

Abstract
Despite the improvement in treatment options, chronic lymphocytic leukemia (CLL) remains an incurable disease and patients show a heterogeneous clinical course requiring therapy for many of them. In the current work, we have built a 20-gene expression (GE)-based risk score predictive for patients overall survival and improving risk classification using microarray gene expression data. GE-based risk score allowed identifying a high-risk group associated with a significant shorter overall survival (OS) and time to treatment (TTT) (P ≤ .01), comprising 19.6% and 13.6% of the patients in two independent cohorts. GE-based risk score, and NRIP1 and TCF7 gene expression remained independent prognostic factors using multivariate Cox analyses and combination of GE-based risk score together with NRIP1 and TCF7 gene expression enabled the identification of three clinically distinct groups of CLL patients. Therefore, this GE-based risk score represents a powerful tool for risk stratification and outcome prediction of CLL patients and could thus be used to guide clinical and therapeutic decisions prospectively.
1. Introduction
Chronic lymphocytic leukemia (CLL), the most common leukemia in the western countries, is characterized by the clonal proliferation and accumulation of neoplastic B lymphocytes in the blood, bone marrow, lymph nodes, and spleen. CLL shows a heterogeneous clinical course, with many patients having an indolent disease while others suffering from rapid disease progression and are in need of early treatment [1]. Clinical staging systems based on physical examination and routine laboratory tests are the first basis for assessing different prognostic subgroups in patients with CLL [1]. However, these staging systems have a limited capacity to predict clinical outcome at an early stage of the disease and do not predict the likelihood of response to treatment in an individual with advanced disease [2].
Several biomarkers have been identified out as prognostic factors in CLL. These include somatic hypermutations in the rearranged variable regions of the immunoglobulin heavy chain (IgVH) genes, which involve around 30–40% of patients. Patients with unmutated IgVH genes had a significantly shorter median overall survival (OS) than those with mutated ones [3]. IgVH mutation status, along with deletions at 11q22-q23 (11q-) and/or 17p13 (17p-), has been identified as independent prognostic factors in CLL patients [4, 5].
Meanwhile, with the advent of microarray technology and gene expression profiling (GEP) analyses, additional markers have been investigated for their potential prognostic impact in CLL. Of these, LPL (Lipoprotein lipase) [6], ZAP70 (zeta-associated protein 70) [7], CLLU1 (Chronic lymphocytic leukemia up-regulated 1) [8], and TCL1A (T-cell leukemia/lymphoma 1A) [9] have been demonstrated to be predictive for clinical outcome. Expression of microRNAs (e.g., miR-29c and miR-223) could be also of prognostic significance in CLL [10]. These markers combined with others were used to develop multigene expression-based prognostic scores. In 2006, Zucchetto et al. constructed a scoring system based on six surface expression molecules [11]. In a study by Rodríguez et al. [12], a predictor model based on the expression of seven genes allowed the characterization of three groups of patients with distinct OS and treatment-free survival (TFS), both in two independent cohorts of patients. In 2010, Kienle et al. identified a four-gene combination, based on ZAP70, TCF7 (Transcription factor 7), DMD (Dystrophin), and ATM (Ataxia telangiectasia mutated) expression, as a predictor of IgVH mutation status in 88% of cases [13]. Stamatopoulos et al. developed a qPCR score, based on the expression of three markers (ZAP70, LPL and miR-29c), that was able to significantly predict OS and TFS by dividing patients into three groups [14]. More recently, Herold et al. developed an eight-gene expression-based risk score which showed additional prognostic value for OS and TFS compared with the established genetic markers and Binet staging [15].
We report here the design of a GE-based risk score, involving 20 genes, whose value is strongly prognostic in 2 independent cohorts of CLL patients.
2. Methods
2.1. Patients
Gene expression microarray data from three independent cohorts of patients diagnosed with CLL were used. Publicly available gene expression data from 2 cohorts with newly diagnosed CLL patients were used to construct GE-based risk score [15]. The first cohort, used as the training cohort, comprised 107 patients, and the second one as the validation cohort comprised 44 patients [15]. Peripheral blood or bone marrow samples were analyzed by Affymetrix oligonucleotide microarrays [15]. A third cohort of 130 newly diagnosed patients, with available Affymetrix gene expression data, was used as validation cohort for time to treatment analyses [16]. Clinical characteristics of patients and number and schedules of treatments were previously published [15, 16]. Interphase FISH data of the training cohort were previously published [17]. Affymetrix gene expression data are publicly available via the online Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE22762, GSE39671, and GSE25571. The data were normalized using the robust multichip average (RMA) method [15, 16].
2.2. Gene Expression Profiling and Statistical Analyses
The statistical significance of differences in overall survival between groups of patients was calculated by the log-rank test. Multivariate analysis was performed using the Cox proportional hazards model. Survival curves were plotted using the Kaplan-Meier method. All these analyses have been done with R.2.10.1 and bioconductor version 2.5.
2.3. Selection of Prognostic Genes on the Training Set
Probe sets were selected for prognostic significance using Maxstat R function (R.2.10.1 and bioconductor version 2.5) and Benjamini Hochberg multiple testing correction [18], yielding 22 significant probe sets in the two independent cohorts of patients with CLL (Table 1).
Table 1.
List of the 22 probe sets associated with a prognostic value in CLL patients.
