

Abstract
Total-internal-reflection fluorescence (TIRF) microscopy provides high optical-sectioning capability and a good signal-contrast ratio for structures near the surfaces of cells. In recent years, several improvements have been developed, such as variable-angle TIRF (VA-TIRF) and spinning TIRF (sp-TIRF), which permit quantitative image analysis and address non-uniform scattering fringes, respectively. Here, we present a dual-color DMD-based shadowless-illuminated variable-angle TIRF (siva-TIRF) system that provides a uniform illumination field. By adjusting the incidence angle of the illuminating laser on the back focal plane (BFP) of the objective, we can rapidly illuminate biological samples in layers of various thicknesses in TIRF or hollow-cone epi-fluorescence mode. Compared with other methods of accomplishing VA-TIRF/sp-TIRF illumination, our system is simple to build and cost-effective, and it provides optimal multi-plane dual-color images. By showing spatiotemporal correlated movement of clathrin-coated structures with microtubule filaments from various layers of live cells, we demonstrate that cortical microtubules are important spatial regulators of clathrin-coated structures. Moreover, our system can be used to prove superb axial information of three-dimensional movement of structures near the plasma membrane within live cells.
OCIS codes: (180.0180) Microscopy, (110.0110) Imaging systems, (050.0050) Diffraction and gratings, (260.6970) Total internal reflection
1. Introduction
Various fluorescence-imaging techniques are used to visualize dynamic movements within live cells. High lateral resolution is achieved by decreasing the focal volume of the excitation illumination using confocal and two-photon microscopy. The axial resolution is further improved by confining the focal volume of the illumination using TIRF, which was first proposed by Axelrod [1]. TIRF microscopy is widely used in visualizing biological events close to the plasma membrane, such as secretory vesicle exocytosis and endocytosis [2, 3]. However, when coherent lasers are used as the light sources for illumination, imperfections in the optics (such as the lasers, mirrors, diachronic mirrors and coverslips) and dust in the light path often lead to the appearance of interference fringes [4, 5]. Organelles within cells exhibit various refraction indices (RIs) that are different from that of the extracellular solution; therefore, these structures transiently perturb the TIRF illumination field and introduce interference fringes upon approaching the plasma membrane [6]. To resolve this problem, it is common to rotate the incidence angle of the illuminating laser around the back focal plane (BFP) of the objective using a wedge, a tip/tilt mirror or an acoustic-optical deflection system (AOD) [4, 5, 7]. The TIRF images then become homogeneous, and fringes are suppressed.
TIRF microscopy can be easily combined with other techniques, such as structure illumination, fluorescence polarization and fluorescence resonance energy transfer (FRET) [8]. Thus it becomes a widely used tool for cell biologist [9]. By combining different lasers to illuminate the sample with one incident angle, a multiple-color TIRF system can monitor spatiotemporal dynamics of different proteins/organelles simultaneously. However, the penetration depths generated by different wavelength are different, which is often ignored in many experiments [2, 10], but lead to large variation in quantitative analysis of FRET efficiency [11]. Therefore, to develop a method that easily match illumination depths of lasers of different color is critical for quantitative analysis and data interpretation in TIRF imaging.
TIRF imaging can provide only two-dimensional information. In 2005, Merrifield et al. combined wide-field illumination with TIRF, and this approach yielded an estimation of the movement of clathrin-coated pits away from the plasma membrane [2]. VA-TIRF microscopy was developed to improve the capability of TIRF to probe Z-dimensional information [12]. In another approach to simultaneously improving the signal-to-noise ratio of fluorophores under illumination and maintaining the three-dimensional sectioning capability, highly inclined laminated optical sheet (HILO) illumination was developed [13]. Later, van’t Hoff et al. developed a programmable light engine that used AOD to dynamically switch the illumination between the TIRF and HILO modes, which could be used to monitor the dynamics of secretory vesicles as they approached and fused with the plasma membrane in live cells [7].
