

Abstract
Background
The adipocyte fatty acid–binding protein (FABP) aP2 is expressed by adipocytes and macrophages and modulates insulin resistance, glucose and lipid metabolism, and atherosclerosis. Insulin sensitivity is improved in obese but not in lean aP2-deficient mice. A second fatty acid–binding protein, mal1, also is expressed in adipocytes and macrophages, and mal1 deficiency produces similar effects on insulin resistance. We tested the hypothesis that combined aP2 and mal1 deficiency would produce synergistic effects on metabolism and reduce atherosclerosis in apolipoprotein E–deficient (apoE−/−) mice.
Methods and Results
Male and female apoE−/− mice null for both aP2 and mal1 (3KO) and apoE−/− controls were fed a low-fat chow diet for 16 or 56 weeks. Lean 3KO mice had significantly lower serum cholesterol and triglycerides as well as improved insulin and glucose tolerance as compared with controls. Analysis of atherosclerotic lesions in the 3KO mice showed dramatic reductions in both early (20 weeks) and late-stage (60 weeks) atherosclerosis. Strikingly, survival in the 3KO mice was improved by 67% as compared with apoE−/− controls when challenged with the Western diet for 1 year.
Conclusions
Combined aP2 and mal1 deficiency improved glucose and lipid metabolism, reduced atherosclerosis, and improved survival in apoE−/− mice, making these proteins important therapeutic targets for the prevention of the cardiovascular consequences of the metabolic syndrome.
Keywords: atherosclerosis, metabolism, syndrome X, macrophages
The metabolic syndrome is the clustering of insulin resistance, hyperinsulinemia, hypertension, dyslipidemia, and impaired fibrinolysis observed primarily in patients with visceral obesity.1 The metabolic syndrome is recognized as a major risk factor for the development of type 2 diabetes mellitus and atherosclerosis.2 In murine models, the adipocyte fatty acid–binding protein aP2 modulates features of the metabolic syndrome, including insulin resistance, triglyceride metabolism, and atherosclerosis.3–5
Cytoplasmic fatty acid–binding proteins (FABPs) are a family of cytoplasmic proteins that bind fatty acid ligands with high affinity. Their functions include shuttling free fatty acids (FFAs) to various intracellular compartments, regulating cellular lipid metabolism and expression of inflammatory cytokines, and modulating gene expression.6 aP2 is highly expressed in adipocytes and macrophages.7–9 Its expression is transcriptionally controlled during adipocyte development and regulated by PPARγ agonists, insulin, and fatty acids.10,11 In the macrophage, aP2 expression is stimulated on exposure to phorbol esters, oxidized low-density lipoproteins, and PPARγ ligands.4,9,12 Both adipocytes and macrophages express a second FABP, mal1 (also known as keratinocyte lipid-binding protein or skin FABP), which is also found in the epidermis, mammary tissue, and testis.4,13
Studies in aP2-deficient mice have shown that aP2 plays a significant role in many aspects of the metabolic syndrome. aP2 deficiency protects mice with dietary or genetic obesity from the development of insulin resistance, hyperglycemia, and hypertriglyceridemia.3,14 Recently, mal1 deficiency also was shown to partially improve glucose homeostasis and insulin sensitivity in obese mice15; however, lean (ie, normal body weight for age) aP2−/− and mal1−/− mice on a standard chow diet show no significant alterations in glucose and cholesterol levels as compared with wild-type mice.3,15
Our previous work demonstrated that aP2 deficiency protects lean apolipoprotein E (apoE)–deficient (apoE−/−) mice from both early and advanced atherosclerosis without significant effects on systemic glucose and lipid metabolism.4,5 Bone marrow transplantation studies showed that macrophage aP2 expression promotes foam cell formation and atherosclerosis.4 Macrophage aP2 deficiency reduces the cellular accumulation of cholesterol esters and inhibits the expression of inflammatory cytokines.4 aP2 deficiency leads to upregulation of mal1 expression in the adipocyte but not in the macrophage.4 Because aP2 and mal1 are coexpressed in adipocytes and macrophages and mal1 is able to compensate for aP2 deficiency, we hypothesized that a combined deficiency of aP2 and mal1 would have synergistic effects on glucose metabolism and atherosclerosis. In the present study, we show that combined aP2 and mal1 deficiency improves glucose and lipid metabolism, reduces atherosclerosis, and, as a result, dramatically improves survival in the apoE−/− mouse model.
