

Abstract
Background
Organ failure in sepsis accounts for significant mortality worldwide. Mitochondrial and metabolic responses are central to the overall response of the cell, and thus of the organ and organism. Adaptive responses in metabolism are critical to the recovery at the cellular level. The purpose of these investigations was to test the hypothesis that sepsis is associated with decreased aerobic respiration and significant metabolic changes in the liver.
Methods
C57BL/6 mice underwent cecal ligation and puncture (CLP) with a 21 gauge needle or an operation without CLP. Mice were euthanized from 0–24 h after the procedure and liver tissue was harvested. Tissue oxygen consumption and mitochondrial complex activity were measured. Global biochemical profiles of 311 metabolites were performed at the 8-h time point (n = 8/group) and analyzed by gas chromatography–mass spectrometry and liquid chromatography tandem mass spectrometry platforms by Metabolon (Durham, North Carolina). The influence of lipopolysaccharide (LPS) on aerobic and anaerobic respiration in primary mouse hepatocytes was also investigated.
Results
CLP in vivo or LPS in vitro resulted in a significant decrease in hepatic oxygen consumption. There was a significant decrease in oxidative phosphorylation measured at 12 h. LPS also resulted in a significant increase in anaerobic respiration in hepatocytes. Interestingly, the metabolomic analysis resulted in a metabolic shift in the liver from carbohydrate-based energy to utilization of fatty acids and amino acids. This included an increase in every tricarboxylic acid cycle intermediate and derivative, suggesting an increased flux into the cycle from fatty acid beta-oxidation and anaplerotic contributions from amino acids.
Conclusions
Sepsis results in a metabolic response and profile consistent with increased anaerobic respiration, which occurs prior to significant changes in hemodynamics. The metabolic responses of cells and organs may be important adaptive responses to prevent organ failure and death.
Keywords: Metabolomic, Carbohydrate, Amino Acid, fatty acid, Lipopolysaccharide
1. Introduction
Sepsis is a continuum of disease comprising a systemic inflammatory response as well as shock resulting from infection. It is a frequently fatal condition, in which mortality is directly related to the extent of organ dysfunction, and can be as high as 70% when three or more organ systems fail [1]. Yet, there is a fundamental lack of understanding of the underlying cellular processes involved in the development of organ dysfunction. A striking feature of failing organs in sepsis is the lack of apoptosis and the conservation of tissue architecture, which differentiates the syndrome from other types of shock [2]. This has inspired increasing interest in cellular adaptive responses involving mitochondrial oxygen utilization and regulation of metabolic activity.
Selective prioritization of cellular energy utilization is a well-known adaptive mechanism in response to injury, as has been demonstrated in hypoxic conditions or during hibernation. This process is highly controlled by the cell and coordinated by mitochondria, given that under physiologic conditions their activity accounts for more than 90% of the cellular consumption of oxygen and the production of ATP [3]. Sepsis is characterized by an alteration in oxygen utilization, clinically evident by increments in mixed venous saturation of oxygen. The etiology of this phenomenon is controversial, but some have proposed that mitochondria are rendered unable to utilize available oxygen and have collectively named this “cytopathic hypoxia” [4,5]. Regardless of the etiology, it is possible that this is a manifestation of cellular adaptation to sepsis.
Maintenance of energy levels within the cell in the setting of disease is fundamental to avoid triggering signaling pathways leading to apoptosis or necrosis. The balance between delivery of oxygen and substrates and demand is fundamental to achieve this; and supporting this notion, others and we have previously shown that levels of ATP in sepsis are stable at least at early time points [6,7]. This may be a reflection of the successful balance between delivery and demand, which may entail a reduction in metabolic activity, as well as appropriate prioritization of energy utilization toward vital-only processes [8].
Further upstream from mitochondria, the tricarboxylic acid or Krebs cycle, which provides the substrate for the electron transport chain in the form of NADH and FADH, feeds from intermediaries derived from the metabolism of carbohydrates, proteins, and lipids, in a process denominated “anaplerotic reactions.” Changes in these metabolic pathways can have potential implications on how energy is processed and utilized at the cellular level, and on how sepsis impacts the organism as a whole. Based on the evidence of ATP-level conservation, absence of apoptosis or necrosis, and concomitant evidence of organ dysfunction, we hypothesize that sepsis results in decreased aerobic respiration and downregulation of hepatic metabolism.
