

Abstract
3’-Azidothymidine (AZT) was the first approved antiviral for the treatment of human immunodeficiency virus (HIV). Reported efforts in clicking the 3’-azido group of AZT have not yielded 1,2,3-triazoles active against HIV or any other viruses. We report herein the first AZT-derived 1,2,3-triazoles with sub-micromolar potencies against HIV-1. The observed antiviral activities from the cytopathic effect (CPE) based assay were confirmed through a single replication cycle assay. Structure-activity-relationship (SAR) studies revealed two structural features key to antiviral activity: a bulky aromatic ring and the 1,5-substitution pattern on the triazole. Biochemical analysis of the corresponding triphosphates showed lower ATP-mediated nucleotide excision efficiency compared to AZT, which along with molecular modeling, suggests a mechanism of preferred translocation of triazoles into the P-site of HIV reverse transcriptase (RT). This mechanism is corroborated with the observed reduction of fold resistance of the triazole analogue to an AZT-resistant HIV variant (9-fold compared to 56-fold with AZT).
Keywords: 3’-[5-Aryl-(1,2,3-triazol-1-yl)]-3’-deoxythymidine; HIV; AZT; click chemistry
Introduction
Nucleoside RT inhibitors (NRTIs) constitute the cornerstone of chemotherapies against HIV infection.1 Typically these inhibitors lack the 3’-OH and act as obligate chain terminators. Such a mechanism of inhibition requires successive phosphorylation by cellular kinases, competitive active site binding against endogenous deoxynucleoside triphosphates (dNTPs) and effective incorporation by RT. Although NRTIs generate a relatively large genetic barrier for viruses to select resistant strains, the common pathway of intracellular phosphorylation, particularly the shared cellular kinases, could easily lead to intra-class drug-drug interactions.2 Among numerous nucleoside antivirals, AZT was the first approved for the treatment and prophylaxis of HIV / AIDS.3,4 Long term clinical use of AZT is unfortunately associated with significant side effects, including myopathy, cardiomyopathy and anemia, presumably due to the sensitivity of γ-DNA polymerase in some cell mitochondria,5 and / or the depletion of thymidine triphosphate6 as AZT can be both a substrate and inhibitor of human thymidine kinases (hTKs).
Mechanistically the unique 3’-azido group of AZT can contribute critically to HIV RT binding.7 Chemically modifying the 3’-azido group may yield novel classes of nucleoside inhibitors with a distinct binding mode and toxicity profile. One convenient channel for the azido modification would be through click chemistry to form 1,2,3-triazoles which can occupy a much bigger binding space with the three consecutive nitrogen atoms taking a profoundly different geometry. The potential of this particular transformation to rapidly provide new opportunities for antiviral discovery has been recognized and pursued by many in the field.8-13 Unfortunately, none of the resulting 1,2,3-triazole analogues were found to inhibit HIV or any other DNA or RNA viruses even at very high concentrations (100—500 μM). Habich et al 10 and Hirota et al 11reported that the AZT-derived 1,2,3-triazoles exhibited no appreciable activity in HIV-1 infected CEM-V and MT-2V cells, nor did they inhibit syncytium formation in infected human peripheral blood monocytes. Herdewijn’s group showed that replacing the 3’-azido group of AZT with other five-membered heterocyclic rings eliminated antiviral activity.12 More recently, Zhou et al13 found that even with a more elaborated side chain, AZT-derived 1,2,3-triazoles showed no inhibitory activity against any of these viruses: parainfluenza type 3, reo type 1, Sindbis, Coxsackie B4, Punta Toro, vesicular stomatitis, respiratory syncytial, herpes simplex, vaccinia, cowpox, and HIV. Notably, AZT-derived 1,2,3-triazoles reported to date typically feature an alkyl substituent at the C-4 site of the triazole (Figure 1, 2). Their lack of antiviral activity is likely due to inefficient cellular activation as 1,2,3-triazole nucleosides are poor substrates for the cytosolic human thymidine kinase 1 (hTK-1) when compared to AZT.14 Intriguingly, when the 4-alkyl group of the triazole is replaced with a substituted phenyl ring, the resulting analogues (1d–1e) showed significantly improved affinity for hTK-2 (Figure 1).15 This observation led us to hypothesize that a bulky aryl substituent on the triazole scaffold may provide crucial binding for the target enzyme, thus fundamentally change the antiviral profile. Based on this hypothesis, we have synthesized both 4-aryl (scaffold 3) and 5-aryl (scaffold 4) 1,2,3-triazoles (Figure 1) and have identified a few analogues with confirmed antiviral activity in the sub-micromolar range. To the best of our knowledge, this represents the first successful attempt of clicking AZT into 1,2,3-triazoles with potent antiviral activity.
Figure 1.
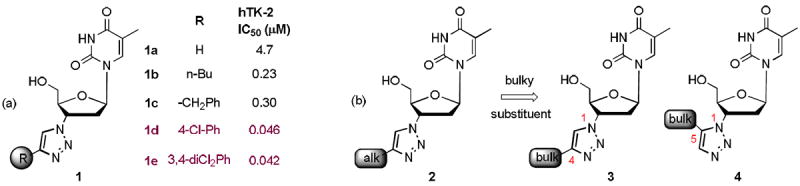
Design of 1,2,3-triazole HIV inhibitors. (a) AZT-derived 1,2,3-triazoles as inhibitors of hTK-2. The C4 aromatic substituent yields significantly improved inhibitory activity (1d–1e); (b) introducing a bulky group at C4 or C5 may turn the inactive 1,2,3-triazoles 2 into potent HIV inhibitor scaffolds 3–4.
Results and Discussion
Chemistry
Click chemistry provides a powerful tool in chemical biology and drug discovery as it allows efficient and clean creation of compounds under extremely mild conditions.16 The Huisgen thermal cycloaddition between an organic azide and an alkyne typically yields a mixture of two regioisomeric 1,2,3-triazoles.17,18 This reaction was rendered clickable through two dramatic Sharpless modifications: the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC)19 which results in the exclusive formation of 1,4-disubstituted 1,2,3-triazoles; and the ruthenium(II)-catalyzed variant (RuAAC)20 that specifically generates 1,5-disubstituted 1,2,3-triazoles. The CuAAC click chemistry has gained particular significance and popularity from its wide range of applications in drug discovery,21 bioconjugation22,23 and material science.24 By contrast, the RuAAC click chemistry remains rather underexplored due to its relatively low applicability and efficiency. As mentioned earlier, reported efforts on clicking AZT have primarily focused on the CuAAC with alkyl alkynes and none of the resulting 1,4-disubstituted 1,2,3-triazoles showed appreciable activity in antiviral assays. A close examination on their inhibition of hTK-2 revealed that a bulkier substituent, preferably a large aromatic ring, may provide key interactions for target binding.15 Toward this end, we were prompted to explore both the CuAAC and RuAAC click reactions to gain access to both 1,4 and 1,5 regioisomers. The reactions were carried out with a broad range of aromatic and aliphatic alkynes as shown in schemes 1 and 2. The propargyl aryl ethers (7a–f) were readily prepared via the alkylation of phenols with propargyl bromide and K2CO3 in good yields (scheme 1).25 The aromatic and aliphatic alkynes were either commercially available or prepared from aldehydes via the Seyferth-Gilbert homologation with the Bestmann reagent26 (14) (scheme 2).
Scheme 1.
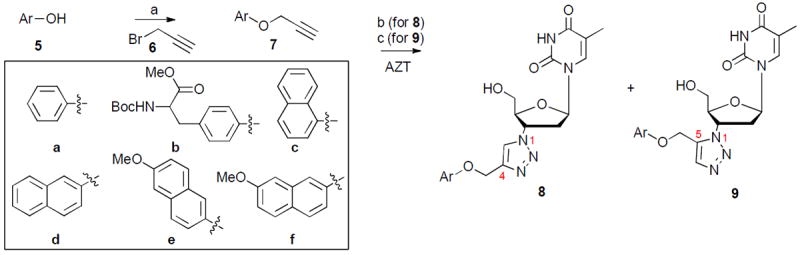
a Synthesis of 1,2,3-triazoles 8 and 9
a Reagents and conditions: a) propargyl bromide, K2CO3, DMF, rt, 12–15 h, 60–76%; b) sodium ascorbate, CuSO4·5H2O, THF/H2O (3:1), rt, 12 h,54–96%; c) Cp*RuCl(PPh3)2, THF, 60 °C, 1–2 d, 24–48%.
Scheme 2.
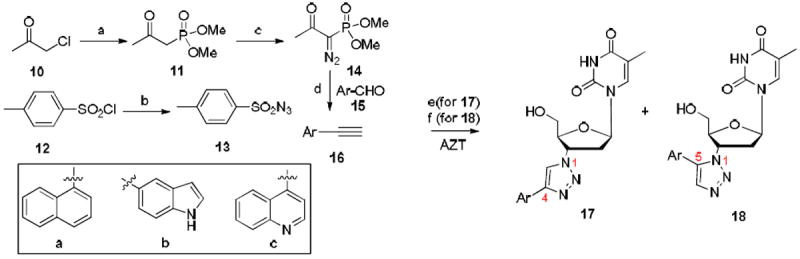
a Synthesis of 1,2,3-triazoles 17 and 18
a Reagents and conditions: a) KI, acetone/CH3CN, rt–50 °C, 12 h, 72%; b) NaN3, acetone/H2O, 0 °C–rt, 14 h, 94%; c) NaH, THF/benzene, then 13, 0 °C–rt, 12h, 65%; d) K2CO3, MeOH, rt, 12 h, 45–90%; e) sodium ascorbate, CuSO4·5H2O, THF/H2O (3:1), rt, 12 h, 54–96%; f) Cp*RuCl(PPh3)2, THF, 60 °C, 1–2 d, 24–48%.
The two versions of the Huisgen–Sharpless cycloaddition proceeded with considerably different efficiencies: the CuAAC with alkynes (7a–f) (scheme 1) and (16a–m) (scheme 2) typically completed within 12 hours under ambient temperature and produced 1,4-disubstituted 1,2,3-triazoles (8a–f) and (17a–m) in good yields, whereas the RuAAC variant with alkynes (7a–f) (scheme 1) and (16a–m) (scheme 2) required longer reaction time and elevated temperature for completion and generated 1,5-disubstituted 1,2,3-triazoles (9a–f) and (18a–m) in only moderate yields. The isolation of 1,4- and 1,5-disubstituted triazoles from reaction mixture proved to be rather straightforward by flash column chromatography. However, caution has to be taken for analogues with a similar Rf to AZT to avoid any AZT contamination which will likely cause false positive in antiviral assays. In our case, the purity of final triazoles was confirmed by NMR and HPLC analysis. The formation of 1,4- and 1,5-disubstituted triazoles were clearly evident from 1H NMR that the 2’-CH2 protons were split into two distinct peaks around 2–3 ppm, which was not observed with AZT.
The preparation of triphosphates for selected nucleoside triazoles is necessitated by the need of biochemical studies. In this event the nucleoside triazoles (18a, 18e–18f) were directly converted to the corresponding triphosphates (21a, 21e–21f) using the one-pot procedure by Eckstein27 (Scheme 3). The triazole nucleotides were purified by reversed-phase HPLC using the C18 reverse phase column and ion exchange chromatography. The triphosphates were obtained in moderate yields (33–43%) and were characterized by HRMS, 1H and 31P NMR.
Scheme 3.
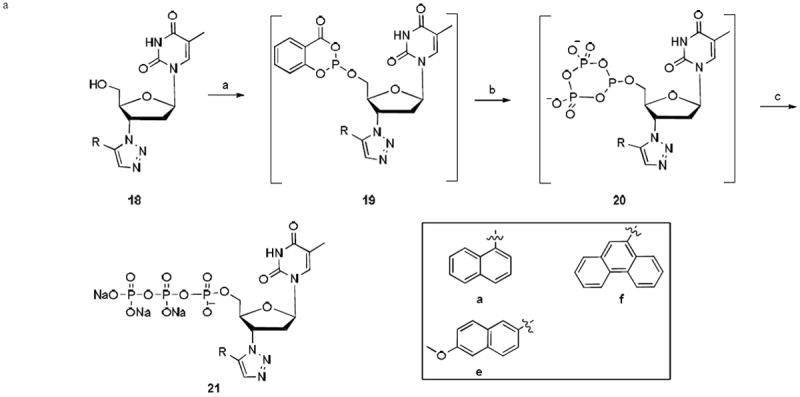
a Synthesis of triazole nucleoside triphosphates 21
aReagents and conditions: a) 2-Chloro-4H-1,3,2-benzodioxaphosphorin-4-one, pyridine/dioxane (1:3), rt, 15 min; b) (Bu3HN)2H2P2O7, Bu3N, DMF, rt, 15 min; c) I2, pyridine/H2O, NH3 (aq), rt, 1 h, Na-ion exchange, 33-43%.
Antiviral Screening
To test the hypothesized beneficial effects of the bulky aromatic group, all synthesized 1,2,3-triazole compounds were screened for antiviral activity. The primary antiviral screening was conducted with a well-established colorimetric cytoprotection assay28,29 based on viral CPE in CME-SS cells. This assay measures cell viability in relation to CPE and requires multiple rounds of viral replication. Initial screening was done at a single concentration (10 μM) and cell viability determined through the addition of (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), a colorless compound that is reduced only by metabolically viable cells to produce a purple compound.30 We first tested the 1,2,3-triazole series where the C4 / C5 bulky substituent is separated from the triazole ring with a methylene ether linkage (scaffolds 8–9). As shown in Table 1, analogues of this series all have good viability (85–100%) under 10 μM. The most striking SAR trend is the observation that most 1,5-substituted 1,2,3-triazoles (9a, 9c–9e) showed significant antiviral activity (42–88% CPE reduction) at 10 μM whereas the 1,4-regio-isomers remained inactive (Table 1), strongly suggesting that the substitution pattern may be crucial to conferring antiviral activity. The relatively flat SAR in regard to the size and positioning of the aromatic ring for 1,5 analogues may simply reflect the deleterious effect of the linkage. Nevertheless, this level of antiviral activity is in stark contrast with all reported AZT-derived 1,2,3-triazoles where no appreciable activity was observed at concentrations up to 500 μM.
Table 1.