Methods
Animal Procedures
aP2-deficient and mal1-deficient mice were generated by using homologous recombination in embryonic stem cells, as described.3,15 We generated aP2−/− mal1−/− apoE−/− (3KO, experimental group) mice by first crossing aP2−/− and mal1−/− mice (both backcrossed >10 generations into C57BL/6J background) to generate aP2−/− mal1−/− mice. The aP2−/− mal1−/− mice were then crossed with apoE−/− animals (all on C57BL/6J background) and the F1 aP2+/− mal1+/− apoE+/− progeny were intercrossed with each other. Age- and sex-matched aP2+/+mal1+/+apoE−/− mice (also on C57BL/6J background) were used as controls. Mice were fed a standard chow diet with 4.5% fat (PMI feeds) ad libitum beginning at 4 weeks of age for 16 weeks. For mouse survival studies, male 3KO and apoE−/− mice were individually caged and maintained on a high-fat atherogenic Western diet (Harlan Teklad, diet No. TD88137: 21% fat, 0.15% cholesterol, 0% cholate) ad libitum beginning at 4 weeks of age for 52 weeks. These animals were not subjected to any experimentation and only survival was recorded for 1 year. At the end of 1 year, the experiment was stopped because of animal facility guidelines, although no deaths occurred in the 3KO group. Animal care and experimental procedures were performed with approval from the animal care committees of Vanderbilt and Harvard Universities.
Serum Measurements
Mice were fasted overnight (12 h) and blood samples were collected by retro-orbital venous plexus puncture under isoflurane (AErrane, Baxter Pharmaceutical Products) anesthesia. Serum cholesterol and triglycerides were determined with reagent kits (Raichem and Sigma-Aldrich) as described.16 Fasting serum glucose was determined by colorimetric assay (Schiapparelli BioSystems). Plasma FFAs were measured by colorimetric assay (Wako Pure Chemical Industries), and plasma adiponectin levels were measured by radio-immunoassay kit (Linco Research). Lipoprotein analysis by fast protein liquid chromatography (FPLC) was performed on fasting serum samples, as previously described.17 The mean peak area of apolipoprotein B (apoB)–containing lipoproteins was calculated as the sum of fractions 14 to 25 (very-low-density lipoprotein and intermediate-density lipoprotein/low-density lipoprotein peaks) for each animal in milligrams per deciliter.
Metabolic Measurements
Insulin tolerance tests (ITT, 0.5 IU/kg body weight) and glucose tolerance tests (GTT, 1.8 g/kg body weight) were performed on conscious mice after a 6-h fast.18 Mesenteric fat from euthanized animals was quantified by determining the total mesenteric fat wet weight in grams for each animal. To measure the portal vein FFA in the fasting and fed states, we anesthetized mice by intraperitoneal administration of 10 mg/kg xylazine (Phoenix Pharmaceuticals) and 100 mg/kg ketamine (Fort Dodge Animal Health), surgically exposed the portal vein, and collected blood samples with a syringe and 28-gauge needle.
Assessment of Arteriosclerosis and Immunohistochemistry
Perfusion fixation, preparation of aortas, and quantification of atherosclerotic lesions in the en face aorta were performed as previously described.19 Lesions in the proximal aorta from serial 10-μm-thick cryosections were stained with Oil Red O, counterstained with hematoxylin, and quantified as described.20,21 For the detection of mal1 protein in proximal aorta lesions, 5-μm serial cryosections from 15-week-old chow-fed aP2−/− apoE−/− or aP2−/− mal1−/− apoE−/− mice were incubated with rabbit antiserum against human mal1 (gift of Dr Rex Parker, Bristol-Myers Squibb). Sections were then incubated with secondary biotinylated anti-rabbit antibodies, visualized with Fast Red TR/Napthol AS-NX substrate (Sigma-Aldrich), and counterstained with hematoxylin.17 Macrophages were detected with rat monoclonal antibody against mouse macrophage marker (MOMA-2, Accurate Chemical and Scientific).17
Statistical Analysis
The mean values for biochemical data from each group were compared by Student t test. The atherosclerosis data were analyzed with a nonparametric Mann-Whitney test. The survival curve data were analyzed by log-rank test. All statistical tests with P<0.05 were considered significant.