| Probe set | Gene symbol | Gene name | Hazard Ratio |
|---|---|---|---|
| 218559_s_at | MAFB | v-maf musculoaponeurotic fibrosarcoma oncogene homolog B | 0.0471 |
| 203413_at | NELL2 | NEL-like 2 | 0.0481 |
| 212761_at | TCF7L2 | transcription factor 7-like 2 | 0.0497 |
| 206157_at | PTX3 | pentraxin 3, long | 0.0537 |
| 219947_at | CLEC4A | C-type lectin domain family 4, member A | 0.0566 |
| 209871_s_at | APBA2 | amyloid beta precursor protein-binding, family A, member 2 | 0.0568 |
| 204526_s_at | TBC1D8 | TBC1 domain family, member 8 | 0.063 |
| 218793_s_at | SCML1 | sex comb on midleg-like 1 | 0.0636 |
| 225924_at | FNIP2 | folliculin interacting protein 2 | 0.0638 |
| 207075_at | NLRP3 | NLR family, pyrin domain containing 3 | 0.0663 |
| 221698_s_at | CLEC7A | C-type lectin domain family 7, member A | 0.0667 |
| 226876_at | FAM101B | family with sequence similarity 101, member B | 0.0691 |
| 212239_at | PIK3R1 | phosphoinositide-3-kinase, regulatory subunit 1 (alpha) | 0.0731 |
| 203397_s_at | GALNT3 | UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 3 | 0.0739 |
| 216037_x_at | TCF7L2 | transcription factor 7-like 2 | 0.0781 |
| 226279_at | PRSS23 | protease, serine, 23 | 0.0833 |
| 229699_at | LOC100129550 | hypothetical LOC100129550 | 0.0841 |
| 244598_at | LCP2 | lymphocyte cytosolic protein 2 | 0.0907 |
| 209183_s_at | C10orf10 | chromosome 10 open reading frame 10 | 14.5055 |
| 203720_s_at | ERCC1 | excision repair cross-complementing rodent repair deficiency, complementation group 1 | 15.0143 |
| 203719_at | ERCC1 | excision repair cross-complementing rodent repair deficiency, complementation group 1 | 15.6883 |
| 221725_at | WASF2 | WAS protein family, member 2 | 16.2394 |
Hazard ratios (HR) are indicated for each gene used to design GE-based risk score in CLL patients. Probe sets are sorted by increasing HR.
2.4. Validation in the Independent Cohort of Patients
The GE-based risk score of CLL patients was individually calculated and patients were grouped according to the prognostic models and cutoffs from the training cohort. The prognostic value of this scoring was evaluated using log-rank statistics and Cox models.
2.5. Gene Set Enrichment Analysis (GSEA)
We compared the gene expression levels from high GE-risk score versus low risk score CLL patients and picked up the genes which had significant different expressions for gene set enrichment analysis (GSEA). Gene set enrichment analysis was carried out by computing overlaps with canonical pathways and gene ontology gene sets obtained from the broad institute [19].
3. Results
3.1. GE-Based Risk Score in CLL
Using Maxstat R function and Benjamini-Hochberg multiple testing correction [18], 22 probe sets were found to have prognostic value for OS (adjusted P value < 0.05) in two independent cohorts of patients with previously-untreated CLL (GSE22762, n = 107 and n = 44 [15]) (Table 1). These 22 probe sets were probed for 20 unique genes and were used to build a GE-based risk score as reported [20]. Figures 1(a) and 1(b) show expression of the 22 prognostic probe sets and GE-based risk score from patients' tumor samples of the training cohort (ranked according to increasing GE-based risk score). When used as a continuous variable, GE-based risk score had a prognostic value in the two cohorts of patients with CLL (P ≤ 10–4, data not shown). Patients of the training cohort (n = 107) were ranked according to increased prognostic score and, for a given score value X, the difference in survival of patients with a GE-based risk score ≤X or >X was computed. A maximum difference in overall survival (OS) was obtained with X = − 32.3, splitting patients into a high-risk group (19.6% of patients, GE-based risk score > −32.3) with a 13.4 months median OS and a low-risk group (80.4% of patients, GE-based risk score ≤ −32.3) with not reached median survival (Figure 2(a)). The prognostic value of GE-based risk score was validated in an independent CLL patient's cohort (n = 44) (Figure 2(b)). Interestingly, a high GE-based risk score is associated with a shorter median time to treatment requirement in two independent cohorts of CLL patients, that is, 2.1 months and 25.2 months for patients with GE score > −32.3 versus 47,7 and 78 months for patients with GE score ≤ −32.3 (P = 7.9E − 9 and P = 0.01, resp.) (Figures 3(a) and 3(b)). In order to investigate the prognostic value of the GE-based risk score in regards to time of first treatment in CLL patients with good prognostic, the analysis was completed in patients without Del17p, without Del11q, and without trisomy 12 known to be associated with a poor prognosis [21]. High GE-based risk score is associated with a shorter time to treatment requirement in patients with cytogenetically defined good prognostic (4.7 months for patients with GE score > −32.3 versus 65.4 for patients with GE score ≤ −32.3, P = 1E − 5) (Figure 3(c)).
Figure 1.
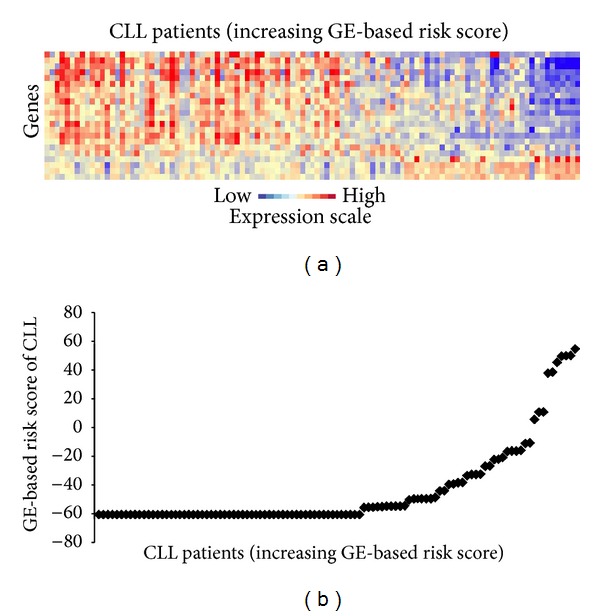
GE-based risk score in CLL patients. (a) Clustergram of genes ordered from best to worst prognosis. The level of the probe set signal is displayed from low (deep blue) to high (deep red) expression. (b) CLL patients (n = 107) were ordered by increasing GE-based risk score.
Figure 2.