Single concentration screening of scaffolds 8 and 9 in the cytoprotection assay against HIV-1 in CEM-SS cells.
| |||
|---|---|---|---|
| Compound | R | Inhibition % @ 10 μM | Viability % @ 10 μM |
| 8a |
|
7 | 85 |
| 9a | 51 | 100 | |
| 8b |
|
0 | 99 |
| 9b | 17 | 90 | |
| 8c |
|
5 | 100 |
| 9c | 42 | 93 | |
| 8d |
|
0 | 100 |
| 9d | 88 | 95 | |
| 8e |
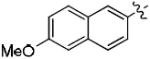
|
0 | 100 |
| 9e | 57 | 100 | |
| 8f |
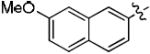
|
0 | 100 |
| 9f | 10 | 99 | |
| 8g | H | 5 | 100 |
| 9g | 0 | 100 | |
Next we tested analogues with the bulky substituent immediately connected to the triazole ring (scaffolds 17–18). Again these triazole analogues typically do not show cytotoxicity at 10 μM (Table 2). As for antiviral activity, 1,5-substituted analogues in general remained superior to their 1,4 regioisomers, though many 1,4 analogues (17b–17f, Table 2) also exhibited discernible antiviral activities (33–44%) at 10 μM, implying that effective bulkiness without a linkage greatly benefits target binding. This bulky group effect is further illustrated within the 1,5-series, where a small cycloalkyl group (18k) confers little inhibitory activity, while large aromatic rings (18a, 18c–18f, 18h–18j) consistently support substantial antiviral activity. Smaller aromatic rings, the phenyl ring (17h, 18h) or substituted phenyl rings (1d, 1e, 18m, 18n), do not appear to provide sufficient target binding, hence the observed less potent antiviral activities. In the meantime, the positioning of the aromatic ring also seems to considerably impact the antiviral activity as evidenced by the much higher potency of the α positioned naphthyl substituent (18a) when compared to the β positioned naphthyl analogues (18d–18e). Furthermore, the highly privileged biphenyl substituent appears to benefit the target binding of both 1,4 (17j) and 1,5 (18j) regioisomers. This beneficial effect for the 1,4 series, however, is drastically reduced when the biphenyl is separated by an O atom (17i vs 17j). Finally, the screening assay initially identified 18l as a promising hit (100% inhibition @ 10 μM). However, this activity was not confirmed upon testing in the single replication cycle assay, in which 18l did not produce significant antiviral activity at concentrations up to 10 μM. Retesting in the screening assay yielded a much reduced percentage inhibition for 18l, corroborating the single replication assay result.
Table 2.
Single concentration screening of scaffolds 17 and 18 in the cytoprotection assay against HIV-1 in CEM-SS cells.
| |||
|---|---|---|---|
| Compound | R | Inhibition % @ 10 μM | Viability % @ 10 μM |
| 17a |
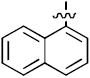
|
1 | 100 |
| 18a | 83 | 100 | |
| 17b |
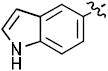
|
35 | 100 |
| 18b | 12 | 82 | |
| 17c |
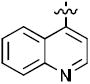
|
44 | 100 |
| 18c | 74 | 94 | |
| 17d |
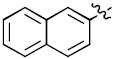
|
33 | 98 |
| 18d | 37 | 100 | |
| 17e |
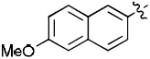
|
35 | 95 |
| 18e | 43 | 100 | |
| 17f |
|
42 | 100 |
| 18f | 100 | 100 | |
| 17g |
|
11 | 100 |
| 18g | 0 | 97 | |
| 17h |
|
24 | 100 |
| 18h | 44 | 100 | |
| 17i |
|
0 | 100 |
| 18i | 67 | 97 | |
| 17j |
|
91 | 88 |
| 18j | 63 | 80 | |
| 17k |
|
1 | 100 |
| 18k | 17 | 100 | |
| 17l |
|
0 | 100 |
| 18l | 100 (43a) | 100 (88a) | |
| 1d |
|
0 | 100 |
| 18m | 23 | 100 | |
| 1e |
|
6 | 100 |
| 18n | 31 | 100 | |
Retest results
Dose-Response Antiviral Potency
The antiviral potential of these novel 1,2,3-triazoles were further assessed in dose-response fashion using the same cytoprotection (CPE reduction) assay with three selected 1,5 analogues (9d, 18a, 18f) and AZT as control. Gratifyingly, all three compounds showed exceptional antiviral activity with low cytotoxicity (Table 3). Of particular significance are analogues 18a and 18f each inhibiting HIV-1 IIIB with submicromolar potency and a very large therapeutic window. The other analogue tested (9d) was also active at low micromolar concentrations. The EC50 of 1.9 μM represents a nearly 30-fold drop in antiviral potency when compared to 18a, strongly suggesting that the bulkiness provided by the aromatic substituent is crucial to target binding and that its separation from the triazole ring through a linkage tremendously compromises the antiviral potency.
Table 3.
Dose-response antiviral testing of selected compounds in two distinct assays.
| Compound | CPE Reduction Assay
|
Single Replication Cycle Assay EC50a (μM) | ||
|---|---|---|---|---|
| EC50a (μM) | CC50b (μM) | TIc | ||
| AZT | 0.0053 | >1.0 | >48 | 0.14 |
| 9d | 1.9 | 70 | 37 | > 5.0 |
| 18a | 0.067 | 61 | 910 | 1.0 |
| 18f | 0.10 | 21 | 210 | > 5.0 |
| 17d | NDd | ND | -- | 7.2 |
| 17f | ND | ND | -- | 4.7 |
| 18e | ND | ND | -- | 4.1 |
Concentration inhibiting virus replication by 50%.
Concentration resulting in 50% cell death.
Therapeutic index, defined by CC50/EC50.
Not determined.
To verify the observed dose-response antiviral potency, we have also tested these compounds in a distinct single replication cycle antiviral assay. This assay quantitatively measures HIV infection in indicator cells (P4R5) through the expression of a Tat-dependent reporter (β-galactosidase) and offers the advantage of quickly determining the infectivity / inhibition with only one replication cycle by colorimetric analysis after incubating with a β-galactosidase substrate. Through this single replication cycle assay the exceptional potency of 18a was confirmed as it inhibited WT HIV-1 in dose response fashion with an EC50 of 1.0 μM (Table 3). While this potency is lower than the submicromolar activity (0.067 μM) observed in the CPE based assay, it may reflect largely the intrinsic difference between these two assays, as AZT also showed considerably different potencies in the two assay methods. Dose response inhibition was also observed with the other two selected analogues (9d and 18f) in low micromolar range (curves not shown), though definite EC50 values were not determined. Significantly, this assay also identified three additional triazole analogues (17d, 17f and 18e) with low micromolar activities (EC50 = 4.1–7.2 μM) against WT HIV-1, further establishing the antiviral potential of these nucleoside triazoles. A few other hits (9c, 9e, 18i, 18j, 18l and 18n) from the screening assay were also tested in the single replication cycle assay, and none showed significant antiviral activity at concentrations up to 10 μM (data not shown).
We then further characterized the antiviral profile of the most active analogue 18a using AZT-resistant and NNRTI-resistant HIV strains. When tested against HIV possessing mutations associated with AZT resistance (D67N/K70R/T215F/K219Q), 18a showed an EC50 of 9.1 μM which amounts to a resistance of 9 fold (Table 4). However, the degree of HIV resistance imparted by these mutations to 18a was substantially less than that to AZT (56 fold). By contrast, testing of 18a against the NNRTI-resistant HIV (L100I/K103N) yielded better potency than against WT HIV (Table 4). Interestingly, the same NNRTIr HIV did not show enhanced susceptibility to AZT (Table 4).
Table 4.
EC50 (μM) values for AZT and AZT-triazole derivative 18a against wild type (WT), AZT-resistant (AZTr), and NNRTI-resistant (NNRTIr) HIVs.
NNRTIr HIV possesses L100I/K103N mutations in RT that confer high-level resistance to most NNRTIs;
Fold resistance relative to WT HIV.
Viral hypersusceptibility of NRTI-resistant HIVs to NNRTIs31,32 and NRTIs33,34 is well known. However, hypersusceptibility of NNRTI-resistant HIVs to other classes of antivirals is rarely reported. That mutations associated with NNRTI resistance confer hypersusceptibility of HIV to our AZT-triazoles is particularly intriguing. Expanded antiviral profiling to include most of our active analogues resulted in 2- to 5-fold enhanced potencies against NNRTI-resistant HIV as compared to WT virus (Table 5). These results substantiate the hypersusceptibility of NNRTI-resistant HIV to our AZT-triazole analogues, though the mechanism involved is presently unclear.
Table 5.
Hypersusceptibility of NNRTIr HIV to AZT-triazole NRTIs
| Compound | EC50 WT HIV (μM) | EC50 NNRTIr HIV (μM) | ratio NNRTIr/WT |
|---|---|---|---|
| 9d | >5.0 | 2.4 | < 0.5 |
| 17d | 7.2 | 2.1 | 0.3 |
| 17f | 4.7 | 1.0 | 0.2 |
| 18a | 1.0 | 0.6 | 0.6 |
| 18e | 4.1 | 1.8 | 0.4 |
AZT-triazole Inhibition of HIV-1 RT-directed DNA Synthesis
AZT-5’-triphosphate is readily used as a substrate by HIV RT, and once incorporated into the viral DNA, acts as a chain terminator to prevent further DNA chain elongation. The anti-HIV activity of the AZT-triazole analogues suggested that they might function in a similar manner. To evaluate this, we carried out biochemical studies with the triphosphate forms (21a and 21f) of the two most potent inhibitors (18a and 18f). The triphosphate derivatives 21a and 21f inhibited HIV RT DNA polymerase activity in vitro with IC50 values (inhibitory concentration required for 50% inhibition of the enzyme activity) of 3.7 and 11.8 μM, respectively. We then focused on 21a for detailed biochemical mechanism of action studies. This analogue was a substrate for HIV RT and was incorporated into DNA albeit with reduced efficiency compared to TTP or AZT-TP (data not shown), consistent with the reduced antiviral potency of 18a compared to AZT (Table 4). HIV RT bound a nucleic acid template/primer terminated with either AZT or 21a with similar affinity (KD values of 5.4 and 10.9 nM, respectively), indicating that reduced dissociation of 21a-terminated template/primer was unlikely to contribute to the observed inhibitory activity.
Pyrophosphorolytic Removal of Incorporated 21a
HIV resistance to AZT arises from RT-catalyzed phosphorolytic removal of the chain-terminating AZT.35,36 The partially reduced sensitivity to 18a seen with HIV containing mutations associated with AZT resistance (Table 4) suggested that, like AZT, incorporated AZT-triazole analogues might also be susceptible to phosphorolytic excision, although with lesser efficiency than AZT. We therefore investigated the efficiency of in vitro ATP-mediated excision catalyzed by AZT-resistant (AZTr) HIV RT for the investigated 21a compound compare to the AZT. As seen in figure 2 (a & b), the rate of nucleotide excision of terminal 21a (0.0126 min-1) was substantially slower than that of terminal AZT (0.024 min-1), consistent with the reduced level of resistance to 18a shown by AZT-resistant HIV (Table 4).
Figure 2.
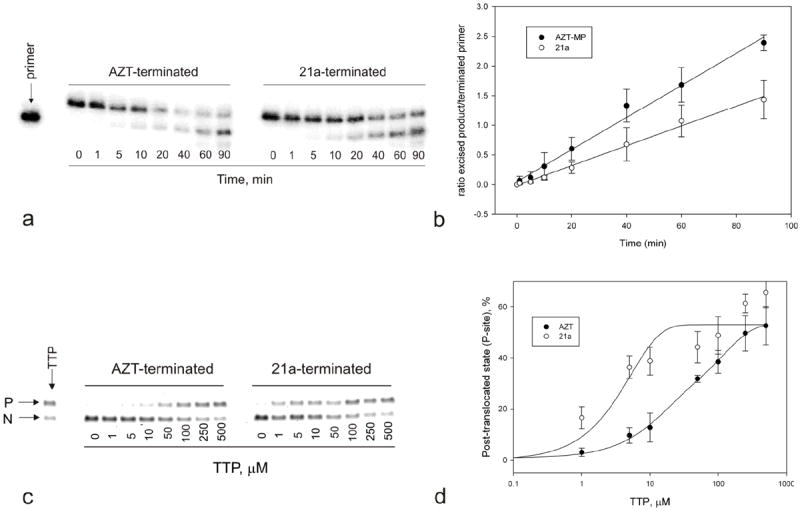
(a) ATP-mediated excision of chain-terminating nucleotides by AZTr RT. Excision reactions were carried out as described in Materials and Methods. Assays were quenched at different times of reaction and monitored by gel electrophoresis. (b) Rate of nucleotide excision for AZT-terminated (●) and 21a-terminated (○) primers. (c). Fe2+- mediated site-specific footprinting assay. Reactions were carried out as described in Materials and Methods. The next incoming nucleotide (TTP) was added in increasing concentrations prior to the Fe2+-mediated cleavage. (d) Graphical representation of data shown in panel (c).
Fe2+-directed Site-specific Footprinting Analysis of 21a-terminated Template/Primers
The efficiency of phosphorolytic removal of chain-terminating nucleotides for the primer 3’-terminus depends on the translocation state of the RT-primer/template complex.37-40 During active DNA synthesis, the primer 3’-terminal nucleotide resides in the P-site (primer site) which allows binding and positioning of the incoming complementary nucleotide-triphosphate for incorporation. Immediately following this incorporation, the new primer 3’-terminal nucleotide occupies the N-site (nucleotide site). To enable further nucleotide incorporation, the primer terminus must translocate to the P-site again. Thus, the N- and P-sites correspond to pre-translocation and post-translocation states, respectively. Phosphorolytic excision of the primer 3’-terminal nucleotide can occur only when this terminal nucleotide is in the N-site.39,40 The relative occupancy of N- and P-sites by any given 3’-terminal nucleotide (translocation equilibrium) will therefore directly impact on the efficiency of phosphorolytic removal of that terminal nucleotide.37-39 The degree of N- and P-site occupancy can be assessed by the technique of Fe2+-mediated site-specific footprinting37 in which Fe2+ bound in the RT RNase H active site under appropriate conditions generates hydroxyl radicals that cleave the template nucleic acid strand at a position directly correlated with the position of the primer terminus in the RT polymerase active site. This technique showed that AZT-terminated primers preferentially occupy the N-site in AZTr-RT37,39, thereby enabling facile phosphorolytic excision of the terminal AZT.