Results
To examine the role of combined aP2 and mal1 deficiency on metabolism and atherosclerosis, we placed 4-week-old male (n=15) and female (n=13) 3KO mice and male and female apoE−/− controls (n=15 and n=12, respectively) on a chow diet for 16 weeks. Fasting levels of serum cholesterol, triglycerides, and glucose at 20 weeks are summarized in Table 1. Mean cholesterol levels were 18% lower in male 3KO mice (P=0.048) and 29% lower in female 3KO mice (P=0.0001) as compared with controls. Similarly, mean fasting triglycerides were 32% lower in male 3KO mice (P=0.009) and 42% lower in female 3KO mice (P=0.0006) as compared with controls. Analysis of the lipoprotein distribution by size exclusion chromatography demonstrated significant quantitative differences in the peak height of the apoB-containing particles between groups (Figure 1). Male 3KO mice (Figure 1A) showed a 13% reduction in the mean peak of the apoB-containing lipoproteins (321.4±40.1 versus 368.5±18.7, mean±SD mg/dL, P=0.026) as compared with apoE−/− mice. Female 3KO mice (Figure 1B) had a 26% reduction in mean peak of apoB-containing lipoproteins (244.6±44.2 versus 330.9±76.2, mean±SD mg/dL, P=0.02) as compared with apoE−/− mice. No significant differences in mean plasma FFA levels were found after a 12-h fast as compared with controls for each sex (Table 1).
TABLE 1.
Fasting Serum Biochemistry in aP2−/− mal1−/− apoE−/− (3KO) and apoE−/− Mice
| Genotype (Sex) | Plasma FFA, mmol/L | Cholesterol, mg/dL | Triglycerides, mg/dL | Glucose, mg/dL |
|---|---|---|---|---|
| ApoE−/− (M) (n=15) | 2.2±0.5 | 395±89 | 130±40 | 70±34 |
| 3KO (M) (n=15) | 2.0±0.3 | 324±91* | 88±24* | 70±31 |
| ApoE−/− (F) (n=12) | 1.4±0.2 | 325±59 | 140±45 | 77±35 |
| 3KO (F) (n=13) | 1.8±0.5 | 230±43* | 81±28* | 40±12* |
Data are shown as mean±SD.
P<0.05 as compared with control.
Figure 1.
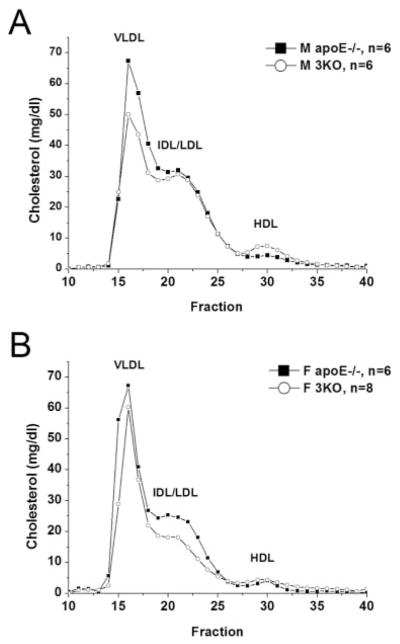
Lipoprotein distribution by FPLC for 3KO and apoE−/− mice. Samples drawn from male (A) and female (B) mice at 20 weeks of age after 12-h fast. Data are average distribution of total cholesterol per fraction in mg/dL for each group. Fractions 14 to 18 contain VLDL; fractions 19 to 25, IDL/LDL; and fractions 26 to 31, HDL. Fractions 32 to 40 contain non–lipoprotein-associated proteins.
Serum glucose levels were virtually identical between the male 3KO mice and controls (Table 1); however, a significant 48% reduction was observed in fasting serum glucose levels of female 3KO mice as compared with controls (40±12 versus 77±35 mg/dL, mean±SD, P=0.0017). Both male and female 3KO groups showed better glucose tolerance than apoE−/− controls (Figure 2C and 2D), which was particularly striking in the female 3KO mice. In accordance with the improved glucose tolerance, both male and female 3KO mice demonstrated greater insulin sensitivity on ITT as compared with controls (Figure 2A and 2B).
Figure 2.

GTT and ITT in 3KO and apoE−/− mice. ITT (0.5 U/kg, IP) shown for male (A) and female (B) mice. GTT (1.8 g/kg, IP) shown for male (C) and female (D) mice. Each data point represents mean glucose level (mg/dL)±SEM *P<0.05, as compared with control group.