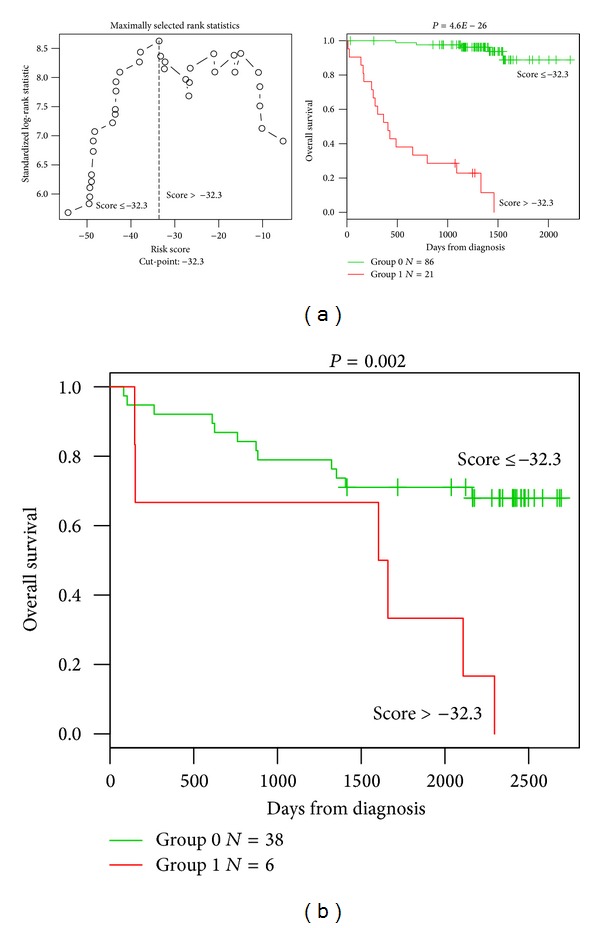
Prognostic value of GE-based risk score in CLL patients. (a) Patients of the training cohort (n = 107) were ranked according to increasing GE-based risk score and a maximum difference in OS was obtained with a score = −32.3, splitting patients into a high risk (19,6%) and a low risk (80,4%) groups. (b) The prognostic value of GE-based risk score was assayed on an independent cohort of 44 patients (validation cohort). The parameters to compute GE-based risk score of patients in the validation cohort and the proportions delineating the 2 prognostic groups were those defined with the training cohort.
Figure 3.
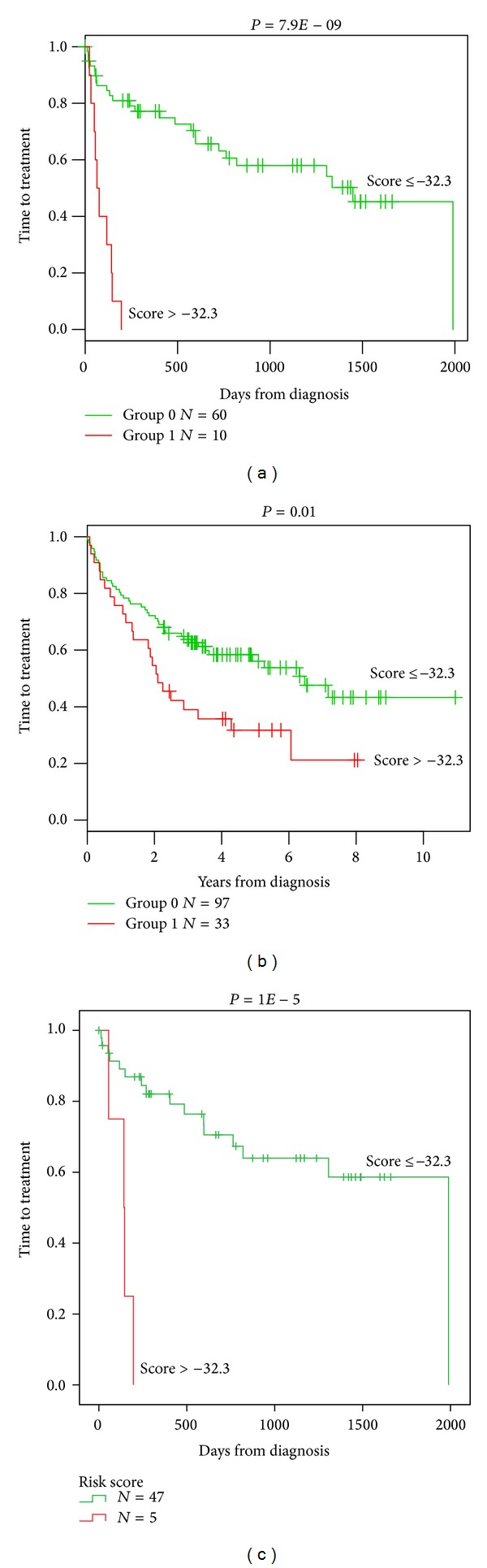
High GE-based risk score is associated with a shorter time to the first treatment in CLL patients. The prognostic value of GE-based risk score was tested in two independent cohorts of CLL patients. A high GE-based risk score is associated with a shorter time to the first treatment in the two independent cohorts ((a) n = 70, P = 7.9E − 9 and (b) n = 130, P = 0.01) and in patients with cytogenetically defined good prognostic ((c) n = 52, P = 1E − 5).
Cox analysis was used to determine whether GE-based risk score provides additional prognostic information compared to previously-identified gene expression-based prognostic markers such as ADAM29 (a disintegrin and metalloprotease domain 29), AKAP12 (a kinase (PRKA) anchor protein 12), DMD, LPL, NRIP1 (Nuclear receptor-interacting protein 1), SET10 (Septin 10), SPG20 (Spastic paraplegia 20), TCF7, TCL1A, TPM1 (Tropomyosin 1), ZAP70 gene expression, the Herold's GEP-based prognostic score (PS8), and Del17p (Table 2) [22–27]. None of these genes were included in the current 20 prognostic genes. Using univariate analyses, GE-based risk score, ADAM29, AKAP12, DMD, LPL, NRIP1, SET10, SPG20, TCF7, TCL1A, TPM1, ZAP70 gene expression, PS8, and Del17p were prognostic (P < 0.05, Table 2(a)). When compared two by two, GE-based risk score tested with NRIP1, SPG20, TCF7, and TPM1 expression, PS8 or Del17p remained significant (P < 0.01, Table 2(b)). When all parameters were tested together, only GE-based risk score, NRIP1, and TCF7 gene expression kept prognostic value (Table 2(c)).
Table 2.
Cox univariate and multivariate analysis of OS in CLL patient's training cohort (n = 107).