2. Methods
2.1. Cell culture
Primary mouse hepatocytes were harvested from C57BL/6 mice as previously described [9]. They were cultured in William E media supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), insulin (0.16 mL), HEPES buffer (7.5 mL) (Gibco, Grand Island, NY), and 5% fetal bovine serum (Gibco) on gel-coated plates. Cells were stimulated with lipopolysaccharide (LPS; 100 ng/mL) to simulate bacterial infection.
2.2. Cecal ligation and perforation
The University of Pittsburgh Institutional Animal Care and Use Committee approved the animal protocols. Experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals. Cecal ligation and perforation (CLP) was performed on C57BL/6 male mice (Jackson Laboratories, Bar Harbor, ME) 6–8 wk in age and weighing 20–25 g. These animals were anesthetized with pentobarbital (70 mg/kg, intraperitoneal). A 1- to 2-cm midline laparotomy was performed, and the cecum was identified. The stool was then manipulated to the tip of the cecum, which was subsequently ligated 1 cm from the tip with a 2-0 silk tie. The cecum was then perforated with a 22 gauge needle and returned into the abdomen. The muscle and skin were closed with a running 2-0 silk suture. Sham-operated animals underwent laparotomy and bowel manipulation without ligation or perforation. Tissue collection occurred at 8 h post CLP. No antibiotics were used, and animals had free access to food and water preoperatively and postoperatively.
2.3. Biochemical profiles
Global biochemical profiles were performed on livers of C57BL/6 mice harvested 8 h after CLP. Tissue was sent to Metabolon (Metabolon Inc, Durham, NC) for analysis using gas chromatography/mass spectrometry or liquid chromatography/tandem mass spectrometry. Classification assays were used to illustrate reproducibility of results. A random forest assay was used. This makes multiple decision trees from a random known sample from either the CLP or sham-operated group. These decision trees are used to apply other unknown samples to the correct group.
2.3.1. Mitochondrial complex IV activity
Complex IV was measured by monitoring the oxidation of ferrocytochrome c at 550 nm. Potassium cyanide was used to determine specificity of oxidation by complex IV.
2.3.2. Oxygen consumption rate and extracellular acidification rate
Hepatocytes were plated at 20,000 cells per well on XF24 cell culture plates (Seahorse Biosciences, North Billerica, MA) in a final volume of 250 mL. Hepatocytes were then treated with LPS 100 ng/mL at various time points. These cells were then rinsed with unbuffered Dulbecco’s modified Eagle’s medium and then placed in a 37°C incubator without CO2 for 60 min and then loaded onto the XF24 instrument. Oxygen consumption rates and extracellular acidification rates were measured. Each condition was run in quadruplicate and each well was read eight times. Experiments were repeated three times.
3. Results
3.1. Experimental sepsis results in changes to macromolecular metabolism
Mice were randomized to sham operation or CLP. Animals were sacrificed 8 h later and values for 311 metabolites were obtained. The overall predictive accuracy of classification was 94% as determined by random forest assay. Biochemical markers of stress and inflammation including corticosterone, citrulline, arachidate, and ascorbate were significantly elevated in the liver of CLP animals, suggesting that the hepatic milieu is consistent with expected changes in sepsis (Fig. 1). When looking at metabolism as a whole, a significant difference (P < 0.05) was seen in 106 metabolites, with 103 of these being increased in experimental sepsis and only 3 being decreased.
Fig. 1.
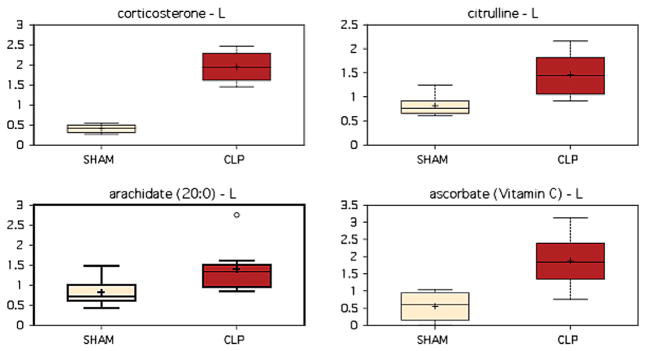
Markers of stress and inflammation are elevated in the liver following CLP, as illustrated by significant elevations in corticosterone (P < 0.001), citrulline (P = 0.0015), arachidate (P = 0.0148), and ascorbate (P = 0.0209). (Color version of figure is available online.)