Here, we present a shadowless-illuminated variable-angle TIRF (siva-TIRF) microscopy method based on a digital micromirror device (DMD). Different grid patterns on the DMD refract laser beams with different orientations and angles. By rapidly alternating the focus of a pair of laser beams among three sets of pre-set positions on the objective BFP during one camera-exposure period, we obtain a uniform illuminated field which is similar to that of a shadowless lamp [Fig. 1(a) ]. By digitally changing the period of the grid patterns on the DMD, we alternately excite fluorescent probes in layers of different penetration depths on a sub-millisecond time scale; this technique can be used to monitor the three-dimensional movement of organelles in live cells in real time. In insulin-secreting INS-1 cells, we imaged 1) the growth of microtubule tips from 150 nm to less than 80 nm below the surface membrane and 2) the rapid motion of clathrin-coated vesicles (CCVs) in the cytosol 500 nm below the surface membrane, and relatively stationary clathrin-coated pits associated with the plasma membrane; By simultaneous dual-color three-dimension imaging, we showed that both rapid movement of CCVs and stationary CCPs were correlated with microtubule filaments. In summary, we implemented a cost-effective, simple-to-build and optimized microscope for high-speed multi-planar dual-color TIRF imaging.
Fig. 1.
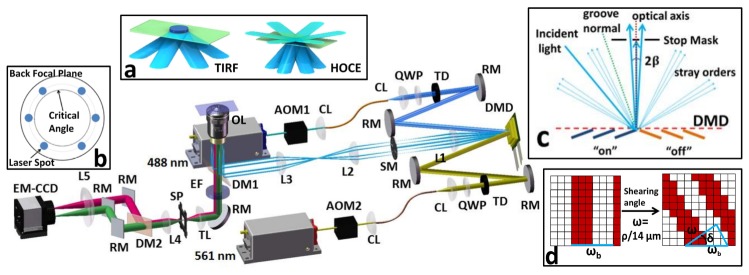
Schematic of the siva-TIRF microscope. AOM: acousto-optical modulator, CL: collimating lens, QWP: quarter-wave plate, TD: tunable diaphragm, RM: reflection mirror, SM: stop mask, OL: objective lens, DM1 and DM2: dichroic mirrors, EF: emission filter, TL: tube lens, SP: split diaphragm. Insert (a) shows the continuously alternating incident laser beams at different positions of the BFP during one camera-exposure cycle, which created uniform illumination in the TIRF and HOCE modes. Insert (b) shows the positions of the focal spots of the incident laser on the BFP. Insert (c) describes the working principle of the operation of a DMD as a grid grating. Insert (d) describes the schematic of the generation of a fine non-integral period of the DMD pattern. ρ is the physical size of one grating period (14 μm is the pixel size of the DMD).The integral ω b consisted of parallel lines of “on” and “off” pixels; the non-integral ω was generated by rotating the basic ω b patterns through various angles δ.
2. Experimental setup and theory
siva-TIRF microscopy. The schematic illustration of our system is shown in Fig. 1. It is based on a commercial inverted fluorescence microscope (IX81, Olympus) equipped with a TIRF objective (UAPON 100XOTIRF, Olympus) and a multiband band-pass filter set (dichroic mirror: Di01-R405/488/561/635-25 × 36, Semrock; emission filter: FF01-446/523/600/677-25, Semrock). Two lasers (Sapphire 488LP and Sapphire 561LP, coherent) were used as the light sources. Two acousto-optical modulators (AOM) were used to switch and control the intensities of the lasers. Collimating lenses (focal length: 10 mm, Lightpath) were used to couple the lasers to single-mode fibers. The output lasers were then collimated using an additional two collimating lenses (collimator, focal length: 75 mm, Thorlabs). A quarter-wave plate and a tunable diaphragm (each from Daheng Optics) were placed in front of each laser to control the polarization and beam size. The lasers were projected onto the active area of a DMD (0.7XGA 12° DDR, Texas Instruments; DMD driver, XDigit technology) through opposite light paths for independent adjustment. Next, the diffraction beams were focused by an achromatic lens, L1 (focal length: 100 mm, Thorlabs), onto the intermediate pupil plane, where a carefully designed stop mask was placed. Two achromatic lenses, L2 (focal length: 60 mm, Thorlabs) and L3 (focal length: 175 mm, Thorlabs), were used to focus the beams onto the BFP of the objective. We used a DMD to generate grid patterns that refracted the incident laser beam into one of zero order along the optical axis and a pair of order ± 1 with two opposite angles symmetric to the optical axis. A 60° or 120° change in the orientation of the grid pattern rotated the position of the diffracted beams of order ± 1 on the BFP but not the beam of zero order. By placing a stop mask at the conjugated plane of the BFP, we blocked the zero-order beam and other stray light [Fig. 1(c)] and focused only beam pairs of order ± 1 onto the objective BFP [Fig. 1(b)]. The interference produced by the beam pairs of order ± 1 were minimized using circularly polarized light and accumulated exposure time during one single camera shot. The emitted fluorescence was collected by the same objective and passed through a dichroic mirror, an emission filter and a tube lens (focal length: 180 mm). Before being captured by an electron-magnifying charge-coupled device (EMCCD iXon 897, Andor), the final fluorescence image was split by an image splitter (Optosplit, Cairn, Kent UK), which consisted of a two-dimensional split diaphragm (SP) that separated the field of view (FOV) of the EMCCD into two non-overlapping regions, a dichroic mirror (DM2, Di560, Semrock), three reflection mirrors and a pair of relay lenses, L4 and L5 (focal length: 150 mm). Raw images in Figs. 3, 4 and 5 were de-convolved using Richardson-Lucy algorithm in DeconvolutionLab plugin in ImageJ to enhance image contrast and remove background from out-of-focus emission.