Mean body weight at 16 weeks of age was ≈10% lower in the male 3KO mice as compared with controls (Table 2); however, no significant difference in mean body weight was observed between female 3KO mice and apoE−/− controls. We also measured plasma adiponectin levels in all 4 groups after a 12-h fast but found no significant differences between groups for either sex (Table 2). We measured mesenteric fat weight and portal vein FFA levels in a subset of male 3KO and apoE−/− mice, as shown in Table 2. No significant difference (P=0.17) was found in mesenteric fat weight in the 3KO mice (n=4) as compared with controls (n=4). No significant difference was detected in portal vein FFA levels in the fed state between groups, but a significant increase in fasting portal vein FFA levels was noted in the 3KO mice as compared with controls (P=0.04).
TABLE 2.
Indices of Adipocyte Function Between aP2−/− mal1−/− apoE−/− (3KO) and apoE−/− Mice
| Genotype (Sex) | Body Weight, g | Plasma Adiponectin, μg/mL | Mesenteric Fat Weight, g | Portal Vein FFA, Fasting State, mmol/L | Portal Vein FFA, Fed State, mmol/L |
|---|---|---|---|---|---|
| ApoE−/− (M) | 30±2.0 | 36.1±8.7 | 0.24±0.06 | 0.74±0.16 | 0.72±0.13 |
| 3KO (M) | 28±1.4* | 35.6±6.6 | 0.16±0.09 | 1.15±0.31* | 0.62±0.20 |
| ApoE−/− (F) | 22±1.6 | 37.6±9.5 | · · · | · · · | · · · |
| 3KO (F) | 23±1.7 | 36.4±8.6 | · · · | · · · | · · · |
Data are shown as mean±SD.
P<0.05 as compared with control.
To assess the effects of combined aP2 and mal1 deficiency on atherosclerosis, we euthanized the mice at 20 weeks for lesion analysis of the proximal and en face aortas. We previously showed that aP2 protein is expressed by macrophages within the atherosclerotic lesions of the proximal aorta in apoE−/− mice.4 mal1 protein also is abundantly expressed within atherosclerotic lesions and colocalizes with the macrophage marker MOMA-2 (Figure 3). Male 3KO mice showed a 53% reduction in mean lesion area in the proximal aorta (58 631±6638 versus 125 165±17 331 μm2/ section, mean±SEM, P=0.003) and a similar 45% reduction in mean lesion area of the en face aorta (0.172±0.030 versus 0.311±0.031%, mean±SEM, P=0.006) as compared with control apoE−/− mice (Figure 4A). Female 3KO mice showed a 37% reduction in proximal aorta lesion area (127 784±14 522 versus 203 284±23 728 μm2/section, mean±SEM, P=0.01) and a 37% lower mean lesion area of the en face aorta (0.259±0.025 versus 0.413±0.063%, mean±SEM, P=0.024). To determine whether the reduction in atherosclerosis would be persistent even in the setting of advanced atherosclerosis, we fed 6 male 3KO mice and 5 male apoE−/− mice a regular chow diet and euthanized them at 60 weeks of age. Dramatic reductions in mean serum cholesterol (291±38 versus 424±55 mg/dL, mean±SD, P=0.002) and triglycerides (61±18 versus 135±21 mg/dL, mean±SD, P=0.0003) were persistent at 60 weeks of age in the 3KO mice as compared with controls. As shown in Figure 4C, a 26% reduction in atherosclerosis of the proximal aorta was observed in 60-week-old male 3KO mice as compared with controls (375 351±22 279 versus 507 218±35 164 μm2/section, mean±SEM, P=0.017). We found an impressive 78% reduction of the en face aorta lesion area (4.11±0.99 versus 18.54±2.57%, mean±SEM, P=0.004) in 60-week-old 3KO mice as compared with controls (Figure 4D and 4E).
Figure 3.

Immunocytochemical detection of macrophages and mal1 expression in atherosclerotic lesions. Proximal aorta atherosclerotic lesions in aP2−/− apoE−/− and aP2−/− mal1−/− apoE−/− mice stained with MOMA-2 to detect macrophages (A, B); mal1 expression (C, D) shown with rabbit antiserum against human mal1 (magnification ×400).
Figure 4.