We used this footprinting approach to compare AZT- and 21a-terminated template/primer positioning in RT (Figure 2 c & d). The latter showed much more facile N- to P-site translocation than did AZT-terminated template/primers. The increased translocation of 21a-terminated primers correlates well with the reduced rate of ATP-mediated phosphorolysis of primer 3’-terminal 21a (Figure 2b), and is consistent with the reduced degree of resistance conferred to the parent nucleoside 18a by AZT-resistance mutations (Table 4).
Computational Modeling
Potential interactions of chain-terminating 18a were assessed by computational modeling approaches. Two different models were constructed: (i) 18a was inserted in place of primer 3’-terminal AZT in the N-site of the RT/nucleic acid complex (PDB 3KLG41), and (ii) 18a was inserted in place of primer 3’-terminal AZT in the P-site of the RT/nucleic acid complex (PDB 3KLH41). In constructing these models, care was taken to ensure that the furanose ring puckering and base glycosidic angle of the 18a conformer corresponded exactly to that of the terminal AZT in the crystal structures (Figure 3). Geometrical constraints applied on the furanose ring puckering and on the spatial orientation of the thymine ring during final geometry optimization resulted in an 18a conformer with the naphthalene ring almost coplanar to the thymine ring (Figure 3).
Figure 3.
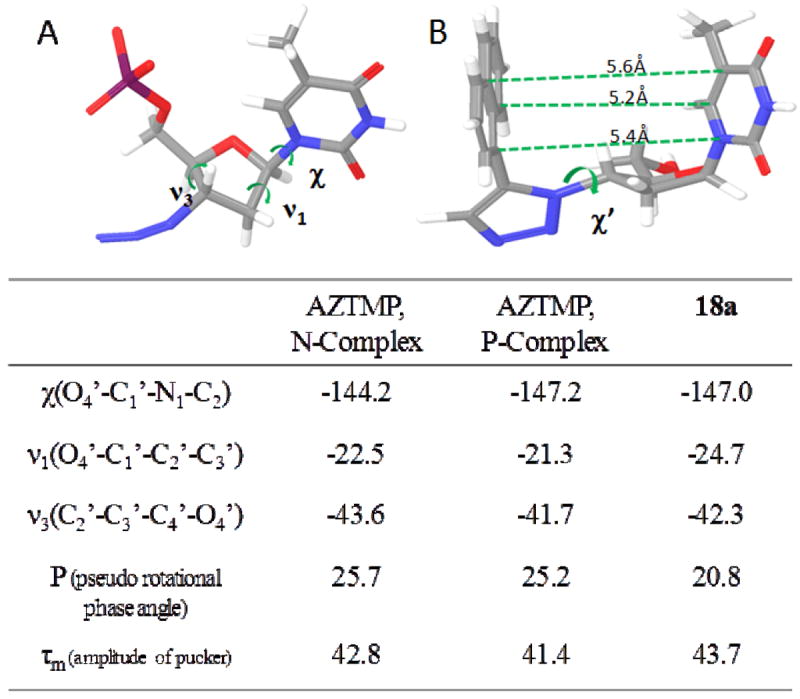
AZTMP and 18a conformational descriptors: (A) furanose ring puckering and glycosidic dihedral angle; (B) Minimized conformer of 18a with furanose ring conformation and glycosidic angle similar to the conformation of AZTMP in the crystal structures.
The triazole and naphthalene ring substituents at C3’ of the furanose ring were allowed free rotation during structural geometry optimization, leading to the generation of a series of rotamers (Figure 4A). The global minimum of χ’ (C2’-C3’-N-N torsional angle) corresponds to that obtained from geometry optimization (χ’= -126°). Nonetheless, we chose several rotamers with χ’ values between -146° and -66° for superimposition over the primer 3’-terminal AZTMP in the N- and P-complexes of HIV-1 RT.
Figure 4.
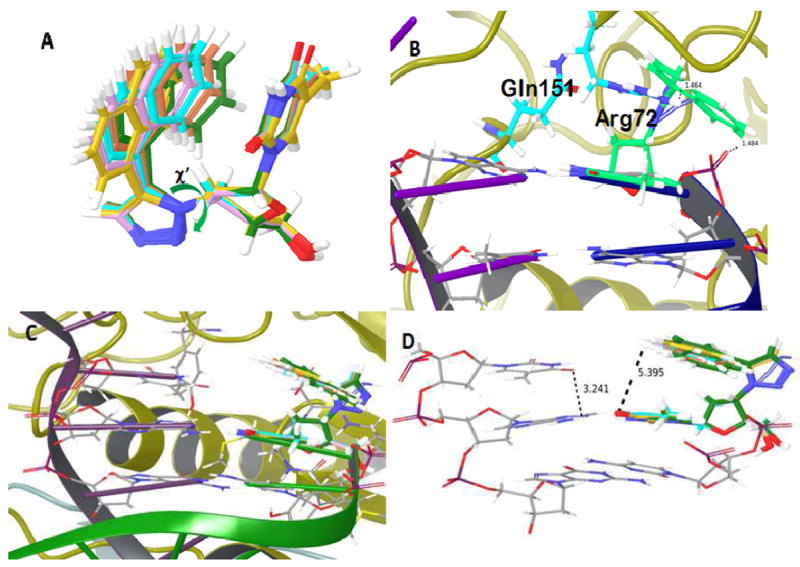
18a modeled in place of primer 3’-terminal AZTMP in the N- and P-sites of HIV RT. (A) Conformational flexibility of the furanose C3’ triazole-naphthalene ring substituents of 18a; (B) Primer 3’-terminal 18a in the N-site may sterically conflict with Gln151 and Arg72, but this conflict can be resolved by rotation of the triazole-naphthalene substituent; (C) No steric conflicts noted for primer 3’-terminal 18a in the P-site; (D) The triazole-naphthalene substituent of primer 3’-terminal 18a in the P-site partially occupies the N-site.
The optimized structure ((χ’= -126°) of primer 3’-terminal 18a in the N-site complex suggests potential steric clashes of the furanose C3’ triazole-naphthalene substituent with the side chains of RT residues Gln151 and Arg72. However, the rotational flexibility of the furanose substituent allows the naphthalene ring to rotate in a manner that minimizes this steric conflict. For example, the 18a rotamer with χ’=-66° is readily accommodated without steric issues (Figure 4B). Interestingly, in this structure, the naphthalene ring “shields” the phosphodiester bond of the primer 3’-terminal 18a. This might interfere with phosphorolytic excision of chain-terminating 18a, consistent with the reduced rate of phosphorolysis noted in our biochemical studies (Figures 2A & B).
In contrast to the N-site complex, the P-site complex with primer 3’-terminal 18a has no steric issues, and in this complex the triazole-naphthalene substituent of 18a can adopt a more energetically favorable torsional angle (χ’ = -126°) (Figure 4C). This more energetically favorable conformation may account in part for the relatively facile translocation of 18a-terminated template/primers (Figures 2C & D). In this structure, the naphthalene ring is coplanar to the thymine ring of 18a. Thus, the P-site complex of primer 3’-terminal 18a mimics two layers of bases occupying both P- and N-sites, with one (the thymine base of 18a) being in the P-site and the other (the naphthalene ring) residing in the N-site, potentially blocking binding of the next incoming nucleotide.
Conclusions
Reported efforts in clicking AZT into 1,2,3-triazoles all failed to achieve any antiviral activities. By introducing a bulky aromatic group at the C5 position of the 1,2,3-triazole, we have identified the first AZT-derived 1,2,3-triazoles with low to sub-micromolar potencies against HIV-1. The observed antiviral activities from CPE based cytoprotection assay were confirmed through a single replication cycle assay with β-gal as the reporter. Mutations associated with AZT resistance also conferred some degree of HIV resistance to the AZT-derived triazoles, whereas mutations associated with nonnucleoside RT inhibitor resistance resulted in viral hypersusceptibility to the compounds. Biochemical analysis of the corresponding triphosphates showed lower ATP-mediated nucleotide excision efficiency than AZT, presumably due to the preferable translocation into the P-site of HIV RT. This finding corroborates the reduced fold resistance (9 for 18a versus 56 for AZT) to AZT-resistant HIV variant. Molecular modeling simulations provided insights into the binding mode and possible residue contacts in N- and P-complexes of HIV-1 RT, and showed that in the N-complex triazole ring substituents shield the 3’-terminal phosphodiester bond from pyrophosphorolysis, consistent with the biochemical observations. AZT-derived 1,2,3-triazoles may provide interesting nucleoside candidates for the future drug development.
Experimental
Chemistry
General Procedures
All commercial chemicals were used as supplied unless otherwise indicated. Dry solvents (THF, Et2O, CH2Cl2 and DMF) were dispensed under argon from an anhydrous solvent system with two packed columns of neutral alumina or molecular sieves. Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All reactions were performed under inert atmosphere of ultra-pure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Mass data were acquired on an Agilent TOF II TOS/MS spectrometer capable of ESI and APCI ion sources. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Phenomenex Gemini,5 micron C18 column (250 mm × 4.6 mm). HPLC conditions: solvent A = H2O, solvent B = MeCN; flow rate = 1.0 mL/min; compounds were eluted with a gradient of 5% MeCN/H2O to 100% MeCN/H2O for 25 min. Purity was determined by total absorbance at 254 nm. All tested compounds have a purity ≥ 96%.
General procedure 1 for the synthesis of 3’-Deoxy-3’-(4-substituted-1H-1,2,3-triazol-1-yl)thymidine derivatives via CuACC
To the mixture of AZT (0.375 mmol, 1.0 equiv.) and alkyne (0.375 mmol, 1.0 equiv.) in 4.0 mL of THF/H2O (3:1) was added freshly prepared 1 M solution of sodium ascorbate (0.1 equiv.) in water, followed by the addition of freshly prepared 1 M solution of CuSO4• 5H2O (0.06 equiv.) in water. The heterogeneous reaction mixture was stirred at room temperature for 12 h and monitored by TLC and MS. After the completion, the reaction was evaporated to dryness. The crude product was purified by column chromatography, eluted with 4-10% MeOH in DCM, yielded the desired 1,4-triazole.
General procedure 2 for the synthesis of 3’-Deoxy-3’-(5-substituted-1H-1,2,3-triazol-1-yl)thymidine derivatives via RuACC
To the mixture of AZT (0.5 mmol, 1.0 equiv.) and alkyne (0.75 mmol, 1.5 equiv.) in 4.0 mL of dry THF was added catalytic amount of Cp*RuCl(PPh3)2 (0.05 equiv.) and stirred at 60 °C for 1-2 days. The reaction was monitored by TLC and MS. The reaction mixture was evaporated to dryness and the crude product was purified by column chromatography, eluted with 4-10% MeOH in DCM, yielded the desired 1,5-triazole.
General Procedure 3 for the Synthesis of deoxynucleoside triphosphates
The deoxynucleoside triazole (0.119 mmol, 1.0 equiv.) and tributylammonium pyrophosphate (0.238 mmol, 2.0 equiv.) were dried under high vacuum for 1 h at ambient temperature in separate round bottom flasks. Throughout the entire experiment, the reaction was maintained under an argon atmosphere. The deoxynucleoside triazole was dissolved in anhydrous pyridine (0.1 mL) and anhydrous dioxane (0.3 mL), a solution of 2-chloro-4H-1,3,2- benzodioxaphosphorin-4-one (0.143 mmol, 1.2 equiv.) in anhydrous dioxane (0.2 mL) was added and stirred at room temperature for 15 min. To the mixture a solution of tributylammonium pyrophosphate (0.213 mmol, 2.0 equiv.) in anhydrous DMF (0.2 mL) was added, which was followed by quick addition of tributylamine (0.596 mmol, 5.0 equiv.) and stirred for 15 min at room temperature. A solution of iodine (1% solution in pyridine/water, 9:1) was then added dropwise till a permanent brown color of iodine was persisted and stirred for 20 min. The excess of iodine was quenched by adding 5% aqueous solution of Na2S2O3. The reaction mixture was evaporated to dryness under vacuum and dissolved in 25% ammonia solution and stirred for 1 h at room temperature. The reaction was monitored by TLC (isopropanol/aq.NH3/H2O = 5:3:2) and MS. The reaction mixture was concentrated under reduced pressure and crude product was purified by reverse-phase preparative HPLC [eluted with a linear gradient of 5% to 40% CH3CN in buffer triethyl ammonium bicarbonate solution (TEAB, 0.1 M, pH=8.0) over 30 min, on a reverse-phase preparative Varian Dynamax Microsorb 100-8 C18 column (250 mm × 44.1 mm, 10 μm) total absorbance at 254 nm at a flow rate of 20.0 mL/min]. The TEAB buffer solution was evaporated by lyophilization afforded the desired 5’-triphosphates triethylammonium salt, which was exchanged into sodium salt by performing Na-ion exchange column chromatography to yield 5’-triphosphates sodium salt as a white solid. The synthesized nucleoside 5’-triphosphates were confirmed by 1H-NMR, 31P-NMR, and HR-MS analyses.