3. Results
Adjustment of the incidence angle of the excitation light and generation of even illumination using the DMD. TIRF illumination generates a thin layer of an evanescent field close to the glass coverslip. The illumination intensity of this evanescent field decays exponentially with increasing distance from the interface, z, as follows:
| (1) |
The penetration depth of this evanescent field (d T) is determined by the illumination wavelength λ and the incidence angle θ as shown in the following equation:
| (2) |
n 1 and n 2 are the reflection indices of the coverslip and the cell culture medium, respectively [Fig. 2(a) , top middle]. A smaller θ will lead to an illumination field with a larger penetration depth d. When θ is decreased to just below the critical angle, a hollow-cone illumination mode (HOCE) replaces the TIRF illumination [Fig. 2(a), top middle and right]. The focal depth of the HOCE (d H) is determined by the incidence angle θ, the laser-beam size D 0 and the objective magnification M, as shown in the following equation:
Fig. 2.

Adjustment of the incidence angle of the excitation light and generation of even illumination using the DMD. (a) Measurement of the correlation between the incidence angle θ and the pattern period ω of the DMD. Upper left: the incidence angle of the excitation light was measured from its path on a glass cube. Lower left: measured correlation between the incidence angle θ and ω. Data were collected for ω from 4.0 lines to 5.0 lines for 488 nm and from 4.5 to 5.5 for 561 nm, which were equivalent to the penetration thickness from 70 nm to 10 μm for both 488 nm and 561 nm. The θ value was the average of the six laser beams created using the same ω but different azimuthal angles, and ∆θ was the largest deviation among the six measurements. Upper middle and right: HOCE and TIRF illuminations. Three lower right plots: calculation of the penetration depths d T in TIRF, d H in HOCE and in the critical phase as a function of θ. (b) Measurement of the laser intensity. Left: diagram of the six laser focal spots on the BFP separated by equal divisions. Right: there were deviations in laser energy of approximately ± 3.5% among the focal spots at the BFP produced using the same ω for the 488 nm laser and corresponding deviations of ± 5% for the 561 nm laser. Different values of ω (and therefore different incidence angles θ) also did not significantly affect the laser intensity at the focal spots. (c) The illumination field became homogeneous when the samples were excited by incident light originating from six focal spots on the BFP. One beam, two beams and six beams of 488 nm and 561 nm light were used to excite the Na-FITC and Rose Bengal samples, respectively. The scale bar in (c) represents 10 μm; the size of the images is 400 × 400 pixels.
| (3) |
In objective-type TIRF microscopy, the value of θ is determined by the radius r from the optical axis of the focal spot of the excitation light on the BFP of the objective, which can be adjusted by changing the integral periodic patterns (ω b) of the DMD in our system. Non-integral patterns can also be generated by digitally rotating the integral pattern on the DMD [Fig. 1(d)], thus permitting fine adjustment of the incidence angles. Therefore, we directly measured the incidence angle θ using a glass cube, and we determined its correlation with the pattern on the DMD (ω). As shown in the top left of Fig. 2(a), adjusting ω precisely changed the incidence angle θ, whereas the variation Δθ was held below 1% of the average measurement. Based on the measured θ, we then calculated d T and d H using the equations shown above and plotted their dependence on θ. In real experiments, we avoided using incidence angles at the “critical phase,” the transition between TIRF illumination and HOCE illumination [achieved via a 0.1° change of θ from 61.4° to 61.5° using a 100 × NA 1.49 objective; see the three graphs in the lower right of Fig. 2(a)].