(a)
| Prognostic variable | Overall survival | |
|---|---|---|
| (n = 107) | ||
| HR | P value | |
| GEP risk score | 45.39 | <0.0001 |
| ADAM29 | 0.40 | 0.04 |
| AKAP12 | 2.60 | 0.02 |
| DMD | 3.39 | 0.004 |
| LPL | 4.19 | 0.001 |
| NRIP1 | 0.12 | <0.0001 |
| SEPT10 | 2.95 | 0.01 |
| SPG20 | 0.28 | 0.006 |
| TCF7 | 0.35 | <0.0001 |
| TCL1A | 4.14 | 0.001 |
| TPM1 | 2.50 | 0.02 |
| ZAP70 | 3.28 | 0.02 |
| PS8 | 10.40 | <0.0001 |
| Del(17p) | 10.13 | <0.0001 |
(b)
| Prognostic variables compared two by two | Overall survival | |
|---|---|---|
| (n = 107) | ||
| HR | P value | |
| GEP risk score | 55.30 | <0.0001 |
| ADAM29 | 1.63 | NS |
|
| ||
| GEP risk score | 46.71 | <0.0001 |
| AKAP12 | 0.93 | NS |
|
| ||
| GEP risk score | 51.78 | <0.0001 |
| DMD | 0.75 | NS |
|
| ||
| GEP risk score | 40.52 | <0.0001 |
| LPL | 1.49 | NS |
|
| ||
| GEP risk score | 8.29 | <0.0001 |
| NRIP1 | 0.035 | 0.003 |
|
| ||
| GEP risk score | 50.95 | <0.0001 |
| SEPT10 | 0.76 | NS |
|
| ||
| GEP risk score | 53.31 | <0.0001 |
| SPG20 | 0.20 | 0.001 |
|
| ||
| GEP risk score | 28.34 | <0.0001 |
| TCF7 | 0.067 | 0.001 |
|
| ||
| GEP risk score | 40.39 | <0.0001 |
| TCL1A | 2.33 | NS |
|
| ||
| GEP risk score | 69.60 | <0.0001 |
| TPM1 | 4.77 | 0.001 |
|
| ||
| GEP risk score | 44.60 | <0.0001 |
| ZAP70 | 1.04 | NS |
|
| ||
| GEP risk score | 10.35 | <0.0001 |
| PS8 | 4.73 | 0.001 |
|
| ||
| GEP risk score | 38.95 | <0.0001 |
| Del(17p) | 3.14 | 0.02 |
(c)
| All prognostic variables | Overall survival | |
|---|---|---|
| (n = 107) | ||
| HR | P value | |
| GEP risk score | 26.23 | 0.002 |
| ADAM29 | 1.04 | NS |
| AKAP12 | 6.89 | NS |
| DMD | 0.58 | NS |
| LPL | 0.54 | NS |
| NRIP1 | 0.017 | 0.01 |
| SEPT10 | 0.58 | NS |
| SPG20 | 0.76 | NS |
| TCF7 | 0.022 | 0.01 |
| TCL1A | 0.29 | NS |
| TPM1 | 2.83 | NS |
| ZAP70 | 1.53 | NS |
| PS8 | 0.63 | NS |
| Del(17p) | 1.94 | NS |
The prognostic factors were tested as single variable (a) or multivariables (b, c) using Cox-model. P values and the hazard ratios (HR) are shown. NS: not significant at a 5% threshold.
Karyotype investigations revealed the association of CLL with del13q14, trisomy 12, del11q22-q23, and del17p13 [1]. Del13q14 is the most frequent alteration that occurs in 50%–60% of cases [1]; trisomy 12 and de11q22-q23 occur in approximately 15% of CLL cases [1] and del17p13 occurs in 5%–10% of untreated CLL patients [28]. Using a cohort of 109 patients with previously untreated CLL (GSE25571) [17], we investigated the association between the GE-based risk score and chromosomal abnormalities. GE-based risk score is significantly higher in patients with del17p13 (Supplementary Figure S1; Supplementary Material is available online at http://dx.doi.org/10.1155/2014/423174).
3.2. Combining Prognostic Information of GE-Based Risk Score and NRIP1 and TCF7 Expression, into a Single Staging
Since GE-based risk score and NRIP1 and TCF7 expression displayed independent prognostic information, the prognostic information of the GE-based risk score was combined with those of TCF7 and NRIP1 gene expression into a single staging. Kaplan-Meier analysis with the 5 patient groups of the training cohort was performed (Figure 4(a)). When 2 consecutive groups showed no prognostic difference, they were merged resulting in a single staging splitting patients into a Group I comprising 72.9% of patients with low GE-based risk score/high TCF7 or NRIP1 expression and low GE-based risk score/high TCF7 and NRIP1 expression, a Group II comprising 11.2% of patients with low GE-based risk score/low TCF7/low NRIP1 expression and high GE-based risk score/high TCF7 and-or high NRIP1 expression, and a Group III comprising 15.9% of patients with high GE-based risk score/low TCF7/low NRIP1 expression (Figure 4(b)). Group I patients had a not reached median OS, patients of groups II and III had, respectively, a median OS of 46.2 months and 10 months (Figure 4(b)).
Figure 4.