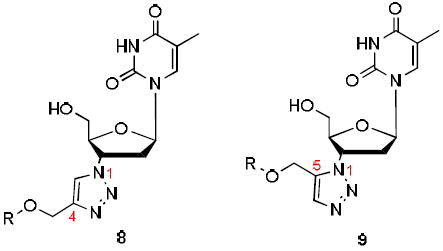
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(phenoxymethyl)-1H-1,2,3-triazol-1yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (8a)
The reaction of AZT (130 mg, 0.486 mmol) with alkyne (64.2 mg, 0.486 mmol) yielded compound 8a (0.18 g, 93%) as a white powder. mp 165–167 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.32 (s, 1H, 3-NH), 8.40 (s, 1H), 7.78 (s, 1H), 7.27 (t, J = 6.2 Hz, 2H), 7.01 (d, J = 6.1 Hz, 2H), 6.93 (t, J = 6.2 Hz, 1H), 6.39 (t, J = 6.6 Hz, 1H), 5.34-5.36 (m, 1H), 5.25 (t, J = 4.8 Hz, 1H, 5’-OH), 5.11 (s, 2H), 4.19-4.20 (m, 1H), 3.58-3.68 (m, 2H), 2.62-2.72 (m, 2H), 1.77 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 158.5, 150.9, 143.4, 136.7, 129.9, 124.7, 121.3, 115.1, 110.1, 84.9, 84.3, 61.4, 61.1, 59.7, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C19H22N5O5 400.1621 [M+H]+, found 400.1641.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(phenoxymethyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (9a)
The reaction of AZT (135 mg, 0.505 mmol) with alkyne (100 mg, 0.758 mmol) yielded compound 9a (93 mg, 48%) as a white solid. mp 182–184 °C; 1H NMR (600 MHz, CD3OD) δ 7.89 (s, 1H), 7.83 (s, 1H), 7.29 (t, J = 7.2 Hz, 2H), 6.96-7.02 (m, 3H), 6.06 (t, J = 6.0 Hz, 1H), 5.40-5.42 (m, 1H), 5.28 (s, 2H), 4.44-4.45 (m, 1H), 3.87 (dd, J = 3.4 Hz, J = 12.6 Hz, 1H), 3.75 (dd, J = 3.0 Hz, J = 12.6 Hz, 1H), 2.68-2.88 (m, 2H), 1.84 (s, 3H, CH3); 13C NMR (150 MHz, CD3OD) δ 166.3, 158.9, 152.2, 138.1, 135.1, 134.9, 130.7, 122.9, 115.8, 111.6, 87.1, 86.6, 62.4, 59.7, 58.8, 39.4, 12.4; HRMS-ESI(+) m/z calcd for C19H22N5O5 400.1621 [M+H]+, found 400.1647.
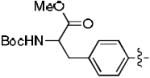
Methyl-2-((tert-butoxycarbonyl)amino)-3-(4-((1-((2S,3S,5R)-2-(hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)propanoate (8b)
The reaction of AZT (300 mg, 1.12 mmol) with alkyne (410 mg, 1.12 mmol) yielded compound 8b (0.58 g, 86%) as a white solid. mp 103–105 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H), 8.40 (s, 1H), 7.79 (s, 1H), 7.24 (d, J = 7.8 Hz, 1H, NH), 7.13 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.40 (t, J = 6.5 Hz, 1H), 5.35-5.36 (m, 1H), 5.25 (m, 1H, OH), 5.08 (s, 2H), 4.18-4.20 (m, 1H), 4.05-4.09 (m, 1H), 3.61-3.69 (m, 1H), 3.58 (s, 3H, OMe), 2.62-2.90 (m, 3H), 2.05 (s, 2H), 1.78 (s, 3H), 1.29 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 173.1, 164.1, 157.2, 155.8, 150.8, 143.4, 136.7, 130.5, 130.3, 124.6, 114.8, 110.1, 84.8, 84.3, 78.7, 61.4, 61.2, 59.7, 55.9, 52.2, 37.6, 36.1, 31.1, 28.5, 12.7; HRMS-ESI(+) m/z calcd for C28H37N6O9 601.2622 [M+H]+, found 601.2620.
Methyl-2-((tert-butoxycarbonyl)amino)-3-(4-((1-((2S,3S,5R)-2-(hydroxymethyl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)-1H-1,2,3-triazol-5-yl)methoxy)phenyl)propanoate (9b)
The reaction of AZT (267 mg, 1.0 mmol) with alkyne (499 mg, 1.5 mmol) yielded compound 9b (18 mg, 30%) as a yellow solid. 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, 3-NH), 7.86 (s,1H), 7.80 (s, 1H), 7.27 (br, 1H), 7.15 (d, J = 8.40 Hz, 1H), 6.94 (d, J = 7.90 Hz, 1H), 6.53 (t, J = 7.2 Hz, 1H), 5.32 (t, J = 5.4 Hz, 1H, 5’-OH), 5.25-5.28 (m, 3H), 4.23-4.25 (m, 1H), 4.05-4.07 (s, 1H), 3.62-3.68 (m, 2H), 3.56 (s, 3H, OMe), 2.73-2.90 (m, 2H), 2.57-2.55 (m, 2H), 1.75 (s, 3H), 1.29 (s, 9H, Boc); 13C NMR (150 MHz, DMSO-d6) δ 178.6, 173.1, 164.1, 156.4, 155.8, 150.9, 145.2, 136.4, 134.6, 133.5, 130.9, 130.6, 114.9, 111.8, 110.1, 85.4, 78.7, 78.7, 61.8, 57.8, 52.2, 46.9, 37.9, 31.1, 28.5, 12.7; HRMS-ESI(+) m/z calcd for C28H37N6O9 601.2622 [M+H]+, found 601.2632.
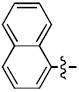
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-((naphthalen-1-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (8c)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (68 mg, 0.375 mmol) yielded compound 8c (156 mg, 93%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, 3-NH), 8.52 (s, 1H), 8.12 (d, J = 6.0 Hz, 1H), 7.85 (d, J = 6.2 Hz, 1H), 7.81 (s, 1H), 7.40-7.51 (m, 4H), 7.17 (d, J = 6.2 Hz, 1H), 6.43 (m, 1H), 5.39-5.41 (m, 1H), 5.34 (s, 2H), 5.25 (t, J = 4.8 Hz, 1H, 5’-OH), 4.23-4.25 (m, 1H), 3.62-3.71 (m, 2H), 2.61-2.76 (m, 2H), 1.79 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 153.9, 150.9, 143.6, 136.7, 134.5, 127.9, 126.9, 126.6, 125.8, 125.3, 124.6, 122.0, 120.8, 110.1, 106.2, 84.9, 84.3, 62.2, 61.2, 59.7, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C23H24N5O5 450.1777 [M+H]+, found 450.1733.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-((naphthalen-1-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (9c)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (150 mg, 0.84 mmol) yielded compound 9c (68 mg, 27%) as a yellow solid. mp 112–115 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.31 (s, 1H, 3-NH), 8.06 (d, J = 6.2 Hz, 1H), 8.00 (s, 1H), 7.85 (d, J = 6.4 Hz, 1H), 7.76 (s, 1H), 7.43-7.52 (m, 4H), 7.16 (d, J = 6.4 Hz, 1H), 6.55 (t, J = 6.0 Hz, 1H), 5.53 (d, J = 12.0 Hz, 1H), 5.47 (d, J = 12.0 Hz, 1H), 5.36-5.38 (m, 1H), 5.27 (t, J = 4.8 Hz, 1H), 4.32-4.34 (m, 1H), 3.59-3.65 (m, 2H), 2.57- 2.67 (m, 2H), 1.70 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 153.3, 150.9, 136.4, 134.7, 133.5, 127.9, 127.1, 126.4, 126.1, 125.1, 121.7, 121.3, 111.9, 110.1, 106.3, 85.3, 84.9, 61.7, 58.8, 58.4, 37.9, 12.7; HRMS-ESI(+) m/z calcd for C23H24N5O5 450.1777 [M+H]+, found 450.1807.
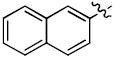
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-((naphthalen-2-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (8d)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (65 mg, 0.375 mmol) yielded compound 8d (141 mg, 84%) as a white solid. mp 233–234 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, 3-NH), 8.46 (s, 1H), 7.81 (s, 1H), 7.80-7.82 (m, 3H), 7.50 (s, 1H), 7.44 (t, J = 7.8 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 7.17 (dd, J = 3.0 Hz, J = 9.6 Hz, 1H), 6.41 (t, J = 7.2 Hz, 1H), 5.36-5.39 (m, 1H), 5.27 (t, J = 5.4 Hz, 1H), 5.25 (s, 2H), 4.21-4.23 (m, 1H), 3.59-3.69 (m, 2H), 2.61-2.75 (m, 2H), 1.78 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 156.3, 150.9, 143.3, 136.7, 134.6, 129.8, 129.1, 127.9, 126.9, 124.8, 124.2, 119.1, 110.1, 107.5, 84.8, 84.3, 61.6, 61.2, 59.7, 53.3, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C23H24N5O5 450.1777 [M+H]+, found 450.1786.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-((naphthalen-2-yloxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (9d)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (102 mg, 0.56 mmol) yielded compound 9d (91 mg, 36%) as a white solid. mp 170–172 °C; 1H NMR (600 MHz, CD3OD) δ 7.90 (s, 1H), 7.86 (s, 1H), 7.76-7.79 (m, 3H), 7.41-7.45 (m, 2H), 7.34 (t, J = 6.2 Hz, 1H), 7.16 (dd, J = 2.1 Hz, J = 12.0 Hz, 1H), 6.60 (t, J = 12.2 Hz, 1H), 5.43-5.46 (m, 1H), 5.41 (s, 2H), 4.47-4.49 (m, 1H), 3.87 (dd, J = 3.6 Hz, J = 12.0 Hz, 1H), 3.76 (dd, J = 3.0 Hz, J = 12.0 Hz, 1H), 2.85-2.89 (m, 1H), 2.69-2.72 (m, 1H), 1.78 (s, 3H, CH3); 13C NMR (150 MHz, CD3OD) δ 164.9, 155.4, 150.8, 136.7, 134.5, 133.6, 129.5, 129.4, 127.2, 126.5, 126.2, 123.8, 117.8, 110.2, 107.2, 85.8, 85.2, 61.2, 58.3, 57.5, 38.2, 11.1; HRMS-ESI(+) m/z calcd for C23H24N5O5 450.1777 [M+H]+, found 450.1804.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(((6-methoxynaphthalen-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (8e)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (79 mg, 0.375 mmol) yielded compound 8e (160 mg, 89%) as a white solid. mp 220–221 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.32 (s, 1H, 3-NH), 8.44 (s, 1H), 7.79 (s, 1H), 7.70-7.72 (m, 2H), 7.43 (s, 1H), 7.25 (s, 1H), 7.11-7.15 (m, 2H), 6.41 (t, J = 6.6 Hz, 1H), 5.36-5.38 (m, 1H), 5.25 (t, J = 5.1 Hz, 1H, 5’-OH), 5.21 (s, 2H), 4.21-4.23 (m, 1H), 3.81 (s, 3H, OMe), 3.61-3.69 (m, 2H), 2.63-2.73 (m, 2H), 1.78 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 156.2, 154.8, 150.9, 143.4, 136.7, 130.0, 129.7, 128.7, 128.6, 124.7 119.3, 111.8, 110.1, 107.9, 106.6, 84.8, 84.3, 61.6, 61.2, 59.7, 55.5, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C24H26N5O6 480.1883 [M+H]+, found 480.1874.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(((6-methoxynaphthalen-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (9e)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (178 mg, 0.83 mmol) yielded compound 9e (80 mg, 30%) as a white solid. mp 228–230 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, 3-NH), 7.99 (s, 1H), 7.80 (s, 1H), 7.72-7.74 (m, 2H), 7.43 (d, J = 6.8 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.12-7.17 (m, 2H), 6.54 (t, J = 6.8 Hz, 1H), 5.39 (s, 2H), 5.30-5.38 (m, 2H, H-3’, 5’-OH), 4.28-4.29 (m, 1H), 3.81 (s, 3H, OMe), 3.63-3.70 (m, 2H), 2.61- 2.64 (m, 2H), 1.74 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 156.4, 154.1, 150.9, 136.5, 134.7, 133.5, 130.3, 129.5, 128.8, 128.6, 119.4, 119.0, 110.1, 108.3, 106.6, 85.4, 85.0, 61.9, 59.0, 58.0, 55.5, 37.9, 12.8; HRMS-ESI(+) m/z calcd for C24H26N5O6 480.1883 [M+H]+, found 480.1899.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(((7-methoxynaphthalen-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione 8f
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (79 mg, 0.375 mmol) yielded compound 8f (154 mg, 86%) as a white solid. mp 237–240 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, 3-NH), 8.45 (s, 1H), 7.79 (s, 1H), 7.71-7.72 (m, 2H), 7.39 (s, 1H), 7.20 (s, 1H), 6.96-7.00 (m, 2H), 6.41 (t, J = 6.6 Hz, 1H), 5.37-5.39 (m, 1H), 5.26 (t, J = 5.4 Hz, 1H, 5’-OH), 5.23 (s, 2H), 4.21-4.22 (m, 1H), 3.84 (s, 3H, OMe), 3.58-3.68 (m, 2H), 2.62-2.74 (m, 2H), 1.79 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 158.2, 156.9, 150.8, 143.4, 136.7, 136.1, 129.6, 129.4, 124.8, 124.3, 116.4, 116.3, 110.1, 107.0, 105.8, 84.8, 84.3, 61.5, 61.2, 59.7, 55.5, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C24H26N5O6 480.1883 [M+H]+, found 480.1874.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(((7-methoxynaphthalen-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (9f)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (177 mg, 0.84 mmol) yielded compound 9f (91 mg, 34%) as a white solid. mp 249-251 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, 3-NH), 7.96 (s, 1H), 7.80 (s, 1H), 7.72-7.74 (m, 2H), 7.39 (s, 1H), 7.22 (s, 1H), 6.98-7.03 (m, 2H), 6.55 (t, J = 6.6 Hz, 1H), 5.41 (s, 2H), 5.30-5.33 (m, 2H), 4.28-4.29 (m, 1H), 3.84 (s, 3H, OMe), 3.66-3.70 (m, 2H), 2.63-2.65 (m, 2H), 1.74 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 158.3, 156.2, 150.9, 136.4, 135.9, 134.7, 133.5, 129.7, 129.5, 124.6, 116.7, 116.0, 110.1, 107.4, 105.8, 85.4, 85.0, 78.5, 61.9, 59.0, 55.5, 37.9, 12.7; HRMS-ESI(+) m/z calcd for C24H26N5O6 480.1883 [M+H]+, found 480.1894.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dion (8g)
The reaction of AZT (130 mg, 0.486 mmol) with alkyne (28 mg, 0.486 mmol) yielded compound 8g (150 mg, 96%) as a white solid. mp 184-185 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.26 (s, 1H), 8.07 (s, 1H), 7.73 (s, 1H), 6.32 (t, J = 6.2 Hz, 1H), 5.26-5.28 (m, 1H), 5.17-5.25 (m, 2H), 4.44 (s, 2H), 4.11-4.13 (m, 1H), 3.61 (dd, J = 3.8 Hz, J = 12.2 Hz, 1H), 3.52 (dd, J = 4.2 Hz, J = 12.2 Hz, 1H), 2.53-2.65 (m, 2H), 1.73 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.9, 148.7, 136.6, 122.7, 110.0, 84.9, 84.3, 61.1, 59.4, 55.4, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C13H18N5O5 324.1308 [M+H]+, found 324.1302.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (9g)
The reaction of AZT (135 mg, 0.505 mmol) with alkyne (42 mg, 0.758 mmol) yielded compound 9g (69 mg, 45%) as a white solid. mp 177–178 °C; 1H NMR (600 MHz, CD3OD) δ 7.92 (s, 1H), 7.65 (s, 1H), 6.61 (t, J = 6.0 Hz, 1H), 5.43-5.42 (m, 1H), 4.74 (s, 2H), 4.38-4.37 (m, 1H), 3.87 (dd, J = 3.0 Hz, J = 12.6 Hz, 1H), 3.78 (dd, J = 3.6 Hz, J = 12.6 Hz, 1H), 2.68-2.92 (m, 2H), 1.89 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.9, 137.4, 136.8, 132.1, 110.2, 85.7, 85.4, 61.1, 58.1, 51.5, 37.8, 11.0; HRMS-ESI(+) m/z calcd for C13H18N5O5 324.1308 [M+H]+, found 324.1306.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(naphthalen-1-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17a)
The reaction of AZT (100 mg, 0.374 mmol) with alkyne (60 mg, 0.375 mmol) yielded compound 17a (139 mg, 89%) as a white solid. mp 238–240 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, 3-NH), 8.90 (s, 1H), 8.39 (s, 1H), 7.99 (s, 2H), 7.94 (d, J = 6.0 Hz, 1H), 7.91 (d, J = 6.4 Hz, 1H), 7.83 (s, 1H), 7.48-7.53 (m, 2H), 6.45 (t, J = 6.5 Hz, 1H), 5.40-5.43 (m, 1H), 5.28 (t, J = 4.8 Hz, 1H, 5’-OH), 4.29-4.31 (m, 1H), 3.66-3.74 (m, 2H), 2.81-2.84 (m, 1H), 2.67-2.71 (m, 1H), 1.80 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.9, 147.1, 136.7, 133.6, 133.0, 129.1, 128.4, 128.1, 127.1, 126.6, 124.0, 121.9, 111.6, 110.1, 84.9, 84.4, 78.6, 61.2, 59.9, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C22H22N5O4 420.1668 [M+H]+, found 420.1664.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(naphthalen-1-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18a)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (128 mg, 0.83 mmol) yielded compound 18a (56 mg, 24%) as a yellow solid. mp 110–112 °C; 1H NMR (600 MHz, CD3OD) δ 8.05 (s, 1H), 8.04 (s, 1H), 7.94-7.99 (m, 2H), 7.90 (s, 1H), 7.81 (s, 1H), 7.55-7.59 (m, 3H), 6.64 (t, J = 6.6 Hz, 1H), 5.39-5.41 (m, 1H), 4.54-4.55 (m, 1H), 3.76 (dd, J = 3.2 Hz, J = 12.0 Hz, 1H), 3.57 (dd, J = 3.0 Hz, J = 12.1 Hz, 1H), 2.82-2.85 (m, 1H), 2.61-2.66 (m, 1H), 1.82 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.9, 138.9, 136.8, 133.6, 133.2, 132.6, 128.9, 128.8, 128.0, 127.5, 127.2, 126.7, 123.4, 110.3, 85.8, 85.3, 61.2, 58.2, 38.2, 10.9; HRMS-ESI(+) m/z calcd for C22H22N5O4 420.1672 [M+H]+, found 420.1700.