To generate a homogeneous averaged illumination field, the different focal spots on the BFP of the objective must have similar energy. As shown in Fig. 2(b), this was indeed the case, which indicates that diffracting the light using DMD patterns of different periods and orientations did not affect the energy of the beams of order ± 1. Next, we tested the illumination field of a current TIRF microscope using homogeneous Na-FITC and Rose Bengal (SPECTRAL Applied Research, F6377 and 198250) solutions, which were excited by 488 nm and 561 nm lasers, respectively. As shown in Fig. 2(c), when only one beam was incident on the sample to generate TIRF illumination, small interference fringes were evident; using two focused beams that were symmetrical on the BFP to illuminate the samples reduced but did not eliminate the fringes. Only when the illumination was rapidly cycled among six focal spots positioned around a ring on the BFP during one EMCCD exposure period were the interference fringes fully eliminated, and the green and red emission channels became homogeneous to the same extent.
Growth of microtubules from 150 nm to less than 80 nm below the surface membrane observed via variable-angle TIRF imaging. We programmed the DMD to sequentially illuminate an INS-1 cell using a 488 nm laser with pattern periods of 4.1, 4.2 and 4.3, which corresponded to TIRF penetration depths of approximately 80 nm, 100 nm and 150 nm, respectively. The cells were transfected with EB3-GFP to label the plus ends of the microtubules. A time-lapse stack that contained images captured at three different illumination depths nearly simultaneously (a 100 ms exposure for each illumination depth, with a total exposure of 330 ms for a cycle) was acquired. The images were then split into three stacks corresponding to different illumination depths. As can be seen from Media 1 (115.3MB, AVI) and Figs. 3(a) , 3(b) and 3(c), we used pseudo-colors to label the different layers. The stacks for the 150 nm penetration depth were colored green; those for 100 nm were colored yellow, and those for 80 nm were colored red. The distributions of the microtubules at different Z positions were clearly different. To visualize the three-dimensional movement of the plus tips of the microtubules, we normalized the intensity of each stack, and merged them together to portray the three-dimensional structure of the EB3 at a given time point. In the merged results presented in Media 1 (115.3MB, AVI) and Fig. 3(d), the movement of the microtubule tips toward the plasma membrane can be seen as a color change from green to yellow to red. In Fig. 3(e), we show an example of the movement of one microtubule within the TIRF zone, as its plus-end tip approached the cell surface, transiently anchored the entire filament to the plasma membrane and finally departed. These data demonstrate that our siva-TIRF method can provide unprecedented axial information near the plasma membrane in comparison with other three-dimensional imaging methods.
Fig. 3.
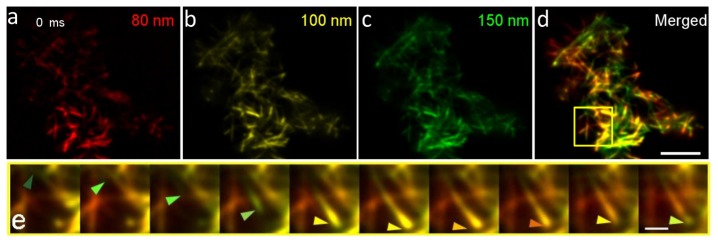
Growth of microtubules from 150 nm to less than 80 nm from the membrane in live INS-1 cell (Media 1 (115.3MB, AVI) ). (a)−(c) Fluorescence images of microtubules labeled with EB3-GFP under TIRF illumination with penetration depths of 80 nm (a), 100 nm (b) and 150 nm (c). (d) Color-merged image of (a)−(c). (a)−(d) are also shown in Media 1 (115.3MB, AVI) . (e) Time-lapse montage of the enlarged region indicated by the yellow box in (d); the arrows show the plus end of a microtubule approaching from deeper than 150 nm to the below 80 nm TIRF zone and then retreating once again. The scale bars in (a)−(d) represent 10 μm, whereas that in (e) represents 2 μm. Each image in the sequence is separated by 2 s from the preceding image.
