

Abstract
Biochemical assays with recombinant human MHC II molecules can provide rapid, quantitative insights into immunogenic epitope identification, deletion, or design1,2. Here, a peptide-MHC II binding assay is scaled to 384-well format. The scaled down protocol reduces reagent costs by 75% and is higher throughput than previously described 96-well protocols1,3-5. Specifically, the experimental design permits robust and reproducible analysis of up to 15 peptides against one MHC II allele per 384-well ELISA plate. Using a single liquid handling robot, this method allows one researcher to analyze approximately ninety test peptides in triplicate over a range of eight concentrations and four MHC II allele types in less than 48 hr. Others working in the fields of protein deimmunization or vaccine design and development may find the protocol to be useful in facilitating their own work. In particular, the step-by-step instructions and the visual format of JoVE should allow other users to quickly and easily establish this methodology in their own labs.
Keywords: Biochemistry, Issue 85, Immunoassay, Protein Immunogenicity, MHC II, T cell epitope, High Throughput Screen, Deimmunization, Vaccine Design
Introduction
Proteins are the fastest growing class of therapeutic agents6, and the rapid expansion of biotherapeutic pipelines has focused increasing attention on the challenges associated with development and use of protein drugs. One unique consideration stems from the fact that, in a healthy and functioning immune system, all extracellular proteins are sampled by antigen presenting cells (APCs). Once internalized by APCs, a protein is cleaved into small peptide fragments, and putative immunogenic segments are loaded into the groove of class II major histocompatibility complex proteins (MHC II). The peptide-MHC II complexes are then displayed on the APC surface, and true immunogenic peptides, termed T cell epitopes, form ternary MHC II-peptide-T cell receptor complexes with cognate CD4 T cell surface receptors7. This critical molecular recognition event initiates a complex signaling cascade, which results in T cell activation, the release of cytokines, CD4 T cell-mediated B cell maturation, and ultimately production of circulating IgG antibodies that bind to and clear the offending exogenous protein. Thus, immunogenic proteins might be deimmunized by identifying constituent T cell epitopes and mutating key residues responsible for MHC II complex formation. It bears noting, however, that T cell epitopes can be numerous and broadly distributed throughout immunogenic proteins, and the majority of epitope-deleting mutations are likely to cause an inadvertent loss of protein function or stability. Therefore, engineering deimmunized biotherapies can be a complex and technically challenging objective, but there exist several examples of successful T cell epitope deletion projects3,5,8-12. Unlike grafting-based "humanization", which is largely restricted to antibody therapeutics, epitope deletion can be applied to essentially any protein target regardless of sequence, structure, function, or the availability of homologous human scaffolds. The first step to implementing such an approach is identification of key peptide epitopes embedded within the target protein sequence.
High throughput biochemical assays using synthetic peptides and recombinant human MHC II molecules can provide rapid preliminary insights into epitope identification and mitigation1,3-5. These ELISA type assays can be a powerful complement to other protein/vaccine design and development tools. For example, one well established experimental approach to epitope mapping relies on time, labor, and resource intensive ex vivo cell proliferation assays 15. Briefly, the primary sequence of a target protein is first divided into a panel of overlapping peptides, often 15-mers with 12 residues overlap between adjacent peptides. The peptide panel is chemically synthesized and the immunogenicity of each peptide is tested in one of several different immunoassays that employ peripheral blood mononuclear cells (PBMC) isolated from human donors13,14. To provide greater confidence in results, peptides are typically tested in replicate with PBMC from 50 or more different donors. In cases where deimmunization is the ultimate objective, the work is compounded further by the need to produce additional panels of mutated peptides and test the new peptide panels in PBMC assays before introducing any deimmunizing mutations into the full length protein for subsequent functional analysis10. While these cellular assays remain the gold standard for assessing immunogenic potential in human patients, the efficiency of such an exhaustive approach might be improved by prefiltering putative immunogenic epitopes using a rapid and high throughput MHC II-peptide binding assay.