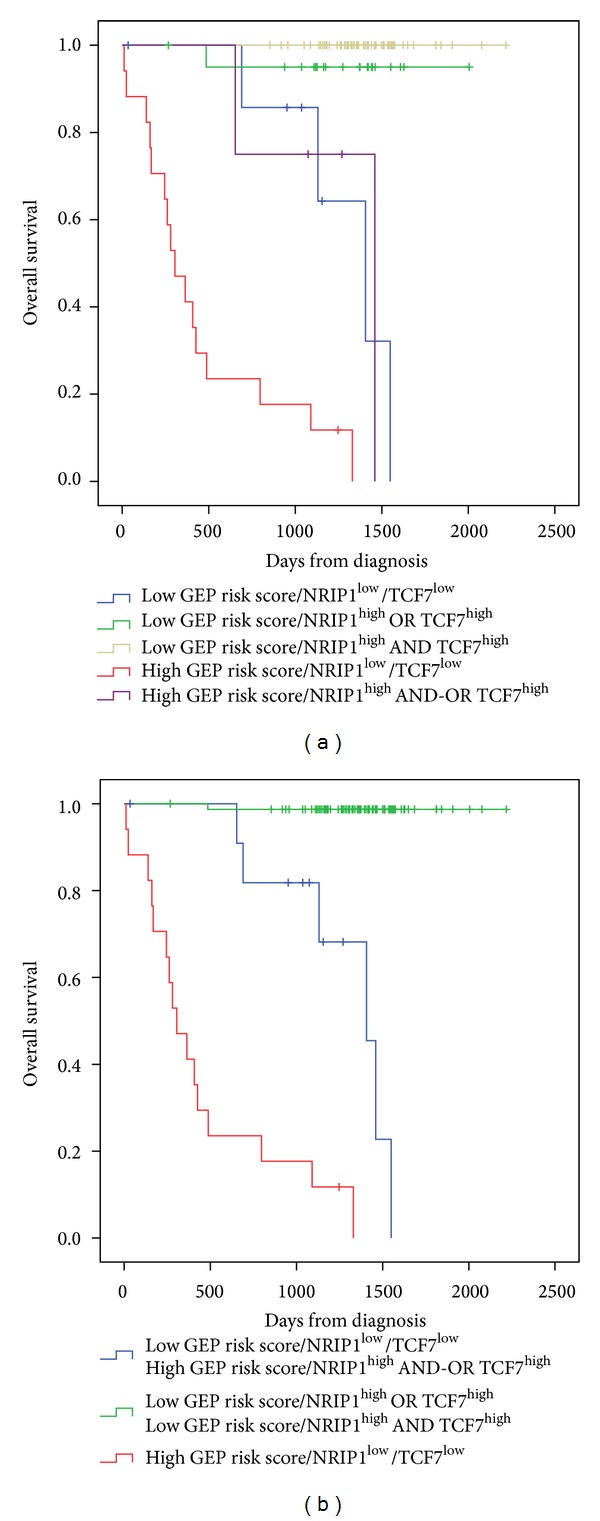
Combination of the prognostic information of GE-based risk score and NRIP1 and TCF7 gene expression. (a) Kaplan-Meier analyses were performed to combine the prognostic information of GE-based risk score and NRIP1 and TCF7 gene expression. Patients were scored from 1 to 5 according to GE-based risk score in TCF7high or Low and NRIP1high or low groups. (b) After merging consecutive groups with no prognostic difference, 3 patient groups with different overall survival (OS) were obtained: I, II, and III (patients of the training cohort, n = 107).
3.3. Tumor Cells Gene Signature in GE-Based High-Risk Group
Gene set enrichment analysis was performed comparing gene expression profiles of CLL patients with high and low GE-based risk score (n = 21 and n = 86, respectively, in the training cohort). Genes downregulated in CLL patients with mutated IgVH chain compared to unmutated (gene set: FAELT_B_CLL_WITH_VH_REARRANGEMENTS_DN, P = 0.001, Supplementary Figure S2 and Table S1) and genes upregulated in CLL patients expressing high levels of lipoprotein lipase (LPL) compared to patients expressing low level of LPL (gene set: BILBAN_B_CLL_LPL_UP, P = 0.001, Supplementary Figure S2 and Table S2) were enriched in GE-based high risk group. Conversely, genes involved in chemokine signaling pathway (gene set: PID_CXCR4_PATHWAY, P = 0.004, and gene set: KEGG_CHEMOKINE_SIGNALING_PATHWAY, P = 0.04, Supplementary Figure S3 and Supplementary Tables S3 and S4) were enriched in GE-based low-risk group.
4. Discussion
Following the introduction of microarray methodology in haematological malignancies research, many studies investigated the prediction of reliable prognostic patient subtypes on the basis of their specific gene expression signatures [20, 29, 30]. CLL, although initially reported as an indolent malignancy, is characterized by a highly heterogeneous clinical course, with many patients eventually progressing and requiring therapy [31]. Several large-scale gene expression-based profiling analyses in this malignancy have led to the identification of prognostic factors [11, 13, 22] and development of prognostic signatures for patients' risk stratification [12, 15]. We report here a new GE-based risk score in CLL specimens based on the expression levels of 20 genes documented by 22 probe sets, splitting patients of two independent cohorts into 2 risk categories. None of the 20 genes constituting the GE-based risk score overlap with the previously published prognostic gene signatures for patients' risk stratification [12, 15]. Interestingly, when compared using multivariate analysis, only the current GE-based risk score and NRIP1 and TCF7 expression, kept prognostic value. NRIP1 gene, known as RIP140, is a nuclear receptor coregulator with important role in energy homeostasis and a potential involvement in breast cancer [23, 32]. Several reports indicate that NRIP1 could either inhibit target gene transcription or act as a transcriptional activator. NRIP1 has been recently described as a novel cell-cycle regulated gene whose expression is directly controlled by E2F transcription factors and increases through their binding to the promoter region [33]. Few studies have analyzed the deregulation of this gene expression in haematological diseases: NRIP1 has been found to be significantly upregulated in acute myeloid leukemia with complex karyotypes and abnormal chromosome 21 [34]. In CLL, NRIP1 was shown to be differentially expressed with regard to IgVH mutational status [22, 35].
TCF7 is a member of a family of HMG box containing factors that are known to associate with β-catenin in the nucleus to mediate Wnt signaling [36]. The canonical Wnt/β-catenin signaling pathway has been shown to play a role in the control of the proliferation, survival, and differentiation of hematopoietic cells [37]. Recent gene expression analyses showed that several members of the Wnt family are overexpressed in CLL cells when compared to their normal counterparts from healthy donors, and this uncontrolled Wnt signaling may contribute to the defect in apoptosis that characterizes this malignancy [38]. The involvement of this pathway in the pathogenesis of several carcinomas, such as colorectal cancer and melanoma, has been also reported [39, 40]. However, there is a significant body of evidence showing that Wnt proteins can function as growth factors for progenitor cells of the B-cell lineage. Indeed, by analyzing the B-cell compartment using LEF1-deficient mice, Reya and colleagues showed a marked reduction of B220+ cells in the fetal liver and perinatal bone marrow caused by both increased apoptosis and decreased proliferation [24]. In the same way, an abnormal B-cell development has been observed in mice knocked out for the Wnt receptor Frizzled 9 [25]. In the present study, low expression of TCF7 with high GEP risk score have been correlated with a poor survival. Mice deficient in the TCF7 gene develop intestinal and mammary adenomas, suggesting a role for TCF7 as a tumor suppressor [26]. Furthermore, TCF7 has been also reported to be expressed in hematopoietic stem cells and that its loss diminishes hematopoietic stem/progenitor cell function [27]. These data suggest that the role of Wnt in B-cell malignancies is controversial, as it may have potential oncogenic, as well as tumor suppressor functions. Moreover, Kienle et al. tested the ability of TCF7 gene to predict the genetic risk in CLL patients, defined by IgHV status, V3-21 usage, 11q-, and 17p-. TCF7 expression provided a high rate of correct assignment of patients at genetic risk [13]. The prognostic impact of our GE-based score associated with NRIP1 and TCF7 genes expression should be tested in the context of IgVH mutational status, ZAP70 protein expression and TP53 mutational status.