Atherosclerosis in proximal and en face aorta in 3KO and apoE−/− mice. Atherosclerosis in 20-week-old male and female mice in proximal aorta (A) and en face aorta (B). Quantification of atherosclerosis for male 60-week-old mice in proximal aorta (C) and en face aorta (D). Data shown as mean lesion area ±SEM for each group. *P<0.05. E, representative pinned-out en face aorta preparations from male 60-week-old mice stained with Sudan-IV, exemplifying dramatic reduction in lesion area in 3KO mice as compared with apoE−/− controls (magnification ×20).
Because of the improvements in lipid and carbohydrate metabolism and atherosclerosis, we hypothesized that combined FABP deficiency would have an impact on longevity in 3KO mice. To evaluate this finding, we placed male 3KO (n=13) and apoE−/− (n=12) mice on a Western diet beginning at 4 weeks of age for 1 year. Survival was dramatically improved at 1 year of age (100% versus 33%, P=0.0004) in 3KO mice as compared with controls (Figure 5).
Figure 5.
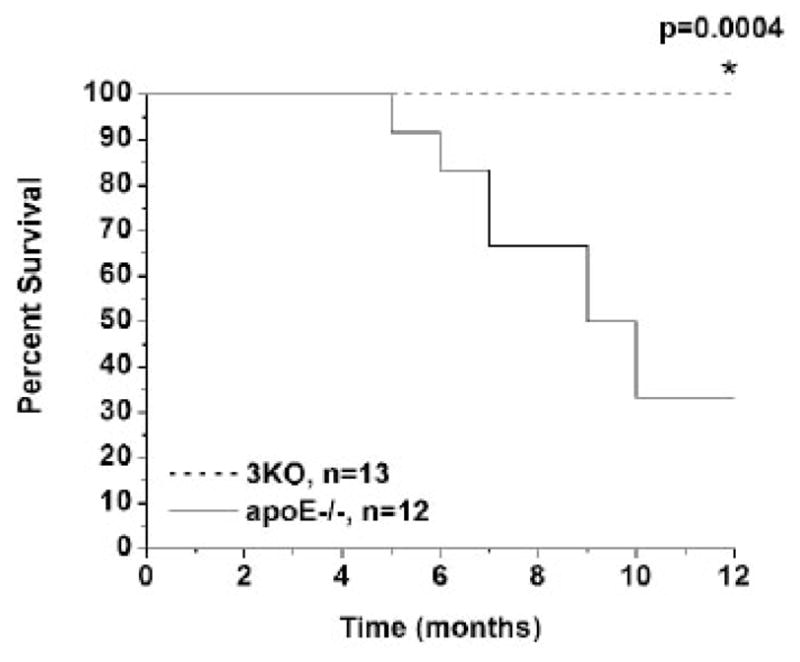
Survival curve for 3KO and apoE−/− mice on high-fat diet. Male 3KO and apoE−/− mice fed high-fat Western diet at 1 month of age for 1 y. 3KO mice show significantly increased survival at 12 months of age.
Discussion
Numerous studies have demonstrated the importance of cellular fatty acid metabolism in the pathogenesis of insulin resistance. Mice with deficiency of the type B scavenger receptor CD36, which is also a fatty acid transporter, show lower plasma glucose and insulin levels on a chow diet but higher serum cholesterol and triglyceride levels than wild-type mice.22 Mice with deficiency of aP2 or mal1 alone demonstrate no significant alterations in insulin sensitivity when they are lean on a chow diet3,15; however, these mice are partially protected from the insulin resistance and hyperglycemia caused by genetic or diet-induced obesity.3,14,15 In the present study, we found a remarkable improvement in insulin sensitivity in lean 3KO mice on a chow diet, indicating a synergistic effect of combined aP2 and mal1 deficiency on glucose metabolism. Adipose tissue from aP2−/− mice have a major upregulation of mal1 expression, which partially compensates for the effects of aP2 deficiency in the adipocyte.3,23 Evidence for this result is demonstrated by the overexpression of mal1 in adipose tissue using a transgenic mouse model, which results in a phenotype of impaired glucose tolerance.15 In 3KO mice, the elimination of mal1 eliminates this compensation for aP2 deficiency, which likely contributes to the dramatic enhancement in insulin sensitivity observed in lean animals.