Likewise, biochemical peptide-MHC II binding assays can be combined with predictive in silico methods to radically accelerate the epitope identification process. There exist a variety of computational tools for T cell epitope prediction; examples include ProPred16, MHCPred17, SVRMHC18, ARB19, SMM-align20, NetMHCIIpan21 as well as proprietary tools such as EpiMatrix by EpiVax22. Likewise, epitope predictors have recently been combined with other bioinformatics and molecular modeling tools to yield integrated protein deimmunization algorithms designed to mitigate the risk that deimmunizing mutations might disrupt protein structure and function23-26. While several epitope predictors have proven to be reasonably accurate27,28, computational results invariably require experimental validation. Rapid, high throughput, and cost effective experimental methods are best suited as a preliminary filter for in silico epitope predictions.
In a similar vein, epitope predictors can drive antigen selection for reverse vaccinology29,30. For example, advances in bioinformatics have yielded whole genome screens that rapidly identify vaccine candidates in the form of whole proteins or peptide epitopes extracted from pathogen proteomes. While this enabling technology is reshaping discovery and development of protective vaccines, it introduces a new challenge in the form of intractably large lists of immunogenic vaccine candidates. High throughput peptide-MHC II binding assays can guide epitope selection by quantifying peptide binding affinity and binding promiscuity among multiple MHC II alleles. As with protein deimmunization, such experimental methods are ultimately required to validate computational prediction of promising vaccine leads.
Here, a peptide-MHC II binding assay scaled to 384-well format is described. The protocol is highly parallelized and reduces reagent costs by 75% compared to previously described 96-well plate formats1,3-5. Using a single liquid handling robot, this method allows one researcher to easily analyze approximately ninety test peptides in triplicate over a range of eight concentrations and four MHC II allele types in less than 48 hr. This article describes the setup of one 384-well ELISA plate for analysis of seven experimental peptides against one MHC II allele, but spread sheet calculators are provided as supplemental material so as to easily scale the experiment to any number of desired peptides and/or MHC II molecules.
Protocol
Four major activities comprise the peptide-MHC II binding assay: 1-Binding: Test peptides compete with labeled control peptides for solution phase binding of soluble MHC II proteins. Binding is measured over a wide range of test peptide concentrations. 2-Capture: After the binding reaction approaches equilibrium, peptide-MHC II complexes are captured and separated from unbound peptide and protein by conformation-dependent recognition with an immobilized antibody. 3-Detection: Captured control peptide is quantitatively detected using time-resolved fluorescence. 4-Analysis: Spectroscopic data are processed, plotted and analyzed to ascertain dose-dependent binding properties of test peptide and MHC II proteins.
At various steps in the procedure below, the use of a liquid handling robot is recommended if one is available. Particularly in a 384-well format, automated dispensing and dilution of liquids minimizes user error, results in more consistent well-to-well volumes, and yields narrower 95% confidence intervals for IC50 values compared to manual 96-well assays (data not shown). If a liquid handling robot is not available, the steps annotated with "(liquid handling robot)" may be done by hand. Similarly, if possible, an automated plate washer that is compatible with the 384-well format is recommended. This effectively standardizes the plate washing process. If a plate washer is not available, the steps annotated with "(plate washer)" may be done by hand.
1. Coat the ELISA Plate (Day 1)
- Dilute L243 antibody stock (0.5 mg/ml) in Borate Buffer to a working concentration of 10 µg/ml.
- To coat a single 384-well plate, add 200 µl of stock antibody to 9.8 ml of Borate Buffer. (See supplemental spreadsheet calculator for alternative scaling).
Add 25 µl of the L243 antibody solution to each well of a 384-well high-binding white ELISA plate. (liquid handling robot)
Seal the plate with a polyester film and incubate overnight at 4 °C. Note: Each 384-well ELISA plate can accommodate up to 15 test peptides at eight dilutions for one MHC II allele, as well as the positive and negative controls. This will ultimately yield triplicate measurements.
2. Make the Test Peptide Dilutions (Day 1)
Starting with a 10 mM stock of each test peptide in DMSO, make the following dilution series in Citrate Phosphate Buffer using 384-well polypropylene plates (Table 1). (liquid handling robot) Note: A full 384-well polypropylene plate requires three separate ELISA plates. One third of a polypropylene plate will require only one ELISA plate (Figure 1).
