconstituted into DC22:1PC/POPG 3:1 LUVs and KcsA reconstituted into DC18:1PC/POPG 3:1 LUVs reflect increased bilayer thickness rather than increased POPG mole fraction. This result is consistent with the preference of KcsA for longer-acyl-chain phospholipids at pH 7 (where the channel is closed) and for shorter acyl chains at pH 4 (where the channel is open) (50). Thus, the bilayer-thickness-dependent changes in fluorescence (KcsA conformation) observed by Williamson et al. (50) may reflect a shift in the distribution between discrete closed and open channel states, as opposed to a gradual change in conformation. The bilayer-thickness-dependent changes in apparent H+ affinity and cooperativity of the noninactivating E71A KcsA suggests that the affected conformational change occurs at the helix-bundle crossing controlled by the H+ sensor (28,55–57), and not at the selectivity filter where inactivation is thought to occur (25,34,54) (Fig. S1).
Conclusions
We have developed a sequential-mixing stopped-flow fluorescence-based assay for exploring ligand-activated ion channel function. The characteristic KcsA gating behavior can be recapitulated using this assay, which validates the method as a bona fide assay for KcsA function. This method should be easily adaptable to other ligand-activated channels for which there is an appropriate indicator ion—using either fluorescence quenching or other fluorometric methods. (Channels that are constitutively active can be studied using the single-mixing mode.) We can thus show that the inactivating WT KcsA and the noninactivating E71A KcsA mutant have indistinguishable activation curves, suggesting that channel activation and inactivation are separable. We further demonstrate that KcsA function is regulated by changes in bulk bilayer properties such as average bilayer thickness, reflecting the energetic coupling between bilayer-spanning proteins and their host bilayer. Taken together with previous results on voltage-dependent calcium channels (61), sodium channels (17,19,62), potassium channels (5), and Cys-loop channels (18), our results underscore the importance of the host bilayer in membrane protein function. The stopped-flow spectrofluorometric assay described here is a powerful tool in the study of both direct-ligand-binding- and bilayer-mediated mechanisms of membrane protein function regulation.
Acknowledgments
We thank Drs. Helgi I. Ingólfsson, David J. Posson, and Jason G. McCoy for helpful discussions.
This work was funded by grants from the National Institutes of Health to O.S.A. (GM021342) and C.M.N. (GM088352).
Supporting Material
References
- 1.Lee A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Andersen O.S., Koeppe R.E., 2nd Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 3.Marsh D. Protein modulation of lipids, and vice-versa, in membranes. Biochim. Biophys. Acta. 2008;1778:1545–1575. doi: 10.1016/j.bbamem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Lundbaek J.A., Collingwood S.A., Andersen O.S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. J. R. Soc. Interface. 2010;7:373–395. doi: 10.1098/rsif.2009.0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt D., MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. USA. 2008;105:19276–19281. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardie R.C., Franze K. Photomechanical responses in Drosophila photoreceptors. Science. 2012;338:260–263. doi: 10.1126/science.1222376. [DOI] [PubMed] [Google Scholar]
- 7.Valiyaveetil F.I., Zhou Y., MacKinnon R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry. 2002;41:10771–10777. doi: 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- 8.Michaelis L., Moore M.J. Location of ubiquinone-10 (CoQ-10) in phospholipid vesicles. Biochim. Biophys. Acta. 1985;821:121–129. doi: 10.1016/0005-2736(85)90162-2. [DOI] [PubMed] [Google Scholar]
- 9.Alvis S.J., Williamson I.M., Lee A.G. Interactions of anionic phospholipids and phosphatidylethanolamine with the potassium channel KcsA. Biophys. J. 2003;85:3828–3838. doi: 10.1016/S0006-3495(03)74797-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Logothetis D.E., Jin T., Rosenhouse-Dantsker A. Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pflugers Arch. 2007;455:83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- 11.Garavito R.M., Ferguson-Miller S. Detergents as tools in membrane biochemistry. J. Biol. Chem. 2001;276:32403–32406. doi: 10.1074/jbc.R100031200. [DOI] [PubMed] [Google Scholar]
- 12.van Dalen A., Hegger S., de Kruijff B. Influence of lipids on membrane assembly and stability of the potassium channel KcsA. FEBS Lett. 2002;525:33–38. doi: 10.1016/s0014-5793(02)03061-2. [DOI] [PubMed] [Google Scholar]
- 13.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 14.Gruner S.M. Lipid membrane curvature elasticity and protein function. In: Peliti L., editor. Biologically Inspired Physics. Plenum Press; New York: 1991. pp. 127–135. [Google Scholar]