Lean and obese aP2-deficient mice show reductions in serum triglycerides but no changes in serum cholesterol levels as compared with wild-type mice.3,14 However, we found that combined FABP deficiency of aP2 and mal1 results in significant improvements in total serum cholesterol and triglycerides despite the presence of severe dyslipidemia resulting from apoE deficiency. In contrast, lean aP2−/− apoE−/− mice demonstrate no significant differences in cholesterol or triglyceride levels as compared with apoE−/− mice on a chow or high-fat Western diet.4,5 Lean chow-fed mal1-deficient mice show modest reductions in serum cholesterol and triglycerides as compared with wild-type mice, but this difference is abolished in the setting of obesity-induced dyslipidemia.15 In addition, mal1−/− apoE−/− mice display no reductions in serum lipids as compared with apoE−/− mice on a chow diet (Jeffrey Boord, MD, unpublished data, 2003). Thus, combined aP2 and mal1 deficiency appears to have additive effects on lipid metabolism in vivo.
Chronic elevations in FFA and visceral obesity have been implicated as contributors to insulin resistance and dyslipidemia in the metabolic syndrome.24 We did find a small reduction in the body weight of the male 3KO mice as compared with controls, but we did not find a significant difference in visceral adipose tissue content as measured by mesenteric fat weight. We found no difference in portal vein FFA levels in the fed state, and we noted a significant increase in portal vein FFA in the fasting state in 3KO mice. Thus, the enhancement in insulin and glucose sensitivity in 3KO mice does not appear to be the result of reduced mesenteric fat content or lower systemic and portal vein FFA levels. aP2-deficient adipose tissue shows striking reductions in the secretion of several inflammatory cytokines, including a dramatic reduction in tumor necrosis factor-α.3 It is likely that interference with this and other inflammatory responses plays at least a partial role in the phenotype of FABP-deficient mice.3,15 Adiponectin, a cytokine produced by adipocytes and suppressed in obesity and type 2 diabetes mellitus, improves insulin sensitivity and reduces serum triglycerides in mice in vivo.25,26 We considered whether increased adiponectin secretion could account for the improvements in glucose and lipid metabolism in 3KO mice, but we found no differences in plasma adiponectin levels between groups.
apoE−/− mice that receive aP2−/− apoE−/− bone marrow are protected from atherosclerosis without changes in lipid or glucose metabolism.4 Thus, macrophage aP2 can promote foam cell formation independent of any effects on insulin resistance. In the present study, combined aP2 and mal1 deficiency had remarkable effects on both insulin sensitivity and lipid metabolism in lean animals. Thus, the observed metabolic changes likely contributed to the protection from both early and advanced atherosclerosis. As a result of the improvements in lipid metabolism that were observed in 3KO mice, it was not possible to directly compare atherosclerotic lesion development with aP2−/− apoE−/− mice, which show no significant improvements in lipids or insulin sensitivity on chow or Western diets.4,5 Because mal1 expression is upregulated in the adipocyte but not in the macrophage with aP2 deficiency, the potential exists for a differential impact of combined aP2 and mal1 deficiency in the adipocyte as compared with the macrophage.4 Future studies will address the relative contributions of aP2 and mal1 expression in the macrophage and the adipocyte to atherogenesis.
The most compelling finding of the present study was the improved survival in 3KO mice as compared with apoE−/− mice on a high-fat diet. Other investigators have reported spontaneous plaque rupture and intraplaque hemorrhage in older apoE−/− mice (>30 weeks old) on both chow and high-fat diets.27,28 Williams et al29 reported that apoE−/− mice on a high-fat diet showed an average time of sudden death at 29 weeks of high-fat feeding. The primary causes of death could not be determined, but plaque rupture in the brachiocephalic artery was noted frequently and some apoE−/− mice experienced myocardial infarctions.29 Johnson and Jackson27 found ruptured atherosclerotic plaques in the brachiocephalic artery in 7 of 8 apoE−/− mice fed a high-fat diet that died suddenly. In our longevity study, the primary cause of death was not determined by necropsy, but we believe cardiovascular events resulting from plaque rupture would be the most likely cause of death in the apoE−/− mice.