| Dilution Number | Volume to take from prior dilution/stock | Volume Citrate Buffer to add | Conc. of Dilution | Conc. in Binding Reaction | Conc. in Neutralized ELISA |
| (μl) | (μl) | (μM) | (μM) | (μM) | |
| 1 | 0.7 | 35.1 | 195.531 | 97.765 | 48.883 |
| 2 | 14.3 | 14.3 | 97.765 | 48.883 | 24.441 |
| 3 | 7.1 | 28.7 | 19.389 | 9.695 | 4.847 |
| 4 | 14.3 | 14.3 | 9.695 | 4.847 | 2.424 |
| 5 | 7.1 | 28.7 | 1.923 | 0.961 | 0.481 |
| 6 | 14.3 | 14.3 | 0.961 | 0.481 | 0.24 |
| 7 | 7.1 | 28.7 | 0.191 | 0.095 | 0.048 |
| 8 | 14.3 | 14.3 | 0.095 | 0.048 | 0.024 |
| Remove and discard 7.1 μl of peptide solution from dilution number 8. |
Table 1. Preparation of test peptide dilution series.
 Figure 1. Plate Map of Peptide Binding Assay. (A) Map of the peptide dilutions in polypropylene 384-well plates. Each polypropylene plate can accommodate three groups of 15 peptides in competition binding reactions against a single MHC II allele. (B) After the binding reaction approaches equilibrium, each peptide group is transferred, in triplicate, to a separate 384-well ELISA plate that has been precoated with anti-MHC II antibody.
Figure 1. Plate Map of Peptide Binding Assay. (A) Map of the peptide dilutions in polypropylene 384-well plates. Each polypropylene plate can accommodate three groups of 15 peptides in competition binding reactions against a single MHC II allele. (B) After the binding reaction approaches equilibrium, each peptide group is transferred, in triplicate, to a separate 384-well ELISA plate that has been precoated with anti-MHC II antibody.
3. Prepare the MHC II Master Mix (Day 1)
- Make the Reaction Buffer
- Add 80 µl of 1 mM Pefa Bloc and 60 mg of octyl-β-D-glucopyranaside to 3,920 µl of Citrate Phosphate Buffer. (See supplemental spreadsheet calculator for alternative scaling).
Dilute selected MHC II stock solutions to 101 nM in Reaction Buffer using a 15 ml conical tube (Table 2). Note: MHC II concentration will ultimately be 50 nM in the binding reaction and 25 nM in the neutralized ELISA assay. The MHC II stock concentrations will vary depending on manufacturer lot number. (See supplemental spreadsheet calculator).
| MHC II Allele DRB1*: | 1501 |
| MHC II Stock Concentration (mg/ml) | 1.3 |
| MHC II Stock Concentration (mM) | 20 |
| Vol. MHC II Stock to Add (ml) | 14.65 |
| Vol. Reaction Buffer to Add (ml): | 2885.35 |
| MHC II Master Mix Concentration (nM) | 101 |
Table 2. Preparation of MHC II master mix.
4. Prepare the Negative and Positive Controls (Day 1)
Add 21.5 µl of Citrate Phosphate Buffer to the "negative control" well of the 384-well polypropylene plate from step 2.1 (Figure 1A).
Add 21.5 µl of Citrate Phosphate Buffer to the "positive control" well of the 384-well polypropylene plate. Note: the "positive control" will be completed in section 5, below.
Remove 49.5 µl of the MHC II Master Mix from step 3.2 and pipette into a 1.5 ml Eppendorf tube.
Add 0.5 µl of Citrate Buffer to the 49.5 µl of the MHC II Master Mix from step 4.3.
Add 21.5 µl of the concentration adjusted MHC II Master Mix from step 4.4 to the "negative control" well of the 384-well polypropylene plate (Figure 1A). Note: this sample represents the zero-signal negative control containing MHC II protein, NO control peptide, and NO test peptide.
5. Add the Appropriate Control Peptide to the MHC II Master Mix (Day 1)
| MHC II Allele DRB1* | Biotinylated Control Peptide | (residues) | Sequence |
| 0101 | Flu-HA-B | (306-318) | Biotin-(Ahx)(Ahx)PRYVKQNTLKLAT-amide |
| 0301 | Myoglobin | (137-148) | Biotin-(Ahx)-(Ahx)-LFRKDIAAKYKE-OH |
| 0401 | YAR-B | (1-14) | Biotin-(Ahx)(Ahx)YARFQSQTTLKQKT-OH |
| 0701 | TetTox-B | (830-843) | Biotin-(Ahx)(Ahx)QYIKANSKFIGITE-OH |
| 1101 | Flu-HA-B | (306-318) | Biotin-(Ahx)(Ahx)PRYVKQNTLKLAT-amide |
| 1501 | MBP-B | (84-102) | Biotin-(Ahx)(Ahx)NPVVHFFKNIVTPRTPPPS-OH |
*We recommend ordering 10 mg scale for economy. Table 3. Control Peptides for Various MHC II Alleles.*
- Dilute the Control peptide (stored at 400 µM in DMSO) 1:20 in fresh DMSO to yield a diluted stock at 20 µM.