Separating clathrin-coated vesicles (CCVs) from clathrin-coated pits (CCPs) using siva-TIRF. Clathrin-dependent endocytosis is the major recycling pathway in both excitable and non-excitable cells, which involves budding and internalization of plasma-membrane-associated CCPs [2]. Clathrin-coated fluorescent structures commonly observed in typical TIRF experiments are often interpreted as CCPs on the plasma membrane [2]. However, it has been shown that endocytosed vesicles sometimes retain clathrin coats and also reside in regions near the plasma membrane [14]. Therefore, it is a challenge to identify the nature of clathrin-coated structures observed under a TIRF microscope; the ability to discriminate among these structures will help to resolve the different physiological functions of surface-membrane-associated CCPs and cytosolic CCVs. As CCPs are typically smaller than 100 nm, we can separate cytosolic CCVs from plasma-membrane CCPs by imaging at penetration depths of 500 nm and 80 nm from the cell surface, respectively. As can be seen from the representative example shown in Fig. 4(c) , most clathrin puncta that could be observed under 80 nm illumination overlapped with the clathrin puncta seen under 500 nm TIRF illumination and were relatively stable throughout the duration of recording. On the other hand, some of the clathrin puncta observed under 500 nm illumination did not co-localize with any puncta observed under 80 nm illumination, and many of them exhibited rapid long-range lateral movement. As illustrated in the insets shown in Fig. 4(d), a clathrin punctum appeared in the cell interior, rapidly approached the cell surface, where a pre-existed CCP was docked, and then disappeared. This phenomenon was observed multiple times, implying that the movement of CCVs within cells may be correlated with plasma-membrane-associated CCPs.
Fig. 4.

Rapid motion of clathrin in a live cell from 500 nm to less than 80 nm from the membrane. (a) and (b) The spatial distribution of clathrin in an INS-1 cell under 80 nm (a), and 500 nm (b) TIRF illumination. (c) Merged images of clathrin puncta observed at different penetration depths. (d) Rapid motion of a clathrin punctum from deep in the cell to the plasma membrane. This montage of time-lapse images corresponds to the region indicated by a yellow frame in (c) with a time interval of 200 ms. The blue arrow represents the moving clathrin. The scale bars in (a)–(c) represents 10 μm, whereas that in (c) represents 500 nm
Distribution and movement of microtubules and clathrin puncta at various penetration depths investigated via dual-color live-cell imaging. Both CCPs and CCVs have been associated with microtubule networks [14, 15]. To explore whether microtubules contributes to the different dynamics of CCPs and CCVs, we performed fast dual-color imaging at different axial planes in INS-1 cells transfected with microtubule plus-end binding protein EB3-EGFP and clathrin-dsRED. By programming the DMD to illuminate an INS-1 cell at penetration depths of 80 nm, 150 nm and 500 nm, we recorded a time-lapse image stack (100 frames dual-color, 100 ms exposure time each color) at three planes, respectively. By combining images from different planes, we visualized three-dimensional spatiotemporal dynamics of microtubule network and clathrin [Figs. 5(a) -5(f)]. As representative examples shown in Fig. 5(g), a stationary CCPs (observed at penetration depth of 80 nm) co-localized with cortical microtubule filament, and exhibited minimal lateral movements during the entire period of recording. On the other hand, a CCV (observed at 500 nm but not 80 nm penetration depths) moved along with filaments within the cell interior. These data confirm that CCVs make long range movement within cells along microtubule [14], and CCPs stabilize on the plasma membrane via its physical interaction with cortical microtubules [15]. Taken together, our imaging data imply a critical role of microtubule in regulating clathrin-mediated endocytosis in INS-1 cells.
Fig. 5.

Distribution of microtubules and clathrin from 100 nm to 500 nm away from the membrane in a live cell, represented by dual-color images. (a)−(c) EB3 in an INS-1 cell under TIRF illumination with penetration depths of 80 nm (a), 150 nm (b), and 500 nm (c). (d)−(f) Clathrin in the same INS-1 cell with penetration depths of 80 nm (d), 150 nm (e), and 500 nm (f). (g) Time-lapse montage of the enlarged region indicated by the yellow box in (a) and (d). The arrows show a CCP that co-localized with a microtubule filament and remained stationary during the time of recording. (h) Time-lapse montage of the enlarged region indicated by the blue box in (c) and (f). The arrows show a CCV that co-localized with a microtubule filament and moved along the filament during the time of recording. Scale bar in (a)−(f) represents 10 μm, whereas that in (g)−(h) represent 1 μm. Each image in the sequence in (g) and (h) is separated by 6 s from the preceding image. Red: clathrin, Green: EB3.