It is possible that combined aP2 and mal1 deficiency contributes to enhanced plaque stability and thus reduces vascular events that lead to sudden death. Other investigators have shown that antiinflammatory therapies such as low-dose aspirin therapy and inhibition of the CD40 signaling pathway30,31 in mouse models can lead to increases in atherosclerotic lesion collagen content consistent with enhanced plaque stability. Preliminary studies in our laboratory suggest that increased collagen deposition is found in 3KO mice as compared with apoE−/− controls after 16 weeks on the Western diet (Jeffrey Boord, MD, unpublished data, 2003). It is important to note that limitations exist in using murine models of plaque stability and rupture. Significant differences in plaque remodeling and hemodynamics are found in murine versus human arteries, and the low incidence of plaque rupture in the proximal aorta in mice makes the study of spontaneous plaque rupture difficult.28,32 Further study of plaque morphology in mice with combined aP2 and mal1 deficiency may provide insight into the effects of lipid metabolism, insulin resistance, and inflammation on plaque stability.
In summary, combined aP2 and mal1 deficiency produces beneficial effects on glucose and lipid metabolism in lean animals and reduces atherosclerosis in vivo. These benefits are persistent even in advanced age, with combined aP2 and mal1 deficiency conferring a significant survival benefit in the setting of apoE deficiency. The increased cardiovascular risk related to the metabolic syndrome in humans results from the combined effects of insulin resistance, hyperinsulinemia, dyslipidemia, and impaired fibrinolysis on the vasculature.1 This action makes modulation of these FABPs attractive therapeutic targets for treating or preventing cardiovascular consequences of the metabolic syndrome in humans.
Acknowledgments
This study was supported by National Institutes of Health grants HL-65405-01 and DK59637-01 (Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt University Mouse Metabolic Phenotyping Center). Dr Boord is supported by a Research Career Development Award from the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. Dr Maeda is supported by a postdoctoral fellowship from Manpei Suzuki Diabetes Foundation. Dr Makowski is supported by a training grant from the National Institute of Environmental Health Sciences (5 T32 ES07155-17).
Footnotes
Disclosure
Dr Hotamisligil receives various types of funding from Biovitrum AB, Sweden, including consultancy fees, honoraria, and research funding. This funding is related both directly and indirectly to the research presented here.
References
- 1.Reusch JEB. Current concepts in insulin resistance, type 2 diabetes mellitus, and the metabolic syndrome. Am J Cardiol. 2002;90:19–26. doi: 10.1016/s0002-9149(02)02555-9. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 3.Hotamisligil GS, Johnson RS, Distel RJ, et al. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 4.Makowski L, Boord JB, Maeda K, et al. Lack of macrophage fatty-acid–binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boord JB, Maeda K, Makowski L, et al. Adipocyte fatty acid–binding protein, aP2, alters late atherosclerotic lesion formation in severe hyper-cholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:1686–1691. doi: 10.1161/01.atv.0000033090.81345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boord JB, Fazio S, Linton MF. Cytoplasmic fatty acid–binding proteins: emerging roles in metabolism and atherosclerosis. Curr Opin Lipidol. 2002;13:141–147. doi: 10.1097/00041433-200204000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Spiegelman BM, Frank M, Green H. Molecular cloning of mRNA from 3T3 adipocytes: regulation of mRNA content for glycerolphosphate dehydrogenase and other differentiation-dependent proteins during adipocyte development. J Biol Chem. 1983;258:10083–10089. [PubMed] [Google Scholar]
- 8.Hunt CR, Ro JH, Dobson DE, et al. Adipocyte P2 gene: developmental expression and homology of 5′-flanking sequences among fat cell-–specific genes. Proc Natl Acad Sci U S A. 1986;83:3786–3790. doi: 10.1073/pnas.83.11.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu Y, Luo N, Lopes-Virella MF. Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res. 2000;41:2017–2023. [PubMed] [Google Scholar]