- Add 25 µl 400 µM control peptide to 475 µl DMSO. Note: this is a large excess, but allows accurate handling of viscous DMSO solutions.
- Further dilute the Control Peptide from step 5.1 (20 mM) 1:100 into the MHC II Master Mix from step 3.2.
- Add 28.8 µl of Control Peptide diluted in step 5.2 to the remaining 2,850.5 µl of the MHC II Master Mix.
Add 21.5 µl of the MHC II Master Mix with control peptide (step 5.2) to the positive control well of the 384-well polypropylene plate (step 4.2) (Figure 1A). Note: this sample represents the high-signal positive control containing MHC II protein, control peptide, and NO test peptide.
6. Make the Binding Reaction (Day 1)
- Add the MHC II Master Mix containing Control Peptide (step 5.2) to each of the Test Peptide dilutions (step 2.1) at a 1:1 ratio to create the Binding Reaction.
- Add 21.5 µl of MHC II Master Mix containing Control Peptide from step 5.2 to 21.5 µl of each Test Peptide dilution from step 2.1. (liquid handling robot)
Seal the Binding Reaction with a polyester film and incubate 12-24 hr in a non-CO2 controlled incubator at 37 °C without shaking.
7. Neutralize and Transfer the Binding Reaction (Day 2)
Remove the 384-well polypropylene plate containing the Binding Reaction (step 6.2) from the 37 °C incubator.
- Dilute the Binding Reaction 1:1 with Neutralization Buffer. (liquid handling robot)
- Add 43 µl of the Neutralization Buffer to each Binding Reaction well.
Remove the ELISA plate (step 1.3) from 4 °C and wash 3x with 60 ml/well of PBS-0.05% Tween 20. (plate washer)
Transfer 25 μl of each neutralized Binding Reaction from step 7.2 into triplicate wells of the antibody-coated 384-well ELISA plate from step 7.3. (Figure 1) (liquid handling robot)
Cover the ELISA plate with a polyester film and place in a 37 °C incubator for 2.5 hr or in a 4 °C refrigerator overnight.
8. ELISA Development (Day 2 or 3)
Remove the ELISA plate from step 7.5, wash 3 times with 60 µl/well of PBS-0.05% Tween. (plate washer)
- Dilute Streptavidin-Europium stock solution (0.1 mg/ml streptavidin, 7 Eu3+/streptavidin) 1,000-fold in the DELFIA Assay Buffer.
- For one 384-well plate, dilute 10 µl Streptavidin-Europium in 10 ml Assay Buffer.
Add 25 µl of the diluted Streptavidin-Europium to each well of the 384-well ELISA plate from step 8.1. (liquid handling robot)
Cover the plate with a polyester film and place in the dark at room temperature for 1 hr. Note: During the 1 hr incubation, remove the Enhancement Solution from the refrigerator and set aside 10 ml/plate in the dark at room temperature.
Following the 1-hr incubation, wash the 384-well ELISA plate from step 8.3 3x with 60 µl/well of PBS-0.05% Tween. (plate washer)
Add 25 µl of Enhancement Solution to each well of the 384-well ELISA plate from step 8.5. (liquid handling robot)
Cover the 384-well ELISA plate with a polyester film and allow the plate to sit in the dark at room temperature for 10-15 min.
Following the 10-15 min incubation, read the fluorescence of the 384-well ELISA plate from step 8.7 using a time resolved fluorescent plate reader with Europium settings (for example, Start Int: 200, Stop Int: 1,000, Ex. 340, Em. 615, Cutoff: None, PMT: Auto, Reads/Well: 50).
9. Data Analysis
Enter data in XY format graph with 3 replicate values in side-by-side columns (X=test peptide concentration; Y=fluorescence measurement).