Interestingly, there were no significant changes or even trends seen toward upregulation or downregulation of carbohydrate metabolism. There was a significant increase in protein metabolism demonstrated by several metabolic pathways (Table 1). There were significant increases in many anaplerotic contributions to the tricarboxylic acid (TCA) cycle as well as increased levels of free amino acids, possibly representing protein breakdown in peripheral tissue to provide an energy source. Similarly, each branched-chain amino acid (valine, leucine, and isoleucine) was upregulated, as were many of their catabolites. Furthermore, there were significant increases in urea cycle intermediates that can be direct contributors to the TCA cycle as well as increased polyamine (ornithine and putrescine) synthesis. These data suggest a possible increased breakdown of protein in the peripheral tissues to provide a source of energy for the liver and organism.
Table 1.
CLP induced increases in amino acid metabolites in liver.
| Super-pathway | Sub-pathway | Biochemical name | Fold change | P value |
|---|---|---|---|---|
| Amino acid | Glycine | Glvcine | 1.28 | 0.0269 |
| Serine | 1.31 | 0.0106 | ||
| Threonine | 1.25 | 0.0175 | ||
| Branched-chain amino acid | Leucine | 1.32 | 0.0078 | |
| Isoleucine | 1.43 | 0.003 | ||
| Valine | 1.42 | 0.004 | ||
| Isobutyrylcarnitine | 2.51 | <0.001 | ||
| Urea cycle | Citruline | 1.79 | 0.0015 | |
| Urea | 4.41 | <0.001 | ||
| Creatine metabolism | Creatine | 1.63 | 0.003 | |
| Glutathione metabolism | Glutathione | 1.83 | 0.003 | |
| Polyamine metabolism | Putrescine | 2.51 | <0.001 | |
| 5-Methylthioadenosine | 1.54 | 0.0232 |
Fat and lipid utilization are also significantly elevated after CLP, possibly reflecting increased mobilization for oxidation by the liver (Table 2). Free fatty acids are increased, providing substrate for beta-oxidation. The ultimate goal of this process is generation of acetyl coenzyme A; and although this is not specifically elevated, there is a significant increase in ketogenesis, suggesting flux through the system.
Table 2.
CLP increases fatty acid metabolites in the liver.
| Super-pathway | Sub-pathway | Biochemical name | Fold change | P value |
|---|---|---|---|---|
| Lipid | Essential fatty acid | Dihomo-linoleate | 1.62 | 0.0171 |
| Docosahexaenoate | 1.30 | 0.0030 | ||
| Long chain fatty acid | Palmitoleate | 1.67 | 0.0183 | |
| Eicosenoate | 2.43 | 0.0089 | ||
| Ketone bodies | 3-Hydroxybutyrate | 1.60 | 0.0062 | |
| Steroid/sterol | Corticosterone | 4.74 | <0.001 |
The products of these aforementioned pathways (glycolysis, fatty acid beta-oxidation, and amino acid catabolism) converge upon the TCA cycle. Not surprisingly, based upon increased fatty acid beta-oxidation and anaplerotic contribution from amino acids, there was a net increase in all TCA cycle intermediates during experimental sepsis (Table 3, Fig. 2).
Table 3.
CLP increases TCA cycle intermediates in liver.
| Super-pathway | Sub-pathway | Biochemical name | Fold change | P value |
|---|---|---|---|---|
| Energy | TCA cycle | Citrate | 4.00 | <0.001 |
| Fumarate | 1.42 | 0.0022 | ||
| Succinylcarnitine | 1.83 | 0.0101 | ||
| Malate | 1.52 | <0.001 | ||
| Oxidative phosphorylation | Phosphate | 1.18 | 0.0057 | |
| Pyrophosphate | 1.32 | 0.082 | ||
| Acetylphosphate | 1.25 | 0.2279 |
Fig. 2.

There was a net increase in hepatic TCA cycle intermediates following CLP. (Color version of figure is available online.)
3.2. Experimental sepsis results in decreased oxidative phosphorylation
We examined markers of aerobic and anaerobic metabolism in primary mouse hepatocytes following LPS treatment as well as in liver tissue following CLP. Both oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured as markers of aerobic and anaerobic respiration, respectively. Hepatocytes showed a significant decrease in OCR at the 12- and 24-h time points following LPS administration, with a trend toward returning to normal levels at the later time points. At the same time points, there was a significant increase in ECAR. Using the OCR-to-ECAR ratio as a measurement of overall aerobic metabolism, we see that this is decreased significantly as early as the 4-h time point in hepatocytes following LPS treatment, but returns toward normal levels by 24 h (Fig. 3A). Activity of mitochondrial complexes I–IV was measured in hepatocytes or liver at the 8-h time point following LPS or CLP, respectively. Although all complexes demonstrated decreased activity following LPS treatment, only complex IV was significantly decreased compared with controls (Fig. 3B). Similarly, there was a significant decrease in the activity of mitochondrial complex II in liver tissue following CLP (Fig. 3C). Again, decreases were seen in all complexes, but not all of them reached statistical significance. Interestingly, levels of adenosine triphosphate (ATP) were not significantly changed in experimental sepsis at the time points examined; however, levels of adenosine 5′-monophosphate (AMP) were significantly increased (1.48-fold; P < 0.001).