1-((2R,4S,5S)-4-(4-(1H-Indol-5-yl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17b)
The reaction of AZT (40 mg, 0.15 mmol) with alkyne (22 mg, 0.15 mmol) yielded compound 17b (51 mg, 81%) as a white solid. mp 235–239 °C; 1H NMR (600 MHz, CD3OD) δ 8.35 (s,1H), 8.01 (s, 1H), 7.92 (s, 1H), 7.57 (d, J = 9.0 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 3.6 Hz, 1H), 6.51 (t, J = 6.0 Hz, 1H), 6.48 (d, J = 3.6 Hz, 1H), 5.47-5.46 (m, 1H), 4.44-4.42 (m, 1H), 3.93 (dd, J = 3.6 Hz, J = 12.0 Hz, 1H), 3.81 (dd, J = 3.0 Hz, J = 12.0 Hz, 1H), 2.99-2.74 (m, 2H), 1.90 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 149.5, 136.8, 128.3, 125.2, 120.9, 119.3, 119.1, 117.3, 111.2, 110.2, 101.3, 85.3, 84.9, 60.7, 59.5, 37.6, 11.0; HRMS-ESI(+) m/z calcd for C20H21N6O4 409.1624 [M+H]+, found 409.1641.
1-((2R,4S,5S)-4-(5-(1H-Indol-5-yl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18b)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (79 mg, 0.56 mmol) yielded compound 18b (65 mg, 41%) as a white solid. mp 140–144 °C; 1H NMR (600 MHz, CD3OD) δ 7.85 (s,1H), 7.77 (s, 1H), 7.68 (s, 1H), 7.57 (d, J = 6.6 Hz, 1H), 7.35 (d, J = 6.4 Hz, 1H), 7.18 (d, J = 6.0 Hz, 1H), 6.68 (t, J = 6.0 Hz, 1H), 6.56 (d, J = 6.0 Hz, 1H), 5.37-5.38 (m, 1H), 4.51-4.52 (m, 1H), 3.76 (dd, J = 3.6 Hz, J = 12.0 Hz, 1H), 3.56 (dd, J = 3.0 Hz, J = 12.0 Hz, 1H), 2.80-2.83 (m, 1H), 2.58-2.63 (m, 1H), 1.85 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 149.5, 136.8, 128.3, 125.2, 120.9, 119.3, 119.1, 117.3, 111.2, 110.2, 101.3, 85.3, 84.9, 60.7, 59.5, 37.6, 11.0; HRMS-ESI(+) m/z calcd for C20H21N6O4 409.1624 [M+H]+, found 409.1758.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(quinolin-4-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17c)
The reaction of AZT (14 mg, 0.052 mmol) with alkyne (8 mg, 0.052 mmol) yielded compound 17c (12 mg, 54%) as a white solid. mp 147–150 °C; 1H NMR (600 MHz, CD3OD) δ 8.73 (s, 1H), 8.63 (s, 1H), 8.13 (s, 1H), 7.93 (s, 1H), 7.82 (s, 2H), 7.66 (t, J = 7.2 Hz, 1H), 6.55 (t, J = 6.6 Hz, 1H), 5.58-5.57 (m, 1H), 5.47 (s, 1H), 4.50 (s, 1H), 3.96 (d, J = 11.4 Hz, 1H), 3.86 (d, J = 11.4 Hz, 1H), 3.04-2.80 (m, 2H), 1.90 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.9, 143.7, 139.8, 136.8, 134.6, 129.8, 128.5, 127.3, 125.6, 124.7, 110.3, 85.4, 84.9, 60.8, 60.1, 37.7, 11.0; HRMS-ESI(+) m/z calcd for C21H21N6O4 421.1624 [M+H]+, found 421.1641.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(quinolin-4-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18c)
The reaction of AZT (40 mg, 0.149 mmol) with alkyne (34 mg, 0.22 mmol) yielded compound 18c (23 mg, 38%) as a white solid. mp 210–212 °C; 1H NMR (600 MHz, CD3OD) δ 9.03 (d, J = 4.2 Hz, 1H), 8.20 (d, J = 6.4 Hz, 1H), 8.04 (s, 1H), 7.89 (t, J = 7.2 Hz, 1H), 7.68-7.71 (m, 2H), 7.62-7.65 (m, 2H), 6.66 (t, J = 6.6 Hz, 1H), 4.96-4.93 (m, 1H), 4.49-4.50 (m, 1H), 3.63 (dd, J = 3.8 Hz, J = 11.8 Hz, 1H), 3.35-3.39 (m, 1H), 2.86-2.90 (m, 1H), 2.51-2.53 (m, 1H), 1.80 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.8, 150.8, 149.6, 147.7, 136.6, 133.4, 130.5, 128.9, 128.2, 127.5, 126.7, 124.4, 123.3, 110.2, 94.1, 85.7, 85.2, 61.1, 58.6, 37.9, 10.9; HRMS-ESI(+) m/z calcd for C21H21N6O4 421.1624 [M+H]+, found 421.1648.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(naphthalen-2-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17d)
The reaction of AZT (100 mg, 0.374 mmol) with alkyne (56 mg, 0.374 mmol) yielded compound 17d (0.128g, 81%) as a white solid. mp 128–131 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, 3-NH), 8.76 (s, 1H), 8.4 (m, 1H), 7.97-7.99 (m, 2H), 7.84 (s, 1H), 7.77 (d, J = 7.2 Hz, 1H,), 7.55-7.59 (m, 3H), 6.48 (t, J = 6.6 Hz, 1H), 5.45-5.48 (m, 1H), 5.28-5.30 (m, 1H, 5’-OH), 4.35-4.36 (m, 1H), 3.69-3.75 (m, 2H), 2.71-2.89 (m, 2H), 1.81 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.9, 146.1, 136.7, 133.9, 130.6, 129.0, 128.8, 127.3, 127.1, 126.5, 126.0, 125.8, 124.1, 110.1, 84.9, 84.3, 61.2, 59.8, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C22H22N5O4 420.1672 [M+H]+, found 420.1680.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(naphthalen-2-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18d)
The reaction of AZT (60 mg, 0.22 mmol) with alkyne (52 mg, 0.33 mmol) yielded compound 18d (25 mg, 27%) as a white solid. mp 110–112 °C; 1H NMR (600 MHz, CD3OD) δ 8.06 (s, 2H), 7.99-7.95 (m, 2H), 7.91 (s, 1H), 7.82 (s, 1H), 7.64-7.56 (m, 3H), 6.65 (t, J = 6.6 Hz, 1H), 5.41-5.40 (m, 1H), 4.56-4.54 (m, 1H), 3.76 (dd, J = 2.4 Hz, J = 11.4 Hz, 1H), 3.58 (dd, J = 3.0 Hz, J = 11.4 Hz, 1H), 2.87-2.62 (m, 2H), 1.83 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.8, 136.8, 133.1, 132.3, 131.6, 128.9, 128.5, 128.4, 128.0, 127.4, 127.1, 126.7, 125.7, 110.2, 85.8, 85.3, 61.1, 58.2, 38.2, 10.9; HRMS-ESI(+) m/z calcd for C22H22N5O4 420.1672 [M+H]+, found 420.1679.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(6-methoxynaphthalen-2-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17e)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (70 mg, 0.375 mmol) yielded compound 17e (0.153 g, 91%) as a white solid. mp >250 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.35 (s, 1H, 3-NH), 8.83 (s, 1H), 8.31 (s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.88 (t, J = 7.2 Hz, 2H), 7.83 (s, 1H), 7.33 (s, 1H), 7.18 (dd, J = 2.6 Hz, J = 9.0 Hz, 1H), 6.45 (t, J = 6.6 Hz, 1H), 5.40-5.42 (m, 1H), 5.30 (t, J = 5.0 Hz, 1H, 5’-OH), 4.28-4.29 (m, 1H), 3.87 (s, 3H, OMe), 3.66-3.75 (m, 2H), 2.69-2.83 (m, 2H), 1.81 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ 164.6, 157.9, 150.9, 147.2, 136.8, 134.4, 130.0, 128.9, 127.9, 125.9, 124.0, 121.3, 119.6, 110.3, 106.4, 84.8, 84.5, 61.4, 61.1, 55.6, 37.5, 12.5; HRMS-ESI(+) m/z calcd for C23H24N5O5 450.1777 [M+H]+, found 450.1775.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(6-methoxynaphthalen-2-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18e)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (143 mg, 0.83 mmol) yielded compound 18e (98 mg, 39%) as a yellow solid. mp 134–136 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.36 (s, 1H, 3-NH), 8.03 (s, 1H), 7.95-7.96 (m, 2H), 7.89 (d, J = 8.4 Hz, 1H), 7.75 (s, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.41 (s, 1H), 7.25 (dd, J = 2.4 Hz, J = 8.8 Hz, 1H), 6.57 (t, J = 6.8 Hz, 1H), 5.21-5.23 (m, 2H), 4.38-4.39 (m, 1H), 3.88 (s, 3H, OMe), 3.56 (dd, J = 1.8 Hz, J = 12.0 Hz, 1H), 3.46 (dd, J = 2.4 Hz, J = 12.0 Hz, 1H), 2.58-2.64 (m, 2H), 1.73 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 158.7, 150.9, 138.6, 136.5, 134.8, 133.4, 130.3, 129.1, 128.5, 128.0, 127.2, 121.6, 120.1, 110.1, 106.3, 85.4, 85.0, 61.8, 58.7, 55.8, 38.2, 12.7; HRMS-ESI(+) m/z calcd for C23H24N5O5 450.1777 [M+H]+, found 450.1811.
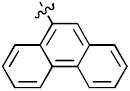
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(phenanthren-9-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17f)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (75 mg, 0.375 mmol) yielded compound 17f (140 mg, 80%) as a white solid. mp >250 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.36 (s, 1H, 3-NH), 8.93 (d, J = 6.0 Hz, 1H), 8.86 (d, J = 6.2 Hz, 1H), 8.82 (s, 1H), 8.49 (d, J = 6.0 Hz, 1H), 8.10 (s, 1H), 8.03 (d, J = 6.6 Hz, 1H), 7.85 (s, 1H), 7.65-7.85 (m, 4H), 6.50 (t, J = 6.2 Hz, 1H), 5.49-5.51 (m, 1H), 5.31 (t, J = 5.4 Hz, 1H, 5’-OH), 4.38-4.36 (m, 1H), 4.06-4.07 (m, 1H), 3.72-3.76 (m, 2H), 2.72-2.92 (m, 2H), 1.81 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.2, 150.9, 146.1, 136.7, 136.1, 131.3, 130.6, 130.2, 129.8, 129.2, 128.2, 127.8, 127.6, 127.5, 127.0, 126.6, 124.4, 123.8, 123.3, 110.1, 84.9, 84.4, 61.3, 59.9, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C26H24N5O4 470.1828 [M+H]+, found 470.1824.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(phenanthren-9-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18f)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (170 mg, 0.83 mmol) yielded compound 18f (92 mg, 35%) as a white solid. mp 165–169 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.30 (s, 1H, 3-NH), 8.96 (d, J = 6.0 Hz, 1H), 8.92 (d, J = 12.0 Hz, 1H), 8.00-8.05 (m, 3H), 7.72-7.79 (m, 3H), 7.62-7.64 (m, 2H), 7.43 (d, J = 6.0 Hz, 1H), 6.62 (t, J = 6.1 Hz, 1H), 4.96 (br, 1H, 5’-OH), 4.74-4.75 (m, 1H), 4.37 (br, 1H), 3.39-3.32 (m, 2H), 2.58-2.64 (m, 1H), 2.36-2.37 (m, 1H), 1.65 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.0, 150.9, 136.3, 136.0, 134.6, 131.2, 130.7, 130.4, 129.6, 128.8, 128.2, 128.0, 127.9, 125.8, 124.0, 123.4, 122.7, 110.1, 85.0, 61.8, 58.9, 55.3, 37.9, 12.6; HRMS-ESI(+) m/z calcd for C26H24N5O4 470.1828 [M+H]+, found 470.1858.