Among the 20 genes we identified, overexpression of ERCC1 correlated with a very poor prognosis (HR = 15.0143 and 15.6883 for 203720_s_at and 203719_s_at probes, resp., Table 1). Since many years, it has been shown that treatment of CLL patients with alkylating agents is associated with low rates of complete remission and no improvement in OS [41]. The ability of CLL cells to efficiently repair alkylator-induced DNA damage through DNA repair genes might explain this lack of response. Indeed, ERCC1 forms with Xpf/ERCC4 an endonuclease complex that is involved in nucleotide excision repair (NER) and in repair of drug-induced crosslinks between two complementary strands of DNA, known as interstrand crosslinking (ICL) [42]. For instance, there is evidence that increased expression of ERCC1 in CLL lymphocytes explains the development of resistance to DNA crosslinking agents, for example, nitrogen mustards [43]. In addition, Clingen et al. demonstrated that sensitivity to SJG-136, a highly efficient ICL agent that reacts with guanine bases in a 5′-GATC-3′ sequence in the DNA minor groove, was dependent to some extent on ERCC1 expression in CHO cells [44]. Fludarabine could enhance the DNA ICL capacity of SJG-136 in primary human CLL cells and thereby offer a rationale for its clinical use in combination with SJG-136 [45]. Furthermore, F11782, a novel dual catalytic inhibitor of topoisomerases I and II, known to be a potent inhibitor of NER could be of therapeutic interest in the GE-based high risk group of CLL patients [46]. More recently, it was demonstrated that a function of PARP in NER DNA repair and clinical grade PARP inhibitors in association with chemotherapy could reverse the resistance of CLL cells to DNA crosslinking agents [47].
Interestingly, GSEA analysis highlighted a significant enrichment of genes downregulated in CLL patients with mutated IgVH chain and genes upregulated in CLL patients expressing high levels of lipoprotein lipase in tumor cells of patients within high risk GE-based score group (Supplementary Figure S2 and Supplementary Tables S1 and S2), in particular already known bad prognosis factors LPL, DMD, AKAP12, and SEPT10 (Supplementary Table S2) [31]. Interestingly, enrichment for IRF4 gene expression was identified in the GE-based high risk group. The t(1,6)(p35.3,p25.2), exclusively found in unmutated CLL, is associated with the involvement of IRF4 (Interferon regulatory factor 4) gene. This translocation is observed with high-risk chromosomal aberrations including deletions of 11q and 17p and appears to be associated with an aggressive clinical course [48]. In CLL tumors with low GE-based risk score, GSEA analysis highlighted an enrichment of genes encoding for chemokine signaling pathways (Supplementary Figure S3 and Tables S3 and S4). Of interest, we identified an enrichment of genes involved in the CXCR4 signaling pathway or in the interactions between the CLL tumor cells and their microenvironment (CCL3, CCL4, and CD49d) (Supplementary Table S3). CLL cells express high levels of functional CXCR4 and signaling through this receptor reduces spontaneous and drug-induced apoptosis and also facilitates CLL cell migration beneath stromal cells [49, 50]. More recently, it was demonstrated that the tyrosine kinase inhibitor Dasatinib inhibits CXCR4 signaling in CLL cells and impairs their migration in response to chemokine stimulation [51]. Dasatinib may constitute a potential therapeutic approach in these subgroups of CLL patients. Activated CLL cells secrete CCL3 and CCL4 for the recruitment of immune cells (T cells and monocytes) for cognate interactions. CD49d integrin (VLA-4), expressed on CLL cells, cooperates with chemokine receptors in establishing cell-to-cell adhesion with stromal cells [52]. These data suggested that tumor CLL cells of the GE-based low risk subgroup are more dependent on the interactions with their microenvironment to support their survival and proliferation.
5. Conclusion
Given the heterogeneity of CLL patients, the current GE-based risk score combined with NRIP1 and TCF7 expression could help in identifying high-risk patients who may benefit from intensive therapeutic strategies and new-targeted treatments.
Supplementary Material
Supplementary Figure S1: Association between the GE-based risk score and chromosomal abnormalities in CLL patients.
Supplementary Figure S2: Top gene sets significantly associated with high GEP-based risk score.
Supplementary Figure S3: Top gene set significantly associated with low GEP-based risk score.
Supplementary TABLE S1: Genes set enrichment analysis revealed a significant overrepresentation of the gene set FAELT_B_CLL_WITH_VH_REARRANGEMENTS_DN in high risk CLL patients compared to low risk patients (P=0.001)
Supplementary TABLE S2: Genes set enrichment analysis revealed a significant overrepresentation of the gene set BILBAN_B_CLL_LPL_UP in high risk CLL patients compared to low risk patients (P=0.001).
Supplementary TABLE S3: Genes set enrichment analysis revealed a significant overrepresentation of the gene set PID_CXCR4_PATHWAY in low risk CLL patients compared to high risk patients (P=0.004).
Supplementary TABLE S4: Genes set enrichment analysis revealed a significant overrepresentation of the gene set KEGG_CHEMOKINE_SIGNALING_PATHWAY in low risk CLL patients compared to high risk patients (P=0.04).
Acknowledgments
This work was supported by Grants from ANR emergence (ETTMM), FEDER (141786 and 42667), ARC (SFI20121205949), and INCa-Canceropole GSO (2012-011 et 2012-E14).
Conflict of Interests
The authors have no conflict of interest to declare.