- 10.Distel RJ, Robinson GS, Spiegelman BM. Fatty acid regulation of gene expression: transcriptional and post-transcriptional mechanisms. J Biol Chem. 1992;267:5937–5941. [PubMed] [Google Scholar]
- 11.Pelton PD, Zhou L, Demarest KT, et al. PPARγ activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochem Biophys Res Commun. 1999;261:456–458. doi: 10.1006/bbrc.1999.1071. [DOI] [PubMed] [Google Scholar]
- 12.Fu Y, Luo N, Lopes-Virella MF, et al. The adipocyte lipid binding protein (ALBP/aP2) gene facilitates foam cell formation in human THP-1 macrophages. Atherosclerosis. 2002;165:259–269. doi: 10.1016/s0021-9150(02)00305-2. [DOI] [PubMed] [Google Scholar]
- 13.Krieg P, Feil S, Fürstenberger G, et al. Tumor specific overexpression of a novel keratinocyte lipid binding protein. J Biol Chem. 1993;23:17362–17369. [PubMed] [Google Scholar]
- 14.Uysal KT, Scheja L, Wiesbrock SM, et al. Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology. 2000;141:3388–3396. doi: 10.1210/endo.141.9.7637. [DOI] [PubMed] [Google Scholar]
- 15.Maeda K, Uysal KT, Makowski L, et al. Role of the fatty acid binding protein mal1 in obesity and insulin resistance. Diabetes. 2003;52:300–307. doi: 10.2337/diabetes.52.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fazio S, Hasty AH, Carter KJ, et al. Leukocyte low density lipoprotein receptor (LDL-R) does not contribute to LDL clearance in vivo: bone marrow transplantation studies in the mouse. J Lipid Res. 1997;38:391–400. [PubMed] [Google Scholar]
- 17.Babaev VR, Fazio S, Gleaves LA, et al. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J Clin Invest. 1999;103:1697–1705. doi: 10.1172/JCI6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uysal KT, Wiesbrock SM, Hotamisligil GS. Functional analysis of TNF receptors in TNF-alpha–mediated insulin resistance in genetic obesity. Endocrinology. 1998;139:4832–4838. doi: 10.1210/endo.139.12.6337. [DOI] [PubMed] [Google Scholar]
- 19.Babaev VR, Patel MB, Semenkovich CF, et al. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor–deficient mice. J Biol Chem. 2000;275:26293–26299. doi: 10.1074/jbc.M002423200. [DOI] [PubMed] [Google Scholar]
- 20.Fazio S, Babaev VR, Burleigh ME, et al. Physiological expression of macrophage apoE in the artery wall reduces atherosclerosis in severely hyperlipidemic mice. J Lipid Res. 2002;43:1602–1609. doi: 10.1194/jlr.m200108-jlr200. [DOI] [PubMed] [Google Scholar]
- 21.Paigen B, Morrow A, Holmes PA, et al. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 22.Febbraio M, Abumrad NA, Hajjar DP, et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 23.Scheja L, Makowski L, Uysal KT, et al. Altered insulin secretion associated with reduced lipolytic efficiency in aP2−/− mice. Diabetes. 1999;48:1987–1994. doi: 10.2337/diabetes.48.10.1987. [DOI] [PubMed] [Google Scholar]
- 24.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocrinol Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 25.Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 26.Maeda K, Okubo K, Shimomura I, et al. cDNA cloning and expression of a novel adipose specific collagen-like factor apM1 (AdiPose Most abundant Gene transcript 1) Biochem Biophys Res Commun. 1996;221:286–289. doi: 10.1006/bbrc.1996.0587. [DOI] [PubMed] [Google Scholar]
- 27.Johnson JL, Jackson CL. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis. 2001;154:399–406. doi: 10.1016/s0021-9150(00)00515-3. [DOI] [PubMed] [Google Scholar]
- 28.Calara F, Silvestre M, Casanada F, et al. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E–deficient and LDL receptor-–deficient mice. J Pathol. 2001;195:257–263. doi: 10.1002/path.915. [DOI] [PubMed] [Google Scholar]
- 29.Williams H, Johnson JL, Carson KG, et al. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2002;22:788–792. doi: 10.1161/01.atv.0000014587.66321.b4. [DOI] [PubMed] [Google Scholar]
- 30.Cyrus T, Sung S, Zhao L, et al. Effect of low-dose aspirin on vascular inflammation, plaque stability, and atherogenesis in low-density lipoprotein receptor–deficient mice. Circulation. 2002;106:1282–1287. doi: 10.1161/01.cir.0000027816.54430.96. [DOI] [PubMed] [Google Scholar]
- 31.Schonbeck U, Sukhova GK, Shimizu K, et al. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci USA. 2000;97:7458–7463. doi: 10.1073/pnas.97.13.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palinski W, Napoli C. Unraveling pleiotropic effects of statins on plaque rupture. Arterioscler Thromb Vasc Biol. 2002;22:1745–1750. doi: 10.1161/01.atv.0000038754.39483.cd. [DOI] [PubMed] [Google Scholar]