- 15.Andersen O.S., Sawyer D.B., Koeppe R.E., II . Modulation of channel function by the host bilayer. In: Easwaran K.R.K., Gaber B., editors. Biomembrane Structure and Function. Adenine Press; Schenectady, NY: 1992. pp. 227–244. [Google Scholar]
- 16.Nielsen C., Andersen O.S. Inclusion-induced bilayer deformations: effects of monolayer equilibrium curvature. Biophys. J. 2000;79:2583–2604. doi: 10.1016/S0006-3495(00)76498-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundbaek J.A., Birn P., Andersen O.S. Capsaicin regulates voltage-dependent sodium channels by altering lipid bilayer elasticity. Mol. Pharmacol. 2005;68:680–689. doi: 10.1124/mol.105.013573. [DOI] [PubMed] [Google Scholar]
- 18.Søgaard R., Werge T.M., Lundbaek J.A. GABAA receptor function is regulated by lipid bilayer elasticity. Biochemistry. 2006;45:13118–13129. doi: 10.1021/bi060734+. [DOI] [PubMed] [Google Scholar]
- 19.Rusinova R., Herold K.F., Andersen O.S. Thiazolidinedione insulin sensitizers alter lipid bilayer properties and voltage-dependent sodium channel function: implications for drug discovery. J. Gen. Physiol. 2011;138:249–270. doi: 10.1085/jgp.201010529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrempf H., Schmidt O., Wagner R. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans. EMBO J. 1995;14:5170–5178. doi: 10.1002/j.1460-2075.1995.tb00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuello L.G., Romero J.G., Perozo E. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 1998;37:3229–3236. doi: 10.1021/bi972997x. [DOI] [PubMed] [Google Scholar]
- 22.Heginbotham L., LeMasurier M., Miller C. Single streptomyces lividans K+ channels: functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 1999;114:551–560. doi: 10.1085/jgp.114.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heginbotham L., Kolmakova-Partensky L., Miller C. Functional reconstitution of a prokaryotic K+ channel. J. Gen. Physiol. 1998;111:741–749. doi: 10.1085/jgp.111.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cordero-Morales J.F., Cuello L.G., Perozo E. Voltage-dependent gating at the KcsA selectivity filter. Nat. Struct. Mol. Biol. 2006;13:319–322. doi: 10.1038/nsmb1070. [DOI] [PubMed] [Google Scholar]
- 25.Cordero-Morales J.F., Cuello L.G., Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 26.Chakrapani S., Cordero-Morales J.F., Perozo E. A quantitative description of KcsA gating I: macroscopic currents. J. Gen. Physiol. 2007;130:465–478. doi: 10.1085/jgp.200709843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakrapani S., Cordero-Morales J.F., Perozo E. A quantitative description of KcsA gating II: single-channel currents. J. Gen. Physiol. 2007;130:479–496. doi: 10.1085/jgp.200709844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson A.N., Posson D.J., Parsa P.V., Nimigean C.M. Molecular mechanism of pH sensing in KcsA potassium channels. Proc. Natl. Acad. Sci. USA. 2008 doi: 10.1073/pnas.0800873105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doyle D.A., Morais Cabral J., MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y., Morais-Cabral J.H., MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 31.Cuello L.G., Jogini V., Perozo E. Design and characterization of a constitutively open KcsA. FEBS Lett. 2010;584:1133–1138. doi: 10.1016/j.febslet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuello L.G., Jogini V., Perozo E. Structural mechanism of C-type inactivation in K+ channels. Nature. 2010;466:203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao L., Mi X., Fan Z. Activation-coupled inactivation in the bacterial potassium channel KcsA. Proc. Natl. Acad. Sci. USA. 2005;102:17630–17635. doi: 10.1073/pnas.0505158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blunck R., Cordero-Morales J.F., Bezanilla F. Detection of the opening of the bundle crossing in KcsA with fluorescence lifetime spectroscopy reveals the existence of two gates for ion conduction. J. Gen. Physiol. 2006;128:569–581. doi: 10.1085/jgp.200609638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuello L.G., Cortes D.M., Perozo E. A molecular mechanism for proton-dependent gating in KcsA. FEBS Lett. 2010;584:1126–1132. doi: 10.1016/j.febslet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ingólfsson H.I., Andersen O.S. Screening for small molecules’ bilayer-modifying potential using a gramicidin-based fluorescence assay. Assay Drug Dev. Technol. 2010;8:427–436. doi: 10.1089/adt.2009.0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore H.P., Raftery M.A. Direct spectroscopic studies of cation translocation by Torpedo acetylcholine receptor on a time scale of physiological relevance. Proc. Natl. Acad. Sci. USA. 1980;77:4509–4513. doi: 10.1073/pnas.77.8.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karpen J.W., Sachs A.B., Hess G.P. Direct spectrophotometric detection of cation flux in membrane vesicles: stopped-flow measurements of acetylcholine-receptor-mediated ion flux. Anal. Biochem. 1983;135:83–94. doi: 10.1016/0003-2697(83)90734-0. [DOI] [PubMed] [Google Scholar]