4. Discussions
Comparison of siva-TIRF with other TIRF systems. In most sp/VA-TIRF systems, the incidence angle of laser is adjusted using a rotating mirror or a movable laser collimator. These configurations involve the mechanical movement of optical components that are slow (several Hz) and prone to vibration [4, 5]. Compared with these configurations, our approach is faster (1 kHz), which exceeds the current sampling speed of EMCCD and sCMOS cameras. van 't Hoff et al. have pioneered the use of two orthogonal AODs to spin and change the angle of the TIRF illumination beam, an approach that has achieved both a uniform illumination field and variable-angle TIRF imaging via the programmable adjustment of the incidence angle [7]. Their approach is faster than our method. However, AOD-based scanning has several disadvantages that limit its application in routine biological imaging. Because the effective aperture of an AOD is small (typically <2 mm), the width of the laser beam diffracted from the AOD is highly restricted. Therefore, a narrow beam is scanned through a cycle to cover the entire field of view (FOV), and this process is likely to affect the illumination intensity. In addition, the diffraction efficiency of an AOD depends on the orientation of the incoming light. In total, the authors have measured a sinusoidal power fluctuation of up to ~20% among different positions on the BFP for a 405 nm laser [7]. In our system, the power deviation among different focal spots on the BFP is much lower [less than ± 6% for different incidence angles for both 488 nm and 561 nm lasers; see Fig. 1(d)]. This lower power deviation can be attributed to the fact that a DMD is a reflective element and to the fact that relatively larger laser beams were used to illuminate the FOV. Typical AODs have a narrow range of working wavelengths, and those with large working wavelength ranges are often expensive. Moreover, the deflection angle and deflection efficiency of an AOD is highly dependent on the wavelength. Therefore, it is difficult to optimize a VA-TIRF system with two excitation wavelengths under TIRF illumination with different penetration depths. In contrast, a DMD reflects very large range of wavelengths (380−750 nm) with a fairly constant efficiency throughout that range. By slightly adjusting the projected angles of light of different colors, dual-color spinning VA-TIRF can be easily optimized, as illustrated in the example shown in Fig. 5. Moreover, the system might be extended to do polarized excitation in the future, as a recent paper showed [16]
Precision and versatility of the DMD-based system. Because all zero-order diffraction light (approximately 50% of the total power) and stray light diffracted by the pixelated pattern (approximately 1% of the total power) is blocked, the efficiency of the excitation-light usage of our system is low, which may be its primary disadvantage. This low efficiency limits the options for excitation-light sources to high-powered lasers, but this limitation no longer represents a significant constraint because the prices of such instruments are continually decreasing at present. A DMD has a fill factor of less than 100%, which will lead to the generation of stray excitation light that may affect the image quality. However, the gap between adjacent pixels on the DMD used in this study was very small (less than 1 μm, equivalent to 7 nm on the imaging plane) and did not significantly affect the image quality, as demonstrated in the images of biological samples presented in this paper. Because it is a pixelated component, another relevant question is whether a DMD can generate a sufficient range of patterns to allow the fine adjustment of the incidence angle. Using “shearing angle” to produce a fine, non-integral period of grid patterns, approximately 1000 different patterns can be produced between two adjacent integral periods, which, in theory, is equivalent to a 0.02 degree adjustment precision of the incidence angle [17]. We demonstrated here that the deviations among the incidence angles diffracted from patterns with the same period but different orientations were of less than 1 degree [Fig. 2(a)], suggesting that our system is precise and robust. The even-illumination TIRF field offered by this system yield more homogeneous images of single molecules, which will lead to more photons collected from individual single molecules on average. This can improve the localization precision of photo-activated localization microscopy (PALM) and stochastic reconstruction microscopy (STORM). Although using partially coherent light sources such as LED source could also significantly reduce the interference fringes in TIRF microscopy, DMD can be programmed to build other types of super-resolution microscopes, such as structured-illumination microscopes (SIMs) [18] and multifocal structured-illumination microscopes (MSIMs) [19].Therefore, by dynamically re-programming the patterns exhibited on the DMD, it is possible to combine multiple modalities of super-resolution imaging in the same system, which will offer a unique opportunity to obtain novel and complementary insights into biological processes.