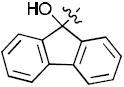
1-((2R,4S,5S)-4-(4-(9-Hydroxy-9H-fluoren-9-yl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17g)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (78 mg, 0.375 mmol) yielded compound 17g (0.156 g, 88%) as a white solid. mp >250 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.31 (s, 1H, 3-NH), 8.19 (s, 1H), 7.77-7.75 (m, 3H), 7.55 (d, J = 7.2 Hz, 2H), 7.37 (t, J = 7.8 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 6.36 (t, J = 6.6 Hz, 1H), 6.34 (s, 1H, OH), 5.29-5.32 (m, 1H), 5.22 (t, J = 4.8 Hz, 1H, 5’-OH), 4.17-4.19 (m, 1H), 3.55-3.65 (m, 2H), 2.58-2.70 (m, 2H), 1.78 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.2, 151.3, 150.8, 149.5, 139.5, 136.7, 129.2, 128.3, 125.6, 122.6, 120.4, 110.1, 84.8, 84.3, 78.4, 61.2, 59.5, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C25H24N5O5 474.1777 [M+H]+, found 474.1770.
1-((2R,4S,5S)-4-(5-(9-Hydroxy-9H-fluoren-9-yl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18g)
The reaction of AZT (150 mg, 0.56 mmol) with alkyne (173 mg, 0.83 mmol) yielded compound 18g (0.103 g, 39%) as a white solid. mp 164–168 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.27 (s, 1H, 3-NH), 7.81 (d, J = 6.2 Hz, 1H), 7.77 (d, J = 6.0 Hz, 1H), 7.67 (s, 1H), 7.52 (s, 1H), 7.39-7.46 (m, 2H), 7.27-7.32 (m, 4H), 6.82 (s, 1H, OH), 6.29 (t, J = 6.4 Hz, 1H), 4.91 (t, J = 4.8 Hz, 1H, 5’-OH), 4.47 (br, 1H), 4.12-4.11 (m, 1H), 3.19-3.20 (m, 1H), 2.73-2.75 (m, 1H), 1.96-1.98 (m, 1H), 1.75 (s, 3H), 1.67-1.68 (m, 1H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.6, 147.5, 141.3, 139.4, 136.0, 132.9, 130.2, 129.1, 109.8, 84.9, 84.8, 78.2, 61.3, 58.2, 37.7, 12.8; HRMS-ESI(+) m/z calcd for C25H24N5O5 474.1777 [M+H]+, found 474.1743.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17h)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (38 mg, 0.375 mmol) yielded compound 17h (0.130 g, 93%) as a white solid. mp 228–231 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, 3-NH), 8.75 (s, 1H), 7.81-7.84 (m, 3H), 7.44 (t, J = 6.2 Hz, 2H), 7.32-7.31 (m, 1H), 6.43 (t, J = 6.6 Hz, 1H), 5.37-5.39 (m, 1H), 5.26-5.27 (m, 1H), 4.25-4.26 (m, 1H), 3.65-3.67 (m, 2H), 2.76-2.79 (m, 2H), 1.79 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.9, 146.9, 136.7, 131.0, 129.3, 128.4, 125.6, 121.4, 110.1, 84.9, 84.3, 61.2, 59.8, 37.5, 12.7; HRMS-ESI(+) m/z calcd for C18H20N5O4 370.1515 [M+H]+, found 370.1513.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-phenyl-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18h)
The reaction of AZT (135 mg, 0.5 mmol) with alkyne (77 mg, 0.75 mmol) yielded compound 18h (72 mg, 39%) as a white solid. mp 195–197 °C; 1H NMR (600 MHz, CD3OD) δ 7.82 (s, 1H), 7.79 (s, 1H), 7.56-7.48 (m, 5H), 6.62 (t, J = 6.0 Hz, 1H), 5.31-5.29 (m, 1H), 4.49-4.48 (m, 1H), 3.76 (dd, J = 3.0 Hz, J = 12.6 Hz, 1H), 3.56 (dd, J = 3.6 Hz, J = 12.6 Hz, 1H), 2.81-2.58 (m, 2H), 1.85 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.8, 138.7, 136.8, 132.4, 129.6, 129.1, 126.1, 110.2, 85.8, 85.3, 61.2, 58.1, 38.1, 11.0; HRMS-ESI(+) m/z calcd for C18H20N5O4 370.1515 [M+H]+, found 370.1561.
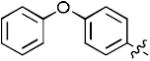
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(4-(4-phenoxyphenyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17i)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (72 mg, 0.375 mmol) yielded compound 17i (0.15 g, 89%) as a white solid. mp 202–203 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.35 (s, 1H, 3-NH), 8.71 (s, 1H),7.83-7.85 (d, J = 6.1 Hz, 2H), 7.82 (s, 1H), 7.40 (t, J = 6.6 Hz, 2H), 7.15 (t, J = 6.4 Hz, 1H), 7.08 (d, J = 6.1 Hz, 2H), 7.03 (d, J = 6.1 Hz, 2H), 6.43 (t, J = 6.6 Hz, 1H), 5.36-5.40 (m, 1H), 5.29 (t, J = 4.8 Hz, 1H, 5’-OH), 4.27-4.29 (m, 1H), 3.64-3.74 (m, 2H), 2.66-2.79 (m, 2H), 1.80 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.4, 157.2, 157.0, 151.1, 146.8, 136.9, 130.8, 127.6, 126.5, 124.4, 121.3, 119.6, 119.5, 110.3, 85.1, 84.5, 61.4, 60.0, 55.6, 37.8, 12.9; HRMS-ESI(+) m/z calcd for C24H24N5O5 462.1777 [M+H]+, found 462.1780.
1-((2R,4S,5S)-5-(Hydroxymethyl)-4-(5-(4-phenoxyphenyl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18i)
The reaction of AZT (135 mg, 0.5 mmol) with alkyne (120 mg, 0.75 mmol) yielded compound 18i (84 mg, 36%) as a white solid. mp 111–114 °C; 1H NMR (600 MHz, CD3OD) δ 7.81 (d, J = 1.2 Hz, 1H), 7.74 (s, 1H), 7.43-7.45 (m, 2H), 7.35-7.38 (m, 2H), 7.15 (t, J = 7.2 Hz, 1H), 7.07-7.08 (m, 2H), 7.02-7.03 (m, 2H), 6.59 (t, J = 7.2 Hz, 1H), 5.30-5.32 (m, 1H), 4.46-4.47 (m, 1H), 3.77 (dd, J = 3.6 Hz, J = 12.0 Hz, 1H), 3.60 (dd, J = 3.0 Hz, J = 12.0 Hz, 1H), 2.58-2.80 (m, 2H), 1.83 (s, 3H, CH3); 13C NMR (150 MHz, CD3OD) δ 164.8, 159.1, 156.0, 150.8, 138.2, 136.8, 132.4, 130.8, 129.7, 124.0, 120.4, 119.3, 118.3, 110.3, 85.8, 85.2, 61.2, 58.0, 38.2, 29.3, 11.1; HRMS-ESI(+) m/z calcd for C24H24N5O5 462.1777 [M+H]+, found 462.1777.
1-((2R,4S,5S)-4-(4-([1,1’-Biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17j)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (100 mg, 0.56 mmol) yielded compound 17j (50 mg, 30%) as a white solid. mp 147–150 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H), 8.81 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.81 (s, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.45 (t, J = 7.8 Hz, 1H), 7.35 (t, J = 6.6 Hz, 1H), 6.43 (t, J = 6.6 Hz, 1H), 5.41-5.37 (m, 1H), 5.28-5.27 (m, 1H), 4.28-4.26 (m, 1H), 3.73-3.63 (m, 2H), 2.81-2.66 (m, 2H), 1.79 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.8, 146.6, 140.0, 139.9, 136.7, 130.1, 129.4, 128.0, 127.6, 126.9, 126.1, 121.5, 110.0, 84.8, 84.3, 61.2, 59.8, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C24H24N5O4 446.1828 [M+H]+, found 446.1827.
1-((2R,4S,5S)-4-(5-([1,1’-Biphenyl]-4-yl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18j)
The reaction of AZT (300 mg, 1.1 mmol) with alkyne (299 mg, 1.6 mmol) yielded compound 18j (220 mg, 43%) as a white solid. mp 129–131 °C; 1H NMR (600 MHz, CD3OD) δ 7.83-7.80 (m, 4H), 7.67 (d, J = 12.0 Hz, 2H), 7.57 (d, J = 12.0 Hz, 2H),7.46 (t, J = 6.2 Hz, 2H), 7.37-7.39 (m, 1H), 6.63 (t, J = 6.2 Hz, 1H), 5.37-5.38 (m, 1H), 5.53 (s, 1H), 3.78 (d, J = 12.0 Hz, 1H), 3.62 (d, J = 6.2 Hz, 1H), 2.78-2.85 (m, 1H), 2.62-2.65 (m, 1H), 1.85 (s, 3H, CH3); 13C NMR (150 MHz, CD3OD) δ 164.5, 150.8, 142.5, 139.7, 138.5, 136.8, 132.5, 129.5, 128.6, 127.6, 127.4, 126.6, 124.9, 117.8, 110.3, 107.2, 85.9, 85.3, 61.2, 58.3, 57.5, 38.2, 11.0; HRMS-ESI(+) m/z calcd for C24H24N5O4 446.1828 [M+H]+, found 446.1818.
1-((2R,4S,5S)-4-(4-Cyclopropyl-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17k)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (25 mg, 0.375 mmol) yielded compound 17k (0.11 g, 89%) as a white solid. mp 207–209 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.32 (s, 1H, 3-NH), 7.99 (s, 1H), 7.78 (s, 1H), 6.36 (t, J = 6.6 Hz, 1H), 5.23-5.25 (m, 2H), 4.14-4.16 (s, 1H), 3.56-3.64 (m, 2H), 2.57-2.68 (m, 2H), 1.90-1.94 (m, 1H), 1.78 (s, 3H, CH3), 0.89-0.88 (m, 2H), 0.68-0.70 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 149.7, 136.7, 134.3, 120.7, 100.9, 84.8, 84.2, 67.5, 61.2, 59.4, 37.5, 12.7, 8.1, 6.9; HRMS-ESI(+) m/z calcd for C15H20N5O4 334.1515 [M+H]+, found 334.1499.
1-((2R,4S,5S)-4-(5-Cyclopropyl-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18k)
The reaction of AZT (135 mg, 0.5 mmol) with alkyne (50 mg, 0.75 mmol) yielded compound 18k (68 mg, 41%) as a white solid. mp 202–205 °C; 1H NMR (600 MHz, CD3OD) δ 7.90 (s, 1H), 7.37 (s, 1H), 6.56 (t, J = 6.0 Hz, 1H), 5.51-5.48 (m, 1H), 4.42-4.41 (m, 1H), 3.90 (dd, J = 3.6 Hz, J = 12.6 Hz, 1H), 3.76 (dd, J = 3.6 Hz, J = 12.6 Hz, 1H), 2.88-2.70 (m, 2H), 1.93-1.90 (m, 4H), 1.10-1.09 (m, 2H), 0.76-0.74 (m, 2H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.8, 140.8, 136.7, 129.8, 110.2, 85.7, 85.1, 61.1, 57.2, 37.4, 11.0, 6.2, 3.0, 2.9; HRMS-ESI(+) m/z calcd for C15H20N5O4 334.1515 [M+H]+, found 334.1551.
1-((2R,4S,5S)-4-(4-Cyclohexyl-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (17l)
The reaction of AZT (100 mg, 0.375 mmol) with alkyne (40 mg, 0.375 mmol) yielded compound 17l (0.130 g, 93%) as a white powder. mp 236–238 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.32 (s, 1H, 3-NH), 8.0 (s,1H), 7.79 (s, 1H), 6.38 (t, J = 6.6 Hz, 1H), 5.24-5.27 (m, 2H, H-3’, 5’-OH), 4.16-4.17 (s, 1H), 3.57-3.65 (m, 2H), 2.59-2.69 (m, 3H), 1.94-1.92 (m, 2H), 1.79 (s, 3H, CH3), 1.23-1.74 (7H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 152.9, 150.8, 136.7, 120.6, 110.1, 84.9, 84.3, 61.2, 59.4, 50.3, 37.5, 35.0, 32.92, 32.90, 26.0, 25.9, 12.6; HRMS-ESI(+) m/z calcd for C18H26N5O4 376.1985 [M+H]+, found 376.1964.
1-((2R,4S,5S)-4-(5-Cyclohexyl-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (18l)
The reaction of AZT (135 mg, 0.5 mmol) with alkyne (82 mg, 0.75 mmol) yielded compound 18l (72 mg, 38%) as a white solid. mp 162–164 °C; 1H NMR (600 MHz, CD3OD) δ 7.86 (s, 1H), 7.53 (s, 1H),6.56 (t, J = 6.0 Hz, 1H), 5.29-5.27 (m, 1H), 4.41-4.39 (m, 1H), 3.86 (dd, J = 3.6 Hz, J = 11.4 Hz, 1H), 3.69 (dd, J = 3.0 Hz, J = 11.4 Hz, 1H), 2.83-2.68 (m, 3H), 1.94-1.75 (m, 8H),1.48-1.39 (m, 5H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.8, 143.5, 136.9, 129.8, 110.2, 85.8, 85.1, 60.9, 57.0, 38.0, 32.7, 32.3, 25.7, 25.3, 11.0; HRMS-ESI(+) m/z calcd for C18H26N5O4 376.1985 [M+H]+, found 376.2201.