Log transform the X values for all data: X=log(X)
Fit the log transformed data with the one-site competitive binding model to extract the IC50 value.
Constrain the bottom value to the average negative control value.
Globally fit the top value and insure that the global fit parameter is below or equal to the positive control.
Representative Results
The mature peptide sequence of Enterobacter cloacae P99 beta-lactamase (BLA) (Genbank ID# X07274.1) was analyzed with ProPred16 for putative peptide binders to MHC II allele DRB1*1501 (Table 4). ProPred identified 117 nonamer peptides with an obligatory P1 anchor residue (i.e. an M, L, I, V, F, Y, or W at position 1, which is required for MHC II binding31). At a 5% threshold, only peptides with scores greater than or equal to 2.6 are likely binders. Thus, at the 5% threshold, only the top 11 peptides are predicted to bind MHC II DRB1*1501.
 Table 4.
Top-scoring ProPred predictions for BLA peptides and MHC II allele DRB1*1501.
Table 4.
Top-scoring ProPred predictions for BLA peptides and MHC II allele DRB1*1501.
A representative panel of predicted epitopes was selected for analysis in our MHC II binding assay (Table 4, bold entries). BLA peptide fragments of fifteen residues were chemically synthesized such that putative nonamer MHC II epitopes were embedded within the synthetic protein fragments. While the MHC II binding groove itself accommodates only nine amino acids, evidence suggests that flanking sequences can influence peptide-MHC II interactions, and synthetic peptides of 15-20 residues are therefore commonly employed15. To represent the complexity of overlapping epitopes that might occur in biologically processed protein fragments, we tested synthetic sequences that contained (i) single predicted binders, (ii) single predicted nonbinders, (iii) multiple predicted binders, (iv) multiple predicted nonbinders, or (v) a mixture of predicted binders and nonbinders (Tables 4 and 5).
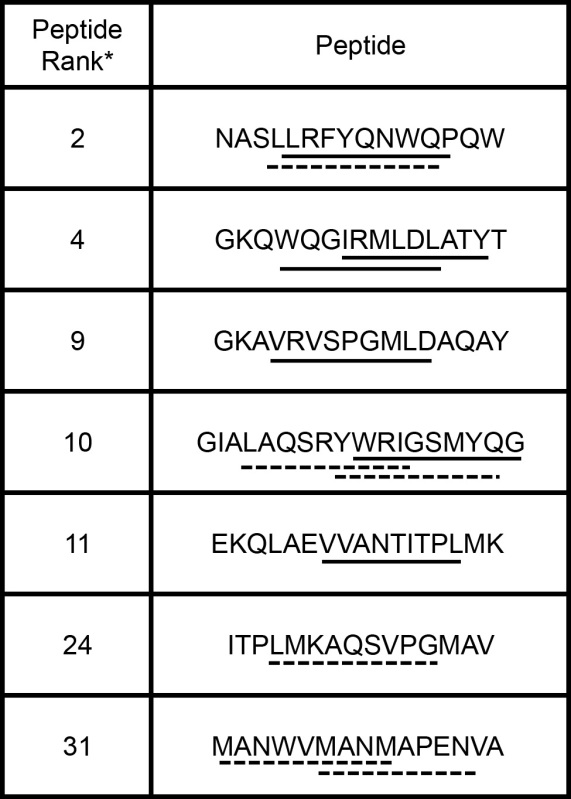 Table 5.
List of Chemically Synthesized Peptides.
Table 5.
List of Chemically Synthesized Peptides.
The capacity of these synthetic peptides to compete with the biotinylated MBP-B control peptide (Table 3) for binding to MHC II DRB1*1501 was analyzed as described in the protocol above. Competitive binding curves for the synthetic peptides are shown in Figure 2. IC50 values were calculated by fitting the log-transformed data using the one-site competitive binding, nonlinear fit function of Prism (Table 6). As seen in Figure 2, the peptides naturally partition into three groups: strong binders with IC50 < 1 μM (peptides 2 and 10); moderate binders with 1 μM ≤ IC50 < 100 μM (peptides 4, 9, 11, and 24); weak binders with IC50 ≥ 100 μM (peptide 31).
 Figure 2. Competitive Binding Curve of BLA Peptides for MHC II allele DRB1*1501. Tight binders are fit with orange lines, moderate binders green lines, and the weak binder a maroon line.