Application of siva-TIRF in biological research. One problem with conventional TIRF microscopes is that they do not provide quantitative information regarding protein/organelle axial movement with respect to the plasma membrane. Theoretically, VA-TIRF can address this problem [12]. However, only a few studies concerning this possibility have been conducted [20–22], most likely because of the slow speed of mechanically changing the incidence angle in conventional TIRF systems and the difficulty in maintaining a stable AOD-based VA-TIRF system for biology labs. Using siva-TIRF, we demonstrated the nearly simultaneous imaging of different depths within live cells, a technique that provides super-resolution axial information and new insights into biological processes. For example, we revealed that the cortical microtubule network close to the plasma membrane is highly dynamic, with filaments continuously growing from the cell interior and retracting from the surface. We were also able to directly discriminate the endosomal population of clathrin puncta from CCPs under TIRF illumination, a task that has previously been performed only via indirectly inference [14]. Rapid, lateral movement of these CCVs was often terminated at pre-existed CCPs on the plasma membrane, suggesting that these intraceullular trafficking events are close correlated with endocytosis. Using fast dual-color multi-plane imaging, we show that both the mobile CCVs and the stationary CCPs are associated with microtubules, implying an important role of microtubule in regulating clathrin-mediated endocytosis. In the above experiments, different Z planes are all acquired within ~500 nm below the plasma membrane, well beyond the axial resolution of modern confocal/two photon microscopy (about 0.8~1 μm). Therefore, Our dual-color multi-plane TIRF imaging method will continue to provide novel insights of microtubule-regulated clathrin-mediated endocytosis in future studies, and it will also be useful for monitoring other endocytic processes such as viral infection in three-dimension in real time.
5. Conclusions
In summary, we designed and built a siva-TIRF microscope based on a DMD. Using this system, we are able to achieve a uniform field of illumination and rapidly switched among planes of different penetration depths under dual-color TIRF illumination. Such systems can be used to analyze three-dimensional spatial-temporal structures and dynamics within live cells, as demonstrated here through an exemplary investigation of spatiotemporal correlation of microtubules and clathrin in insulin-secreting cell lines. Our implementation provides a cost-effective, simple-to-build and highly optimized microscope for high-speed multi-plane dual-color imaging and quantitative analysis of cellular dynamics.
Acknowledgments
The authors would like to thank Dr. Xindong Huang for his help on using DMD device. The authors would also like to thank Liqiang Tang and COLD SPRING SCIENCE Corporation for technique support of microscope. The work was supported by grants from the Major State Basic Research Program of China (2013CB531200, 2012CB518200 and 2011CB809100), the National Key Technology R&D Program (SQ2011SF11B01041), the National Science Foundation of China (31221002, 81222020 and 31271205), and the Beijing Natural Science Foundation (7121008) and the Basic and Clinical Research Fund of Capital Medical University (No. 13JL19).
References and links
- 1.Axelrod D., “Cell-substrate contacts illuminated by total internal reflection fluorescence,” J. Cell Biol. 89(1), 141–145 (1981). 10.1083/jcb.89.1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merrifield C. J., Perrais D., Zenisek D., “Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells,” Cell 121(4), 593–606 (2005). 10.1016/j.cell.2005.03.015 [DOI] [PubMed] [Google Scholar]
- 3.Steyer J. A., Horstmann H., Almers W., “Transport, docking and exocytosis of single secretory granules in live chromaffin cells,” Nature 388(6641), 474–478 (1997). 10.1038/41329 [DOI] [PubMed] [Google Scholar]
- 4.Fiolka R., Belyaev Y., Ewers H., Stemmer A., “Even illumination in total internal reflection fluorescence microscopy using laser light,” Microsc. Res. Tech. 71(1), 45–50 (2008). 10.1002/jemt.20527 [DOI] [PubMed] [Google Scholar]