- 39.Garcia A.M., Miller C. Channel-mediated monovalent cation fluxes in isolated sarcoplasmic reticulum vesicles. J. Gen. Physiol. 1984;83:819–839. doi: 10.1085/jgp.83.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LeMasurier M., Heginbotham L., Miller C. KcsA: it’s a potassium channel. J. Gen. Physiol. 2001;118:303–314. doi: 10.1085/jgp.118.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peyron M., Pierens G.K., Stewart R.C. The Modified Stretched-Exponential Model for Characterization of NMR Relaxation in Porous Media. J. Magn. Reson. A. 1996;118:214–220. [Google Scholar]
- 42.Berberan-Santos M., Bodunov E., Valeur B. Mathematical functions for the analysis of luminescence decays with underlying distributions 1. Kohlrausch decay function (stretched exponential) Chem. Phys. 2005;315:171–182. [Google Scholar]
- 43.Garcia A.M. Determination of ion permeability by fluorescence quenching. Methods Enzymol. 1992;207:501–510. doi: 10.1016/0076-6879(92)07035-m. [DOI] [PubMed] [Google Scholar]
- 44.Martinus N., Vincent C.A. Viscosity of aqueous solutions of TlNO3, Tl2SO4 and TlOH at 25°C. J. Chem. Soc., Faraday Trans. 1. 1976;72:2505–2511. [Google Scholar]
- 45.Kolthoff I.M., Chantooni M.K. Transfer activity coefficients in various solvents of several univalent cations complexed with dibenzo-18-crown-6. Anal. Chem. 1980;52:1039–1044. [Google Scholar]
- 46.Cordero-Morales J.F., Jogini V., Perozo E. A multipoint hydrogen-bond network underlying KcsA C-type inactivation. Biophys. J. 2011;100:2387–2393. doi: 10.1016/j.bpj.2011.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwamoto M., Oiki S. Amphipathic antenna of an inward rectifier K+ channel responds to changes in the inner membrane leaflet. Proc. Natl. Acad. Sci. USA. 2013;110:749–754. doi: 10.1073/pnas.1217323110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marius P., Zagnoni M., Lee A.G. Binding of anionic lipids to at least three nonannular sites on the potassium channel KcsA is required for channel opening. Biophys. J. 2008;94:1689–1698. doi: 10.1529/biophysj.107.117507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marius P., Alvis S.J., Lee A.G. The interfacial lipid binding site on the potassium channel KcsA is specific for anionic phospholipids. Biophys. J. 2005;89:4081–4089. doi: 10.1529/biophysj.105.070755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williamson I.M., Alvis S.J., Lee A.G. Interactions of phospholipids with the potassium channel KcsA. Biophys. J. 2002;83:2026–2038. doi: 10.1016/S0006-3495(02)73964-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sperotto M.M., Mouritsen O.G. Lipid enrichment and selectivity of integral membrane proteins in two-component lipid bilayers. Eur. Biophys. J. 1993;22:323–328. doi: 10.1007/BF00213555. [DOI] [PubMed] [Google Scholar]
- 52.Dumas F., Sperotto M.M., Mouritsen O.G. Molecular sorting of lipids by bacteriorhodopsin in dilauroylphosphatidylcholine/distearoylphosphatidylcholine lipid bilayers. Biophys. J. 1997;73:1940–1953. doi: 10.1016/S0006-3495(97)78225-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beaven A.H., Pastor R.W., Im W. Exploring protein-lipid interactions using gramicidin a as a model system. Biophys. J. 2013;104:432a–433a. [Google Scholar]
- 54.Cuello L.G., Jogini V., Perozo E. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature. 2010;466:272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perozo E., Cortes D.M., Cuello L.G. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nat. Struct. Biol. 1998;5:459–469. doi: 10.1038/nsb0698-459. [DOI] [PubMed] [Google Scholar]
- 56.Perozo E., Cortes D.M., Cuello L.G. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y.S., Sompornpisut P., Perozo E. Structure of the KcsA channel intracellular gate in the open state. Nat. Struct. Biol. 2001;8:883–887. doi: 10.1038/nsb1001-883. [DOI] [PubMed] [Google Scholar]
- 58.Sumino A., Sumikama T., Oiki S. The open gate structure of the membrane-embedded KcsA potassium channel viewed from the cytoplasmic side. Sci Rep. 2013;3:1063. doi: 10.1038/srep01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rubin M.M., Changeux J.P. On the nature of allosteric transitions: implications of non-exclusive ligand binding. J. Mol. Biol. 1966;21:265–274. doi: 10.1016/0022-2836(66)90097-0. [DOI] [PubMed] [Google Scholar]
- 60.Maer A.M., Nielsen C., Andersen O.S. Gramicidin channels in bilayers formed from phosphatidylcholine mixtures with a constant average number of methylene groups per lipid molecule. Biophys. J. 1999;76:A213. [Google Scholar]
- 61.Lundbaek J.A., Birn P., Andersen O.S. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 62.Morris C.E., Juranka P.F. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys. J. 2007;93:822–833. doi: 10.1529/biophysj.106.101246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.