Figure 2. Competitive Binding Curve of BLA Peptides for MHC II allele DRB1*1501. Tight binders are fit with orange lines, moderate binders green lines, and the weak binder a maroon line.
| Peptide Rank | IC50 (μM) | 95% C.I. for IC50 (μM) | Quality of Fit (R2) |
| 2 | 0.1199 | 0.09404-0.1529 | 0.98 |
| 4 | 15.1 | 11.83-19.28 | 0.87 |
| 9 | 17.11 | 13.39-21.88 | 0.81 |
| 10 | 0.2743 | 0.2149-0.3502 | 0.98 |
| 11 | 10.49 | 8.252-13.34 | 0.98 |
| 24 | 4.787 | 3.769-6.082 | 0.96 |
| 31 | 190.6 | 137.7-263.9 | 0.80 |
Table 6. Calculated IC50 values of BLA peptide fragments for DRB1*1501.
Discussion
Biotherapeutics have established themselves as a cornerstone of modern medicine, representing four of the top five selling drugs in 201232. The biopharmaceutics sector has shown sustained growth for several years6, and the ongoing development of novel agents as well as the emergence of biosimilars has expanded biopharmaceutical pipelines. Looking to the future, assessing and mitigating the immunogenicity of protein therapeutics will become an integral part of early stage biotherapeutic development. To facilitate this process, biotechnologists may avail themselves of computational methods for epitope prediction16-22 as well as integrated protein design algorithms that seek to reduce immunogenicity while maintaining function3,23-26. Likewise, vaccine design and development may capitalize on the power of predictive algorithms4, and the continued maturation of reverse vaccinology is expected to yield protective vaccines for a broad array of infectious agents that have eluded previous efforts33. While these computational tools have the capacity to radically accelerate drug and vaccine development processes, the results of in silico predictions must ultimately be filtered further so as to identify a tractable number of top performing candidates. Recent advances in quantitative peptide-MHC II analysis have included the development of engineered yeast cell surface display systems34 and microbead based Luminescent Oxygen Channeling Immunoassays35, but in vitro ELISA type assays remain the workhorse for quantifying binding between specific MHC II and peptide pairs. Here, a microliter scale procedure for rapid, quantitative, and cost effective analysis of peptide epitope binding to human MHC II immune molecules is described. These methods may be used as an early stage experimental tool in the funneling process.
For the purposes of demonstration, the sequence of BLA was analyzed for putative MHC II binding peptides using the ProPred web server. Seven of these peptides were chemically synthesized and experimentally tested for binding to soluble human MHC II DRB1*1501. Similar to other analyses of predictor accuracy 28, ProPred binding prediction correlated reasonably well with experimentally measured IC50 for the DRB1*1501 MHC II allele. It is also important to note that results are sensitive to variations in concentrations and care should be taken to ensure accurate dilutions of MHC, control peptides, and test peptides. Likewise, a limitation of this method is that the IC50 values are relative to the concentrations of MHC and control peptides. Among the BLA peptide test set, the highest ranked ProPred epitope exhibited the strongest binding to recombinant DRB1*1501. Similarly, all other predicted epitope binders exhibited moderate to high MHC II affinities. As expected, synthetic peptides containing overlapping predicted binding epitopes and nonbinding epitopes were found to bind MHC II in all cases. Peptide 31, which spanned two of the lowest ranked predicted epitopes, proved to be the weakest MHC II binder. Peptide 24, however, was found to bind MHC II with moderate affinity despite the fact that its only highly ranked constituent epitope (epitope 24) was not a predicted binder at the 5% threshold. Interestingly, epitope 24 was identified in a previous study aimed at deimmunizing BLA10. In those experiments, synthetic peptide 24 was shown to be highly immunogenic in human PBMC assays. Thus, in this one case, our MHC II binding assay proved to be predictive of immunogenicity while ProPred did not. In aggregate the results demonstrate the utility of computational predictions in guiding researchers towards likely immunogenic epitopes, but they also underscore the importance of high throughput experimental methods as an integral component of the protein/vaccine design process.