1-((2R,4S,5S)-4-(4-(4-Chlorophenyl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione 1d
The reaction of AZT (100 mg, 0.37 mmol) with alkyne (50 mg, 0.37 mmol) yielded compound 1d (135 mg, 90%) as a white solid. mp >250 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, NH), 8.80 (s, 1H), 7.81-7.86 (m, 3H), 7.50 (d, J = 8.4 Hz, 2H), 6.43 (t, J = 6.3 Hz, 1H), 5.39-5.38 (m, 1H), 5.29 (br s, 1H), 4.27 (br s, 1H), 3.64-3.73 (m, 2H), 2.72-2.79 (m, 1H), 2.67-2.71 (m, 1H), 1.80 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.2, 150.9, 145.9, 136.6, 132.8, 129.9, 129.4, 127.3, 121.8, 110.1, 84.8, 84.3, 61.2, 59.9, 37.6, 12.7; HRMS-ESI(+) m/z calcd for C18H18ClN5O4 404.1126 [M+H]+, found 404.1125.
1-((2R,4S,5S)-4-(5-(4-Chlorophenyl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione 18m
The reaction of AZT (100 mg, 0.37 mmol) with alkyne (76 mg, 0.55 mmol) yielded compound 18m (75 mg, 50%) as a white solid. mp 210–214 °C; 1H NMR (600 MHz, CD3OD) δ 7.74 (s, 1H), 7.73 (s, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 1H), 6.52 (t, J = 6.1 Hz, 1H), 5.18-5.19 (m, 1H), 4.39-4.40 (m, 1H), 3.67 (dd, J = 3.6 Hz, J = 12.0 Hz, 1H), 3.48 (dd, J = 3.0 Hz, J = 12.0 Hz, 1H), 2.68-2.74 (m, 1H), 2.53-2.57 (m, 1H), 1.77 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 164.9, 150.8, 137.7, 136.8, 135.8, 132.6, 130.7, 129.1, 124.9, 110.3, 85.9, 85.2, 61.1, 58.2, 38.1, 11.0; HRMS-ESI(+) m/z calcd for C18H18ClN5O4 404.1126 [M+H]+, found 404.1126.
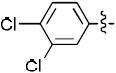
1-((2R,4S,5S)-4-(4-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione 1e
The reaction of AZT (100 mg, 0.37 mmol) with alkyne (63 mg, 0.37 mmol) yielded compound 1e (149 mg, 92%) as a white solid. mp 90–95 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, 3-NH), 8.90 (s, 1H), 8.06 (d, J = 1.8 Hz, 1H), 7.81-7.84 (m, 2H), 7.70 (d, J = 8.4 Hz, 1H), 6.43 (t, J = 6.1 Hz, 1H), 5.41-5.40 (m, 1H, 5’-OH), 5.29-5.30 (m, 1H), 4.26-4.27 (m, 1H), 3.67-3.78 (m, 2H), 2.68-2.82 (m, 1H), 2.65-2.72 (m, 1H), 1.80 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.2, 150.9, 144.8, 136.6, 132.2, 131.6, 130.7, 127.2, 125.5, 122.4, 110.1, 84.9, 84.4, 61.2, 60.0, 55.4, 37.5, 12.6; HRMS-ESI(+) m/z calcd for C18H18Cl2N5O4 438.0736 [M+H]+, found 438.0728.
1-((2R,4S,5S)-4-(5-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-1-yl)-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione 18n
The reaction of AZT (100 mg, 0.37 mmol) with alkyne (95 mg, 0.55 mmol) yielded compound 18n (102 mg, 63%) as a white solid. mp 110–115 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, 3-NH), 7.98 (s, 1H), 7.88 (br s, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.75 (s, 1H), 7.54(d, J = 12.8 Hz, 1H), 6.53 (t, J = 6.3 Hz, 1H), 5.22-5.23 (m, 1H, 5’-OH), 5.12-5.13 (m, 1H), 4.32-4.35 (m, 1H), 3.48-3.60 (m, 2H), 2.64-2.68 (m, 1H), 2.57-2.60 (m, 1H), 1.76 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.1, 150.9, 136.6, 136.2, 133.9, 133.0, 132.3, 131.6, 130.0, 127.4, 110.1, 85.2, 85.0, 61.7, 58.9, 55.3, 38.0, 12.7; HRMS-ESI(+) m/z calcd for C18H18Cl2N5O4 438.0736 [M+H]+, found 438.0733.
Trisodium mono(((2S,3S,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(5-(naphthalen-1-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)methyl triphosphate (21a)
Following the general procedure 3 for the synthesis of 5’-triphosphate, nucleoside 18a yielded compound 21a (43%) as a white solid. 1H NMR (600 MHz, D2O) δ 8.04 (d, J = 12.0 Hz, 1H), 7.93 (d, J = 12.0 Hz, 1H), 7.84 (s, 1H),7.72 (dd, J = 3.2 Hz, J = 6.2 Hz, 1H), 7.62-7.68 (m, 1H), 7.50 (br, 1H), 7.32-7.38 (m, 1H), 6.83-6.87 (m, 2H), 6.32 (t, J = 6.2 Hz, 1H), 4.86 (br, 2H), 4.02 (br, 1H), 3.77 (br, 1H), 2.54-2.58 (m, 1H), 2.22 (br, 1H), 1.72 (s, 3H, CH3); 13C NMR (150 MHz, D2O) δ 175.4, 166.2, 159.5, 151.2, 136.8, 133.8, 133.1, 131.4, 130.8, 128.6, 127.6, 123.9, 122.4, 119.2, 117.9, 116.1, 111.5, 85.5, 82.8, 37.3, 11.4; 31P-NMR (162 MHz, D2O) δ - 23.21 (t, J = 18.79 Hz, 1P), -12.05 (d, J = 18.95 Hz, 1P), -10.63 (d, J = 20.08 Hz, 1P); HRMS-ESI(-) m/z calcd for C22H23N5O13P3 658.0511 [M-3Na+3H]-, found 658.0502.
Trisodium mono(((2S,3S,5R)-3-(4-(6-methoxynaphthalen-2-yl)-1H-1,2,3-triazol-1-yl)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)tetrahydrofuran-2-yl)methyl triphosphate (21e)
Following the general procedure 3 for the synthesis of 5’-triphosphate, nucleoside 18e yielded compound 21e (41%) as a white solid. 1H NMR (600 MHz, D2O) δ 8.25 (br, 1H), 7.84 (br, 1H), 7.76-7.75 (m, 1H), 7.39 (br, 1H), 7.14 (br, 1H), 6.99 (br, 1H), 6.91-6.87 (m, 1H), 6.55 (br,1H), 5.63 (br, 1H), 5.21 (s, 1H), 4.23-4.34 (m, 2H), 3.67 (s, 3H), 3.06 (br, 1H), 2.75-2.83 (m, 2H), 1.90 (s, 3H, CH3); 31P-NMR (162 MHz, D2O) δ - 23.30 (t, J = 12.79 Hz, 1P), -11.82 (d, J = 13.93 Hz, 1P), -10.98 (d, J = 12.79 Hz, 1P); HRMS-ESI(-) m/z calcd for C23H25N5O14P3 688.0616 [M-3Na+3H]-, found 688.0610.
Trisodium mono(((2S,3S,5R)-5-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3-(5-(phenanthren-9-yl)-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)methyl triphosphate) (21f)
Following the general procedure 3 for the synthesis of 5’-triphosphate, nucleoside 18f yielded compound 21f (33%) as a white solid. 1H NMR (600 MHz, D2O) δ 8.23-8.57(m, 3H), 7.76-7.79 (m, 3H), 7.48-7.60 (m, 3H), 7.32-7.38 (m, 2H), 6.09 (br, 1H), 4.83-4.89 (m, 2H), 4.03 (br, 1H), 3.80 (br, 1H), 2.45 (br, 1H), 2.08 (br, 1H), 1.45 (br, 3H, CH3); 31P-NMR (162 MHz, D2O) δ - 22.35 (t, J = 19.90 Hz, 1P), -10.31 (d, J = 19.92 Hz, 1P), -8.92 (d, J = 21.06 Hz, 1P); HRMS-ESI(-) m/z calcd for C26H25N5O13P3 708.0667 [M-3Na+3H]-, found 708.0676.
Biology
Materials and Methods
Reagents
Wild type (WT) and AZT-resistant HIV-1 RT (AZTr) with the mutations D67N/K70R/T215F/K219Q were expressed and purified as previously described.42 Unlabeled dNTPs were purchased from Promega (Madison, WI), adenosine 5’-triphosphate (ATP) from Sigma-Aldrich (St.Lois, MO) and [γ-32P]-ATP was obtained from MD Biosciences (St.Paul, MN). AZT-TP and oligonucleotides were obtained from Trilink Biotechnologies (San Diego, CA). All the other reagents were of the highest quality available and were used without further purification.
Cytoprotection Antiviral Assay.30
The HIV Cytoprotection assay used CEM-SS cells and the IIIB strain of HIV-1. Briefly virus and cells were mixed in the presence of test compound and incubated for 6 days. The virus was pre-titered such that control wells exhibit 70 to 95% loss of cell viability due to virus replication. Therefore, antiviral effect or cytoprotection was observed when compounds prevent virus replication. Each assay plate contained cell control wells (cells only), virus control wells (cells plus virus), compound toxicity control wells (cells plus compound only), compound colorimetric control wells (compound only) as well as experimental wells (compound plus cells plus virus). Cytoprotection and compound cytotoxicity were assessed by MTS (CellTiter® 96 Reagent, Promega, Madison WI) and the EC50 (concentration inhibiting virus replication by 50%), CC50 (concentration resulting in 50% cell death) and a calculated TI (therapeutic index CC50/EC50) were provided. Each assay included the HIV RT inhibitor AZT as a positive control.
Single Replication Cycle Antiviral Assay
HIV replication was evaluated in single replication cycle HIV assays, using P4R5 indicator cells that express CD4, CXCR4 and CCR5 as well as a β-galactosidase reporter gene under the control of an HIV LTR promoter.43 Cells were maintained in DMEM/10% FBS supplemented with puromycin (0.5 g/ml). Viral infectivity was assessed in 96-well microplate assays seeded with P4R5 cells at a density of 5 ×103 cells/well. Cells were exposed to varying concentrations of the drugs for 16h, then inoculated with 25 ng HIV-1 p24/well and the extent of infection was evaluated 48 h post-infection using a fluorescence-based β-galactosidase detection assay as previously described.44
RT Polymerase Activity
HIV RT polymerase activity was determined as described previously45 in 50 μL total volume with 10nM wt HIV-1 RT, 10 μM [H3] TTP and 40nM homopolymeric template/primer poly(rA)-oligo(dT)16 in 50 mM Tris-HCl, pH8.0, 60 mM KCl and 10 mM MgCl2 for 20 min at 37°C. RT solution was incubated with inhibitors in DMSO (1% final concentration of DMSO) for 5 min prior adding primer/template and substrate. Reactions were quenched by 200 μl ice cold 10% TCA containing 20 mM sodium pyrophosphate and filtered using a 1.2μm glass fiber filter 96-well plates (Millipore) followed by sequentially wash with 10% TCA and ethanol. The extent of radionucleotide incorporation was determined by liquid scintillation spectrometry.
Phosphorolytic Nucleotide Excision Assay
40-nt RNA template with the sequence 5’-AGG UGA GUG AGA UGA UAA CAA AUU UGG GAG CCC CAG AUG C and DNA primer 5’-GCA TCT GGG GCT CGG AAA TTT G were obtained from TriLink Biotechnologies (San Diego, CA). Primer was 5’-end labeled using [γ-P32] ATP and T4 polynucleotide kinase (New England BioLabs, Ipswich, MA) according to suppliers’ instructions and then annealed to the template in 1:1.5 molar ratio. Prepared DNA/RNA duplex was chain terminated by AZT-TP and 21a for following experiments.
The phosphorolytic removal of 3’-AZT-MP or 21a was assessed by incubating AZTr RT with the chain-terminated template/primer in 2:1 ratio in 50 mM Tris-HCl pH 8.0, 50 mM KCl. Reactions were initiated by the addition of 6 mM MgCl2 and 3 mM ATP. ATP was previously treated by inorganic pyrophosphatase (0.01 U; Sigma) to ensure removal of any contaminating PPi in the ATP preparation. Reactions were incubated at 37°C and stopped at appropriate time by adding 1 volume of 2×gel loading dye (98% deionized formamide, 10 mM EDTA and 1 mg/ml bromphenol blue). Products were separated on 12% denaturing PAGE containing 7M Urea and analyzed by phosphorimaging (Typhoon 9400, Amersham Biosciences).
Site-Specific Footprinting
Fe2+-mediated site-specific footprints were obtained using following oligonucleotides: 3’-end Cy3 labeled template: Cy3-CGT TGG GAG TGA ATT AGC CCT TCC AGT CCC CCC TTT TCT TTT AAA AAG TGG CTA AGA, and unlabeled primer AGG GGG GAC TGG AAG GGC TAA (TriLink Biotechnologies). Reactions were conducted as described37 with some modifications. Briefly, 50nM of primer/template duplex was incubated with 300nM RT in buffer containing 120mM Na cacodylate, pH7.0, 20mM NaCl, 6mM MgCl2, and 100μM AZT-TP or 21a for 20 min at 37°C to ensure incorporation of AZT-MP and 21a. After that, various concentrations of TTP (next incoming nucleotide) were added and incubated for 10 min followed by treatment with 500 μM Fe(NH4)2SO4. The samples were separated and analyzed by denaturing gel electrophoresis as described before.
Computational Modeling
All modeling studies were carried out employing Schrodinger Small Molecule Drug Discovery Suite 2012 (Schrodinger LLC, New York, NY). Coordinates of two HIV-1 RT structures crosslinked to AZTMP-terminated DNA (pdb access codes 3KLH (P-site complex) and 3KLG (N-site complex) were retrieved from Protein Data Bank and subjected to Protein Preparation Wizard Workflow46 (Schrodinger Suite 2012 Protein Preparation Wizard; Epik version 2.3; Impact version 5.8; Prime version 3.1) for addition of hydrogen atoms, fixing missing atoms and loops, protonation state assignment, and energy minimization with OPLS2005 force field. Refined protein structures were further used to generate docking grids for further docking experiments using Glide47 application (version 5.8). Docking grids were centered on the residues in the closest proximity to AZTMP (Arg72, Tyr115, and Phe160 for the N-site complex, and the YMDD binding motif—Tyr183, Met184, Asp185, Asp186 for the P-site complex). Both grids were made to accommodate ligands within 12Å in length.