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. The New England Journal of Medicine. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stamatopoulos K, Belessi C, Moreno C, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood. 2007;109(1):259–270. doi: 10.1182/blood-2006-03-012948. [DOI] [PubMed] [Google Scholar]
- 4.Rassenti LZ, Huynh L, Toy TL, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. The New England Journal of Medicine. 2004;351(9):893–901. doi: 10.1056/NEJMoa040857. [DOI] [PubMed] [Google Scholar]
- 5.Rassenti LZ, Jain S, Keating MJ, et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood. 2008;112(5):1923–1930. doi: 10.1182/blood-2007-05-092882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. Journal of Molecular Medicine. 2002;80(12):753–769. doi: 10.1007/s00109-002-0384-9. [DOI] [PubMed] [Google Scholar]
- 7.Orchard JA, Ibbotson RE, Davis Z, et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. The Lancet. 2004;363(9403):105–111. doi: 10.1016/S0140-6736(03)15260-9. [DOI] [PubMed] [Google Scholar]
- 8.Josefsson P, Geisler CH, Leffers H, et al. CLLU1 expression analysis adds prognostic information to risk prediction in chronic lymphocytic leukemia. Blood. 2007;109(11):4973–4979. doi: 10.1182/blood-2006-11-054916. [DOI] [PubMed] [Google Scholar]
- 9.Herling M, Patel KA, Weit N, et al. High TCL1 levels are a marker of B-cell receptor pathway responsiveness and adverse outcome in chronic lymphocytic leukemia. Blood. 2009;114(21):4675–4686. doi: 10.1182/blood-2009-03-208256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stamatopoulos B, Meuleman N, Haibe-Kains B, et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood. 2009;113(21):5237–5245. doi: 10.1182/blood-2008-11-189407. [DOI] [PubMed] [Google Scholar]
- 11.Zucchetto A, Bomben R, Dal Bo M, et al. A scoring system based on the expression of six surface molecules allows the identification of three prognostic risk groups in B-cell chronic lymphocytic leukemia. Journal of Cellular Physiology. 2006;207(2):354–363. doi: 10.1002/jcp.20570. [DOI] [PubMed] [Google Scholar]
- 12.Rodríguez A, Villuendas R, Yáñez L, Gómez ME, Díaz R, Pollán M. Molecular heterogeneity in chronic lymphocytic leukemia is dependent on BCR signaling: clinical correlation. Leukemia. 2007;21:1984–1991. doi: 10.1038/sj.leu.2404831. [DOI] [PubMed] [Google Scholar]
- 13.Kienle D, Benner A, Läufle C, Winkler D, Schneider C, Bühler A. Gene expression factors as predictors of genetic risk and survival in chronic lymphocytic leukemia. Haematologica. 2010;95:102–109. doi: 10.3324/haematol.2009.010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stamatopoulos B, Meuleman N, de Bruyn C, et al. A molecular score by quantitative PCR as a new prognostic tool at diagnosis for chronic lymphocytic leukemia patients. PLoS ONE. 2010;5(9):1–12. doi: 10.1371/journal.pone.0012780.e12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herold T, Jurinovic V, Metzeler KH, et al. An eight-gene expression signature for the prediction of survival and time to treatment in chronic lymphocytic leukemia. Leukemia. 2011;25(10):1639–1645. doi: 10.1038/leu.2011.125. [DOI] [PubMed] [Google Scholar]
- 16.Chuang H-Y, Rassenti L, Salcedo M, Licon K, Kohlmann A, Haferlach T. Subnetwork-based analysis of chronic lymphocytic leukemia identifies pathways that associate with disease progression. Blood. 2012;120:2639–2649. doi: 10.1182/blood-2012-03-416461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herold T, Jurinovic V, Mulaw M, et al. Expression analysis of genes located in the minimally deleted regions of 13q14 and 11q22-23 in chronic lymphocytic leukemia-unexpected expression pattern of the RHO GTPase activator ARHGAP20. Genes Chromosomes and Cancer. 2011;50(7):546–558. doi: 10.1002/gcc.20879. [DOI] [PubMed] [Google Scholar]
- 18.Kassambara A, Hose D, Moreaux J, et al. Genes with a spike expression are clustered in chromosome (sub) bands and spike (sub) bands have a powerful prognostic value in patients with multiple Myeloma. Haematologica. 2012;97(4):622–630. doi: 10.3324/haematol.2011.046821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bret C, Klein B, Moreaux J. Gene expression-based risk score in diffuse large B-cell lymphoma. Oncotarget. 2013;3:1700–1710. doi: 10.18632/oncotarget.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenquist R, Cortese D, Bhoi S, Mansouri L, Gunnarsson R. Prognostic markers and their clinical applicability in chronic lymphocytic leukemia: where do we stand? Leukemia & Lymphoma. 2013;54:2351–2364. doi: 10.3109/10428194.2013.783913. [DOI] [PubMed] [Google Scholar]
- 22.van’t Veer MB, Brooijmans AM, Langerak AW, et al. The predictive value of lipoprotein lipase for survival in chronic lymphocytic leukemia. Haematologica. 2006;91(1):56–63. [PubMed] [Google Scholar]
- 23.Nautiyal J, Steel JH, Mane MR, Oduwole O, Poliandri A, Alexi X. The transcriptional co-factor RIP140 regulates mammary gland development by promoting the generation of key mitogenic signals. Development. 2013;140:1079–1089. doi: 10.1242/dev.085720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reya T, O’Riordan M, Okamura R, et al. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity. 2000;13(1):15–24. doi: 10.1016/s1074-7613(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 25.Ranheim EA, Kwan HCK, Reya T, Wang Y-K, Weissman IL, Francke U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood. 2005;105(6):2487–2494. doi: 10.1182/blood-2004-06-2334. [DOI] [PubMed] [Google Scholar]
- 26.Roose J, Huls G, van Beest M, et al. Synergy between tumor suppressor APC and the β-catenin-Tcf4 target Tcf1. Science. 1999;285(5435):1923–1926. doi: 10.1126/science.285.5435.1923. [DOI] [PubMed] [Google Scholar]
- 27.Huls G, van Es J, Clevers H, de Haan G, van Os R. Loss of Tcf7 diminishes hematopoietic stem/progenitor cell function. Leukemia. 2013;7:1613–1614. doi: 10.1038/leu.2012.354. [DOI] [PubMed] [Google Scholar]
- 28.Zenz T, Kröber A, Scherer K, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112(8):3322–3329. doi: 10.1182/blood-2008-04-154070. [DOI] [PubMed] [Google Scholar]