Importantly, this 384-well experimental design is highly parallelized and permits rigorous analysis of up to 15 peptides and one MHC II allele per 384-well ELISA plate. Using a single liquid handling robot, this method allows one researcher to analyze ninety test peptides against four MHC II allele types in less than 48 hours. This design includes eight different test peptide concentrations and triplicate measurement at each concentration. Therefore, the user is assured robust and reproducible quantitative results. Others working in the fields of protein therapeutics or vaccine design and development may find this protocol to be useful in facilitating their work in protein deimmunization or immunogenicity analysis.
| Citrate Phosphate Buffer (22.2 mM Citric Acid, 55.6 mM Dibasic Na2HPO4, pH 5.4) |
| 222 ml 0.1 M citric acid |
| 278 ml 0.2 M dibasic Na2HPO4 |
| 500 ml MilliQ H2O |
| Mix citric acid, sodium phosphate, and 450 ml of MilliQ H2O. Adjust pH to 5.4 and q.s. to 1 L. |
| This solution is stable at room temperature. |
| Binding Reaction Buffer |
| Citrate Phosphate Buffer, pH 5.4 with |
| 2% v/v 1 mM PefaBloc |
| 1.5% w/v Octyl-β-D-glucopyranaside |
| Use right away. |
| 1 mM PefaBloc |
| 1 ml MilliQ H2O |
| 25 mg PefaBloc SC (powder) |
| Make aliquots and store at -20 ˚C. |
| Borate Buffer Formula (12.5 mM Sodium tetraborate decahydrate, pH 8.2) |
| 1.19 g sodium tetraborate decahydrate (Sigma) |
| 250 ml MilliQ H2O |
| Dissolve sodium tetraborate decahydrate in 225 ml of MilliQ H2O. Adjust pH to 8.2 and q.s. to 250 ml. |
| This solution is stable at room temperature. |
| PBS-Tween (2.7 mM KCl, 1.5 mM KH2PO4, 136.9 mM NaCl, 8.9 mM Na2HPO4•7H2O, 0.05% Tween, pH 7.4) |
| 4.5 L MilliQ H2O |
| 500 ml 10x Dulbecco’s PBS (DPBS) |
| 2.5 ml Tween 20 |
| This solution is stable at room temperature. |
| Neutralization Buffer (50 mM Trizma HCl, pH 8.0) |
| 475 ml MilliQ H2O |
| 25 ml 1 M Trizma HCl |
| Mix and adjust pH to 8.0 |
| This solution is stable at 4 °C. |
Table 7. Buffer Recipes.
Disclosures
Leonard Moise is employed by and holds stock options in EpiVax, Inc., a privately owned biotechnology company located in Providence, RI. The work contained in this research report is free of any bias that might be associated with the commercial goals of the company.
Acknowledgments
This work was supported by NIH grants R01-GM-098977 and R21-AI-098122 to CBK and KEG. RSS was supported in part by a Luce Foundation Fellowship and in part by a Thayer Innovation Program Fellowship from the Thayer School of Engineering.
References
- Steere AC, et al. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J. Exp. Med. 2006;203:961–971. doi: 10.1084/jem.20052471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddrizzani L, et al. Different modes of peptide interaction enable HLA-DQ and HLA-DR molecules to bind diverse peptide repertoires. J. Immunol. 1997;159:703–711. [PubMed] [Google Scholar]
- Osipovitch DC, et al. Design and analysis of immune-evading enzymes for ADEPT therapy. Protein Eng. Design. 2012;25:613–623. doi: 10.1093/protein/gzs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L, et al. In silico-accelerated identification of conserved and immunogenic variola/vaccinia T-cell epitopes. Vaccine. 2009;27:6471–6479. doi: 10.1016/j.vaccine.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L, et al. Effect of HLA DR epitope de-immunization of Factor VIII in vitro and in vivo. Clin. Immunol. 2012;142:320–331. doi: 10.1016/j.clim.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal SR. What's fueling the biotech engine-2011 to 2012. Nat. Biotechnol. 2012;30:1191–1197. doi: 10.1038/nbt.2437. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Ann. Rev. Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- Warmerdam PAM, et al. Elimination of a human T-cell region in staphylokinase by T-cell screening and computer modeling. Thrombosis Haemostasis. 2002;87:666–673. [PubMed] [Google Scholar]
- Jones TD, et al. Identification and removal of a promiscuous CD4+T cell epitope from the C1 domain of factor VIII. J. Thrombosis Haemostasis. 2005;3:991–1000. doi: 10.1111/j.1538-7836.2005.01309.x. [DOI] [PubMed] [Google Scholar]