- 5.Mattheyses A. L., Shaw K., Axelrod D., “Effective elimination of laser interference fringing in fluorescence microscopy by spinning azimuthal incidence angle,” Microsc. Res. Tech. 69(8), 642–647 (2006). 10.1002/jemt.20334 [DOI] [PubMed] [Google Scholar]
- 6.Beuthan J., Minet O., Helfmann J., Herrig M., Müller G., “The spatial variation of the refractive index in biological cells,” Phys. Med. Biol. 41(3), 369–382 (1996). 10.1088/0031-9155/41/3/002 [DOI] [PubMed] [Google Scholar]
- 7.van ’t Hoff M., de Sars V., Oheim M., “A programmable light engine for quantitative single molecule TIRF and HILO imaging,” Opt. Express 16(22), 18495–18504 (2008). 10.1364/OE.16.018495 [DOI] [PubMed] [Google Scholar]
- 8.Axelrod D., Omann G. M., “Combinatorial microscopy,” Nat. Rev. Mol. Cell Biol. 7(12), 944–952 (2006). 10.1038/nrm2062 [DOI] [PubMed] [Google Scholar]
- 9.Mattheyses A. L., Simon S. M., Rappoport J. Z., “Imaging with total internal reflection fluorescence microscopy for the cell biologist,” J. Cell Sci. 123(21), 3621–3628 (2010). 10.1242/jcs.056218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zenisek D., Steyer J. A., Almers W., “Transport, capture and exocytosis of single synaptic vesicles at active zones,” Nature 406(6798), 849–854 (2000). 10.1038/35022500 [DOI] [PubMed] [Google Scholar]
- 11.Lin J., Hoppe A. D., “Uniform total internal reflection fluorescence illumination enables live cell fluorescence resonance energy transfer microscopy,” Microsc. Microanal. 19(2), 350–359 (2013). 10.1017/S1431927612014420 [DOI] [PubMed] [Google Scholar]
- 12.Lanni F., Waggoner A. S., Taylor D. L., “Structural organization of interphase 3T3 fibroblasts studied by total internal reflection fluorescence microscopy,” J. Cell Biol. 100(4), 1091–1102 (1985). 10.1083/jcb.100.4.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tokunaga M., Imamoto N., Sakata-Sogawa K., “Highly inclined thin illumination enables clear single-molecule imaging in cells,” Nat. Methods 5(2), 159–161 (2008). 10.1038/nmeth1171 [DOI] [PubMed] [Google Scholar]
- 14.Keyel P. A., Watkins S. C., Traub L. M., “Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy,” J. Biol. Chem. 279(13), 13190–13204 (2003). 10.1074/jbc.M312717200 [DOI] [PubMed] [Google Scholar]
- 15.Montagnac G., Meas-Yedid V., Irondelle M., Castro-Castro A., Franco M., Shida T., Nachury M. V., Benmerah A., Olivo-Marin J. C., Chavrier P., “αTAT1 catalyses microtubule acetylation at clathrin-coated pits,” Nature 502(7472), 567–570 (2013). 10.1038/nature12571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson D. S., Toledo-Crow R., Mattheyses A. L., Simon S. M., “Polarization-Controlled TIRFM with Focal Drift and Spatial Field Intensity Correction,” Biophys. J. 106(5), 1008–1019 (2014). 10.1016/j.bpj.2013.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiolka R., Beck M., Stemmer A., “Structured illumination in total internal reflection fluorescence microscopy using a spatial light modulator,” Opt. Lett. 33(14), 1629–1631 (2008). 10.1364/OL.33.001629 [DOI] [PubMed] [Google Scholar]
- 18.Dan D., Lei M., Yao B., Wang W., Winterhalder M., Zumbusch A., Qi Y., Xia L., Yan S., Yang Y., Gao P., Ye T., Zhao W., “DMD-based LED-illumination super-resolution and optical sectioning microscopy,” Sci. Rep. 3, 1116 (2013). 10.1038/srep01116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.York A. G., Parekh S. H., Dalle Nogare D., Fischer R. S., Temprine K., Mione M., Chitnis A. B., Combs C. A., Shroff H., “Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy,” Nat. Methods 9(7), 749–754 (2012). 10.1038/nmeth.2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohrbach A., “Observing secretory granules with a multiangle evanescent wave microscope,” Biophys. J. 78(5), 2641–2654 (2000). 10.1016/S0006-3495(00)76808-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loerke D., Stühmer W., Oheim M., “Quantifying axial secretory-granule motion with variable-angle evanescent-field excitation,” J. Neurosci. Methods 119(1), 65–73 (2002). 10.1016/S0165-0270(02)00178-4 [DOI] [PubMed] [Google Scholar]
- 22.Olveczky B. P., Periasamy N., Verkman A. S., “Mapping fluorophore distributions in three dimensions by quantitative multiple angle-total internal reflection fluorescence microscopy,” Biophys. J. 73(5), 2836–2847 (1997). 10.1016/S0006-3495(97)78312-7 [DOI] [PMC free article] [PubMed] [Google Scholar]