LigPrep48 (version 2.5) was employed to generate the 3D structure of 18a and carry out preliminary energy minimization with the OPLS2005 force field. Several low energy furanose ring conformations were generated and the conformation similar to the furanose ring puckering in crystral structure AZTMP was selected for further treatment. Final refinement of 18a structure was carried out with MacroModel49 (version 9.9) and the quantum chemistry module Jaguar50 (version 7.9) applications. Electronic structure calculations were carried out employing Density Functional Theory level of approximation with the hybrid functional B3LYP and 6-31G basis set. The vibrational analysis (Jaguar) was performed on optimized structure and the absence of negative frequencies indicates that the obtain structure not a transition state on potential energy surface. MacroModel49 coordinate scan was used to carry out the rotation around C-N bond with 10° increments. The calculated rotamers vary in the dihedral angle which we designated as χ’ (C2’-C3’-N-N). We calculated potential energy (OPLS2005 force field) for all obtained rotamers. 18a conformers were first superimposed over AZTMP using the Superposition tool implemented in Maestro51 (version 9.3) then Glide47 (version 5.8) was employed for standard precision scoring in place to estimate 18a interactions with surrounding RT residues.
Supplementary Material
Acknowledgments
This research was supported in part by the Research Development and Seed Grant Program of the Center for Drug Design, University of Minnesota, and by grants from the National Institutes of Health (AI076119 and AI079801). We thank Roger Ptak at Southern Research Institute for the cytoprotection antiviral assay data.
ABBREVIATIONS USED
- AZT
3’-azidothymidine
- HIV
human immunodeficiency virus
- CPE
cytopathic effect
- SAR
structure-activity-relationship
- RT
reverse transcriptase
- NRTIs
nucleoside RT inhibitors
- dNTP
deoxynucleoside triphosphate
- hTK
human thymidine kinase
- CuAAC
copper(I)-catalyzed azide–alkyne cycloaddition
- RhAAC
Ruthenium(II)-catalyzed azide–alkyne cycloaddition
Footnotes
Supporting Information Available. Synthesis and 1H NMR data of all alkyne intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cihlar T, Ray AS. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 2010;85:39–58. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 2.Ray AS. Intracellular interactions between nucleos(t)ide inhibitors of HIV reverse transcriptase. AIDS Rev. 2005;7:113–125. [PubMed] [Google Scholar]
- 3.Broder S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res. 2010;85:1–18. doi: 10.1016/j.antiviral.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright K. AIDS therapy. First tentative signs of therapeutic promise. Nature. 1986;323:283. doi: 10.1038/323283a0. [DOI] [PubMed] [Google Scholar]
- 5.Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial Myopathy Caused by Long-Term Zidovudine Therapy. New Engl J Med. 1990;322:1098–1105. doi: 10.1056/NEJM199004193221602. [DOI] [PubMed] [Google Scholar]
- 6.Lynx MD, Mckee EE. 3 ’-azido-3 ’-deoxythymidine (AZT) is a competitive inhibitor of thymidine phosphorylation in isolated rat heart and liver mitochondria. Biochem Pharmacol. 2006;72:239–243. doi: 10.1016/j.bcp.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camerman A, Mastropaolo D, Camerman N. Azidothymidine: crystal structure and possible functional role of the azido group. Proc Natl Acad Sci USA. 1987;84:8239–8242. doi: 10.1073/pnas.84.23.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy V, Obikhod A, Zhang HW, Coats SJ, Herman BD, Sluis-Cremer N, Agrofoglio LA, Schinazi RF. Synthesis and anti-HIV evaluation of 3’-triazolo nucleosides. Nucleosides Nucleotides Nucleic Acids. 2011;30:264–270. doi: 10.1080/15257770.2011.580291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazrek HB, Taourirte M, Oulih T, Barascut JL, Imbach JL, Pannecouque C, Witrouw M, De Clercq E. Synthesis and anti-HIV activity of new modified 1,2,3-triazole acyclonucleosides. Nucleosides Nucleotides Nucleic Acids. 2001;20:1949–1960. doi: 10.1081/NCN-100108325. [DOI] [PubMed] [Google Scholar]
- 10.Habich D, Barth W, Rosner M. Synthesis of 3’-(1,2,3-Triazol-1-Yl)-3’-Deoxythymidines. Heterocycles. 1989;29:2083–2088. [Google Scholar]
- 11.Hirota K, Hosono H, Kitade Y, Maki Y, Chu CK, Schinazi RF, Nakane H, Ono K. Synthesis and Anti-Human-Immunodeficiency-Virus (Hiv-1) Activity of 3’-Deoxy-3’-(Triazol-1-Yl)Thymidines and 2’,3’-Dideoxy-3’-(Triazol-1-Yl)Uridines, and Inhibition of Reverse-Transcriptase by Their 5’-Triphosphates. Chem Pharm Bull. 1990;38:2597–2601. doi: 10.1248/cpb.38.2597. [DOI] [PubMed] [Google Scholar]
- 12.Wigerinck P, Vanaerschot A, Janssen G, Claes P, Balzarini J, Declercq E, Herdewijn P. Synthesis and Antiviral Activity of 3’-Heterocyclic Substituted 3’-Deoxythymidines. J Med Chem. 1990;33:868–873. doi: 10.1021/jm00164a063. [DOI] [PubMed] [Google Scholar]
- 13.Zhou L, Amer A, Korn M, Burda R, Balzarini J, De Clercq E, Kern ER, Torrence PF. Synthesis and antiviral activities of 1,2,3-triazole functionalized thymidines: 1,3-dipolar cycloaddition for efficient regioselective diversity generation. Antivir Chem Chemother. 2005;16:375–383. doi: 10.1177/095632020501600604. [DOI] [PubMed] [Google Scholar]
- 14.Lin J, Roy V, Wang L, You L, Agrofoglio LA, Deville-Bonne D, McBrayer TR, Coats SJ, Schinazi RF, Eriksson S. 3’-(1,2,3-Triazol-1-yl)-3’-deoxythymidine analogs as substrates for human and Ureaplasma parvum thymidine kinase for structure-activity investigations. Bioorg Med Chem. 2010;18:3261–3269. doi: 10.1016/j.bmc.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Poecke S, Negri A, Gago F, Van Daele I, Solaroli N, Karlsson A, Balzarini J, Van Calenbergh S. 3’-[4-Aryl-(1,2,3-triazol-1-yl)]-3’-deoxythymidine analogues as potent and selective inhibitors of human mitochondrial thymidine kinase. J Med Chem. 2010;53:2902–2912. doi: 10.1021/jm901532h. [DOI] [PubMed] [Google Scholar]
- 16.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Huisgen R. Kinetics and mechanism of 1,3-dipolar cycloadditions. Angew Chem Int Ed Engl. 1963;2:633–645. [Google Scholar]
- 18.Huisgen R. 1,3-Dipolar Cycloadditions. Past and Future. Angew Chem Int Ed Engl. 1963;2:565–598. [Google Scholar]
- 19.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Chen X, Xue P, Sun HH, Williams ID, Sharpless KB, Fokin VV, Jia G. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J Am Chem Soc. 2005;127:15998–15999. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]
- 21.Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 22.Presolski SI, Hong VP, Finn MG. Copper-Catalyzed Azide-Alkyne Click Chemistry for Bioconjugation. Curr Protoc Chem Biol. 2011;3:153–162. doi: 10.1002/9780470559277.ch110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nwe K, Brechbiel MW. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother Radiopharm. 2009;24:289–302. doi: 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binder WH, Kluger C. Azide/alkyne-“click” reactions: Applications in material science and organic synthesis. Curr Org Chem. 2006;10:1791–1815. [Google Scholar]
- 25.Wang YZ, Ji KG, Lan S, Zhang LM. Rapid Access to Chroman-3-ones through Gold-Catalyzed Oxidation of Propargyl Aryl Ethers. Angew Chem Int Edit Engl. 2012;51:1915–1918. doi: 10.1002/anie.201107561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Müller S, Liepold B, Bestmann HJ. An Improved One-pot Procedure for the Synthesis of Alkynes from Aldehydes. Synlett. 1996;6:521–522. [Google Scholar]
- 27.Ludwig J, Eckstein F. Rapid and Efficient Synthesis of Nucleoside 5’-O-(1-Thiotriphosphates), 5’-Triphosphates and 2’,3’-Cyclophosphorothioates Using 2-Chloro-4h-1,3,2-Benzodioxaphosphorin-4-One. J Org Chem. 1989;54:631–635. [Google Scholar]
- 28.Schols D, Pauwels R, Vanlangendonck F, Balzarini J, De Clercq E. A highly reliable, sensitive, flow cytometric/fluorometric assay for the evaluation of the anti-HIV activity of antiviral compounds in MT-4 cells. J Immunol Methods. 1988;114:27–32. doi: 10.1016/0022-1759(88)90149-4. [DOI] [PubMed] [Google Scholar]
- 29.Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods. 1988;20:309–321. doi: 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
- 30.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 31.Shulman NS, Delgado J, Bosch RJ, Winters MA, Johnston E, Shafer RW, Katzenstein DA, Merigan TC. Nonnucleoside reverse transcriptase inhibitor phenotypic hypersusceptibility can be demonstrated in different assays. J Acquir Immune Defic Syndr. 2005;39:78–81. doi: 10.1097/01.qai.0000159517.78100.7b. [DOI] [PubMed] [Google Scholar]
- 32.Whitcomb JM, Huang W, Limoli K, Paxinos E, Wrin T, Skowron G, Deeks SG, Bates M, Hellmann NS, Petropoulos CJ. Hypersusceptibility to non-nucleoside reverse transcriptase inhibitors in HIV-1: clinical, phenotypic and genotypic correlates. AIDS. 2002;16:F41–47. doi: 10.1097/00002030-200210180-00002. [DOI] [PubMed] [Google Scholar]
- 33.Michailidis E, Ryan EM, Hachiya A, Kirby KA, Marchand B, Leslie MD, Huber AD, Ong YT, Jackson JC, Singh K, Kodama EN, Mitsuya H, Parniak MA, Sarafianos SG. Hypersusceptibility mechanism of Tenofovir-resistant HIV to EFdA. Retrovirology. 2013;10:65. doi: 10.1186/1742-4690-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grant PM, Taylor J, Nevins AB, Calvez V, Marcelin AG, Wirden M, Zolopa AR. International cohort analysis of the antiviral activities of zidovudine and tenofovir in the presence of the K65R mutation in reverse transcriptase. Antimicrob Agents Chemother. 2010;54:1520–1525. doi: 10.1128/AAC.01380-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. Phenotypic mechanism of HIV-1 resistance to 3’-azido-3’-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry. 1998;37:15908–15917. doi: 10.1021/bi981200e. [DOI] [PubMed] [Google Scholar]
- 36.Meyer PR, Matsuura SE, Mian AM, So AG, Scott WA. A mechanism of AZT resistance: An increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol Cell. 1999;4:35–43. doi: 10.1016/s1097-2765(00)80185-9. [DOI] [PubMed] [Google Scholar]
- 37.Marchand B, Gotte M. Site-specific footprinting reveals differences in the translocation status of HIV-1 reverse transcriptase - Implications for polymerase translocation and drug resistance. J Biol Chem. 2003;278:35362–35372. doi: 10.1074/jbc.M304262200. [DOI] [PubMed] [Google Scholar]
- 38.Marchand B, White KL, Ly JK, Margot NA, Wang R, McDermott M, Miller MD, Gotte M. Effects of the translocation status of human immunodeficiency virus type 1 reverse transcriptase on the efficiency of excision of tenofovir. Antimicrob Agents Chemother. 2007;51:2911–2919. doi: 10.1128/AAC.00314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarafianos SG, Clark AD, Das K, Tuske S, Birktoft JJ, Ilankumaran P, Ramesha AR, Sayer JM, Jerina DM, Boyer PL, Hughes SH, Arnold E. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 2002;21:6614–6624. doi: 10.1093/emboj/cdf637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu X, Das K, Han Q, Bauman JD, Clark AD, Jr, Hou X, Frenkel YV, Gaffney BL, Jones RA, Boyer PL, Hughes SH, Sarafianos SG, Arnold E. Structural basis of HIV-1 resistance to AZT by excision. Nat Struct Mol Biol. 2010;17:1202–1209. doi: 10.1038/nsmb.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fletcher RS, Holleschak G, Nagy E, Arion D, Borkow G, Gu ZX, Wainberg MA, Parniak MA. Single-step purification of recombinant wild-type and mutant HIV-1 reverse transcriptase. Protein Expr Purif. 1996;7:27–32. doi: 10.1006/prep.1996.0004. [DOI] [PubMed] [Google Scholar]
- 43.Parikh UM, Koontz DL, Chu CK, Schinazi RF, Mellors JW. In vitro activity of structurally diverse nucleoside analogs against human immunodeficiency virus type 1 with the K65R mutation in reverse transcriptase. Antimicrob Agents Chemother. 2005;49:1139–1144. doi: 10.1128/AAC.49.3.1139-1144.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abram ME, Parniak MA. Virion instability of human immunodeficiency virus type 1 reverse transcriptase (RT) mutated in the protease cleavage site between RT p51 and the RT RNase H domain. J Virol. 2005;79:11952–11961. doi: 10.1128/JVI.79.18.11952-11961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arion D, Sluis-Cremer N, Min KL, Abram ME, Fletcher RS, Parniak MA. Mutational analysis of Tyr-501 of HIV-1 reverse transcriptase - Effects on ribonuclease H activity and inhibition of this activity by N-acylhydrazones. J Bio Chem. 2002;277:1370–1374. doi: 10.1074/jbc.M110254200. [DOI] [PubMed] [Google Scholar]
- 46.Schrödinger Suite 2012 Protein Preparation Wizard; Epik version 2.3, Schrödinger, LLC, New York, NY, 2012; Impact version 5.8, Schrödinger, LLC, New York, NY, 2012; Prime version 3.1. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 47.Glide, version 5.8. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 48.LigPrep, version 2.5. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 49.MacroModel, version 9.9. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 50.Jaguar, version 7.9. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 51.Maestro, version 9.3. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.