Fig. 3.

(A) Overall aerobic metabolism was measured using the OCR-to-ECAR ratio in hepatocytes following LPS administration. This ratio decreased significantly at 4 and 12 h after LPS, but showed some return toward normal by 24 (H) Additionally, mitochondrial complex activity was decreased in hepatocytes after LPS administration (B) as well as in liver tissue following CLP (C). (Color version of figure is available online.)
4. Discussion
In summary, sepsis is associated with decreased oxidative phosphorylation and increased anaerobic metabolism. Although there are no consistent trends in alteration of carbohydrate metabolism, sepsis induces categorical increases in both protein and fatty acid metabolism.
This is consistent with prior literature supporting increased use of hepatic lipogenesis and decreased use of glycogen as substrates in sepsis [10–12]. Consequently, the respiratory quotient, an indirect measure of basal metabolic rate (RQ = CO2 eliminated/O2 consumed) decreases concomitant with an increased oxygen utilization [13,14]. As mentioned previously, these experiments did not show significant changes in ATP levels but did show increases in AMP. Maintaining ATP levels may be part of an adaptive metabolic pathway whereby metabolism shifts, as well as oxygen utilization, to lead to no significant net change in ATP. However, changes in AMP may function as the signaling molecule to further regulate metabolic pathways as well as inflammation. Examples of AMP regulating metabolism would include the activation of phosphofructokinase and pyruvate kinase. Furthermore, AMP regulates AMP-activated protein kinase signaling to influence both metabolism and inflammation [8,15,16].
In contrast to the idea of sepsis as a hypermetabolic state, there is literature supporting a state of cellular dormancy or hibernation as an adaptive means of survival [17,18]. In this setting, organ dysfunction could be understood as an adaptive response to decrease energy utilization, avoiding further injury and cell death. However, the complexity of the disease makes interpretation of oxygen utilization and mitochondrial activity studies in sepsis difficult, and thus proof or disproof of this hypothesis is still lacking [19]. Animal and human studies have repetitively shown that mitochondrial function is impaired in sepsis [20,21]. In agreement with this, our data demonstrated a decrease in mitochondrial complex activity as well as in OCR. This could potentially argue in favor of metabolic downregulation as a protective response. However, we also found a concomitant increase in ECAR. Furthermore, we found that sepsis induced a preferential utilization of proteins and lipids for anaplerotic reactions without downregulation of the use of carbohydrates, suggesting that the Krebs cycle is still active, consuming intermediaries and supplying substrates to the electron transport chain. Altogether, these findings do not support the theory of an orchestrated metabolic downregulation and hibernation-like response. In fact, our data provide evidence that in the early stages of sepsis, metabolism is active; that it is supplied by protein and lipid catabolism; and that oxygen consumption may decrease as a result of a shift toward anaerobic metabolism rather than due to an intended blockade of the respiratory transport chain.
Additionally, changes in metabolism demonstrated in the liver tissues from these in vivo experiments represent one point in time. Although there are changes seen in metabolites within the liver, suggesting a drastic change in substrates utilized for energy production, there is still an overall decrease in hepatic mitochondrial complex activity. These changes in oxidative phosphorylation may be independent of and extend beyond changes in substrate availability and utilization. Sepsis and other stress responses can potentially lead to alterations in mitochondrial mass and activity that could account for overall decreases in aerobic respiration [18–21].
Though the hemoproteins were not specifically measured in this dataset, the concomitant increase in succinyl coenzyme A and glycine suggest a mechanism for increased heme synthesis known to occur in sepsis [22,23]. Heme-oxygenase 1 has been shown to be dramatically induced in the liver during sepsis and is integral to the induction of adaptive responses such as autophagy in order to prevent cell death [24]. There is increasing evidence that metabolic activity heavily regulates other cellular processes, including inflammation. Mitochondria function as vital signaling organelles that regulate signaling pathways and nuclear gene expression for a host of cellular pathways not classically related to metabolism [25]. Understanding the integration of metabolism and inflammation is likely to yield insight into multiple disease states. This includes sepsis as well as chronic inflammatory conditions.