- Harding FA, et al. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol. Cancer. 2005;4:1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- Mazor R, et al. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E3597–E3603. doi: 10.1073/pnas.1218138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, et al. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern F, LiPira G, Gratama JW, Manca F, Roederer M. Measuring Ag-specific immune responses: understanding immunopathogenesis and improving diagnostics in infectious disease, autoimmunity and cancer. Trends Immunol. 2005;26:477–484. doi: 10.1016/j.it.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Li Pira G, Ivaldi F, Moretti P, Manca F. High throughput T epitope mapping and vaccine development. J. Biomed. Biotechnol. 2010;2010:325720. doi: 10.1155/2010/325720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeister B, et al. Mapping T cell epitopes by flow cytometry. Methods. 2003;29:270–281. doi: 10.1016/s1046-2023(02)00349-3. [DOI] [PubMed] [Google Scholar]
- Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–1237. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- Guan P, Doytchinova IA, Zygouri C, Flower DR. MHCPred: bringing a quantitative dimension to the online prediction of MHC binding. Appl. Bioinformatics. 2003;2:63–66. [PubMed] [Google Scholar]
- Wan J, et al. SVRMHC prediction server for MHC-binding peptides. BMC Bioinformatics. 2006;7:463. doi: 10.1186/1471-2105-7-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui HH, et al. Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics. 2005;57:304–314. doi: 10.1007/s00251-005-0798-y. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Justesen S, Lund O, Lundegaard C, Buus S. NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immun. Res. 2010;6:9. doi: 10.1186/1745-7580-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot AS, Knopp PM, Martin W. De-immunization of therapeutic proteins by T-cell epitope modification. Dev. Biol. 2005;122:171–194. [PubMed] [Google Scholar]
- Choi Y, Griswold KE, Bailey-Kellogg C. Structure-based redesign of proteins for minimal T-cell epitope content. J. Comput. Chem. 2013;34:879–891. doi: 10.1002/jcc.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker AS, Choi Y, Griswold KE, Bailey-Kellogg C. Structure-guided deimmunization of therapeutic proteins. J. Comput. Biol. 2013;20:152–165. doi: 10.1089/cmb.2012.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker AS, Griswold KE, Bailey-Kellogg C. Optimization of therapeutic proteins to delete T-cell epitopes while maintaining beneficial residue interactions. J. Bioinform. Comput. Biol. 2011;9:207–229. doi: 10.1142/s0219720011005471. [DOI] [PubMed] [Google Scholar]
- Parker AS, Zheng W, Griswold KE, Bailey-Kellogg C. Optimization algorithms for functional deimmunization of therapeutic proteins. BMC Bioinformatics. 2010;11:180. doi: 10.1186/1471-2105-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot AS, Martin W. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin. Immunol. 2009;131:189–201. doi: 10.1016/j.clim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Wang P, et al. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A, Rappuoli R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity. 2010;33:530–541. doi: 10.1016/j.immuni.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L, Cousens L, Fueyo J, De Groot AS. Harnessing the power of genomics and immunoinformatics to produce improved vaccines. Expert Opin. Drug Discov. 2011;6:9–15. doi: 10.1517/17460441.2011.534454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturniolo T, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat. Biotechnol. 1999;17:555–561. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]
- Biologic drugs set to top 2012 sales. Nat. Med. 2012;18:636. doi: 10.1038/nm0512-636a. [DOI] [PubMed] [Google Scholar]
- Seib KL, Zhao X, Rappuoli R. Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin. Microbiol. Infect. 2012;18:109–116. doi: 10.1111/j.1469-0691.2012.03939.x. [DOI] [PubMed] [Google Scholar]
- Jiang W, Boder ET. High-throughput engineering and analysis of peptide binding to class II MHC. Proc. Natl. Acad. Sci. U.S.A. 2010;107:13258–13263. doi: 10.1073/pnas.1006344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justesen S, Harndahl M, Lamberth K, Nielsen L-LB, Buus S. Functional recombinant MHC class II molecules and high-throughput peptide-binding assays. Immun. 2009;5:1–20. doi: 10.1186/1745-7580-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]