- 29.Bou Samra E, Klein B, Commes T, Moreaux J. Development of gene expression-based risk score in cytogenetically normal acute myeloid leukemia patients. Oncotarget. 2012;3:824–832. doi: 10.18632/oncotarget.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreaux J, Klein B, Bataille R, et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica. 2011;96(4):574–582. doi: 10.3324/haematol.2010.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sevov M, Rosenquist R, Mansouri L. RNA-based markers as prognostic factors in chronic lymphocytic leukemia. Expert Review of Hematology. 2012;5(1):69–79. doi: 10.1586/ehm.11.80. [DOI] [PubMed] [Google Scholar]
- 32.Docquier A, Harmand P-O, Fritsch S, Chanrion M, Darbon J-M, Cavaillès V. The transcriptional coregulator RIP140 represses E2F1 activity and discriminates breast cancer subtypes. Clinical Cancer Research. 2010;16(11):2959–2970. doi: 10.1158/1078-0432.CCR-09-3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Docquier A, Augereau P, Lapierre M, Harmand P-O, Badia E, Annicotte J-S. The RIP140 gene is a transcriptional target of E2F1. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0035839.e35839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baldus CD, Liyanarachchi S, Mrózek K, et al. Acute myeloid leukemia with complex karyotypes and abnormal chromosome 21: amplification discloses overexpression of APP, ETS2, and ERG genes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(11):3915–3920. doi: 10.1073/pnas.0400272101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haslinger C, Schweifer N, Stilgenbauer S, et al. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. Journal of Clinical Oncology. 2004;22(19):3937–3949. doi: 10.1200/JCO.2004.12.133. [DOI] [PubMed] [Google Scholar]
- 36.Wu JQ, Seay M, Schulz VP, et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genetics. 2012;8(3) doi: 10.1371/journal.pgen.1002565.e1002565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annual Review of Cell and Developmental Biology. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 38.Lu D, Zhao Y, Tawatao R, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):3118–3123. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Etet PFS, Vecchio L, Nwabo Kamdje AH. Interactions between bone marrow stromal microenvironment and B-chronic lymphocytic leukemia cells: any role for Notch, Wnt and Hh signaling pathways? Cellular Signalling. 2012;24(7):1433–1443. doi: 10.1016/j.cellsig.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 40.Ge X, Wang X. Role of Wnt canonical pathway in hematological malignancies. Journal of Hematology & Oncology. 2010;3(33) doi: 10.1186/1756-8722-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbott BL. Chronic lymphocytic leukemia: recent advances in diagnosis and treatment. The Oncologist. 2006;11(1):21–30. doi: 10.1634/theoncologist.11-1-21. [DOI] [PubMed] [Google Scholar]
- 42.Weeda G, Donker I, de Wit J, et al. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Current Biology. 1997;7(6):427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 43.Geleziunas R, McQuillan A, Malapetsa A, et al. Increased DNA synthesis and repair-enzyme expression in lymphocytes from patients with chronic lymphocytic leukemia resistant to nitrogen mustard. Journal of the National Cancer Institute. 1991;83(8):557–564. doi: 10.1093/jnci/83.8.557. [DOI] [PubMed] [Google Scholar]
- 44.Clingen PH, de Silva IU, McHugh PJ, et al. The XPF-ERCC1 endonuclease and homologous recombination contribute to the repair of minor groove DNA interstrand crosslinks in mammalian cells produced by the pyrrolo[2,1-c][1,4]benzodiazepine dimer SJG-136. Nucleic Acids Research. 2005;33(10):3283–3291. doi: 10.1093/nar/gki639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pepper C, Lowe H, Fegan C, et al. Fludarabine-mediated suppression of the excision repair enzyme ERCC1 contributes to the cytotoxic synergy with the DNA minor groove crosslinking agent SJG-136 (NSC 694501) in chronic lymphocytic leukaemia cells. British Journal of Cancer. 2007;97(2):253–259. doi: 10.1038/sj.bjc.6603853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barret JM, Cadou M, Hill BT. Inhibition of nucleotide excision repair and sensitisation of cells to DNA cross-linking anticancer drugs by F 11782, a novel fluorinated epipodophylloid. Biochemical Pharmacology. 2002;63(2):251–258. doi: 10.1016/s0006-2952(01)00835-8. [DOI] [PubMed] [Google Scholar]
- 47.King BS, Cooper KL, Liu KJ, Hudson LG. Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. The Journal of Biological Chemistry. 2012;287:39824–39833. doi: 10.1074/jbc.M112.393504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michaux L, Wlodarska I, Rack K, et al. Translocation t(1;6)(p35.3;p25.2): a new recurrent aberration in “unmutated” B-CLL. Leukemia. 2005;19(1):77–82. doi: 10.1038/sj.leu.2403543. [DOI] [PubMed] [Google Scholar]
- 49.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94(11):3658–3667. [PubMed] [Google Scholar]
- 50.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 51.McCaig AM, Cosimo E, Leach MT, Michie AM. Dasatinib inhibits CXCR4 signaling in chronic lymphocytic leukaemia cells and impairs migration towards CXCL12. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0048929.e48929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivina M, Hartmann E, Kipps TJ, et al. CCL3 (MIP-1α) plasma levels and the risk for disease progression in chronic lymphocytic leukemia. Blood. 2011;117(5):1662–1669. doi: 10.1182/blood-2010-09-307249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Association between the GE-based risk score and chromosomal abnormalities in CLL patients.
Supplementary Figure S2: Top gene sets significantly associated with high GEP-based risk score.
Supplementary Figure S3: Top gene set significantly associated with low GEP-based risk score.
Supplementary TABLE S1: Genes set enrichment analysis revealed a significant overrepresentation of the gene set FAELT_B_CLL_WITH_VH_REARRANGEMENTS_DN in high risk CLL patients compared to low risk patients (P=0.001)
Supplementary TABLE S2: Genes set enrichment analysis revealed a significant overrepresentation of the gene set BILBAN_B_CLL_LPL_UP in high risk CLL patients compared to low risk patients (P=0.001).
Supplementary TABLE S3: Genes set enrichment analysis revealed a significant overrepresentation of the gene set PID_CXCR4_PATHWAY in low risk CLL patients compared to high risk patients (P=0.004).
Supplementary TABLE S4: Genes set enrichment analysis revealed a significant overrepresentation of the gene set KEGG_CHEMOKINE_SIGNALING_PATHWAY in low risk CLL patients compared to high risk patients (P=0.04).