The response to sepsis is a complex and dynamic one, differing between stages of the disease and among the organs involved. Further investigations will be needed to understand the interplay of metabolism and inflammation as well as the initiation and regulation of cellular adaptive signaling.
References
- 1.Hebert PC, Drummond AJ, Singer J, Bernard GR, Russell JA. A simple multiple system organ failure scoring system predicts mortality of patients who have sepsis syndrome. Chest. 1993;104:230. doi: 10.1378/chest.104.1.230. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 3.Taylor CT. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J. 2008;409:19. doi: 10.1042/BJ20071249. [DOI] [PubMed] [Google Scholar]
- 4.Fink MP. Cytopathic hypoxia: mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:219. doi: 10.1016/s0749-0704(05)70161-5. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz DR, Malhotra A, Fink MP. Cytopathic hypoxia in sepsis: an overview. Sepsis. 1999;2:279. [Google Scholar]
- 6.Chaudry IH, Wichterman KA, Baue AE. Effect of sepsis on tissue adenine nucleotide levels. Surgery. 1979;85:205. [PubMed] [Google Scholar]
- 7.Singer M. Mitochondrial function in sepsis: acute phase versus multiple organ failure. Crit Care Med. 2007;35:S441. doi: 10.1097/01.CCM.0000278049.48333.78. [DOI] [PubMed] [Google Scholar]
- 8.Liu TF, Brown CM, El Gazzar M, et al. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92:499. doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West MA, Keller GA, Cerra FB, Simmons RL. Killed Escherichia coli stimulates macrophage-mediated alterations in hepatocellular function during in vitro coculture: a mechanism of altered liver function in sepsis. Infect Immun. 1985;49:563. doi: 10.1128/iai.49.3.563-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Askanazi J. Influence of total parenteral nutrition on fuel utilization in injury and sepsis. Ann Surg. 1980;191:40. doi: 10.1097/00000658-198001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoner HB. The effect of sepsis on the oxidation of carbohydrate and fat. Br J Surg. 1983;70:32. doi: 10.1002/bjs.1800700113. [DOI] [PubMed] [Google Scholar]
- 12.De Vasconcelos PR, Kettlewell MG, Williamson DH. Time course of changes in hepatic metabolism in response to sepsis in the rat: impairment of gluconeogenesis and ketogenesis in vitro. Clin Sci. 1987;72:683. doi: 10.1042/cs0720683. [DOI] [PubMed] [Google Scholar]
- 13.Nanni G, Siegel JH, Coleman B, Fader P, Catiglione R. Increased lipid fuel dependence in the critically ill septic patient. J Trauma. 1984;24:14. doi: 10.1097/00005373-198401000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Giovannini I, Boldrini G, Castagneto M, et al. Respiratory quotient and patterns of substrate utilization in human sepsis and trauma. JPEN. 1983;7:226. doi: 10.1177/0148607183007003226. [DOI] [PubMed] [Google Scholar]
- 15.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardie DG. AMP-activated protein kinase – an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Boxel GI, Doherty WL, Parmar M. Cellular oxygen utilization in health and sepsis. Contin Educ Anaesth Crit Care Pain. 2012;12:207. [Google Scholar]
- 18.Singer M. Cellular dysfunction in sepsis. Clin Chest Med. 2008;29:655. doi: 10.1016/j.ccm.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Karl IE. Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis. JAMA. 1992;267:1503. [PubMed] [Google Scholar]
- 20.Brealey D, Singer M. Mitochondrial dysfunction in sepsis. Curr Infect Dis Rep. 2003;5:365. doi: 10.1007/s11908-003-0015-9. [DOI] [PubMed] [Google Scholar]
- 21.Brealey D, Karyampudi S, Jacques TS, et al. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. 2004;286:R491. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 22.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557. [PubMed] [Google Scholar]
- 23.Wunder C, Potter RF. The heme oxygenase system: its role in liver inflammation. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:199. doi: 10.2174/1568006033481410. [DOI] [PubMed] [Google Scholar]
- 24.Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053. doi: 10.1002/hep.24324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayir H, Kagan VE. Bench-to-bedside review: mitochondrial injury, oxidative stress and apoptosis – there is nothing more practical than a good theory. Crit Care. 2008;12:206. doi: 10.1186/cc6779. [DOI] [PMC free article] [PubMed] [Google Scholar]