

Abstract
Fatty acid binding proteins (FABPs) are members of a highly conserved family of proteins with the task of protecting a cell’s delicate lipid balance. Yet they fail when faced with metabolic or inflammatory stress, turning the cytosol into an inhospitable environment with less than ideal outcomes. This review will focus on how FABPs direct lipid traffic and simultaneously control inflammatory and metabolic pathways under the pressures of the Metabolic Syndrome.
Keywords: fatty acid binding protein (FABP), Metabolic Syndrome, inflammation, obesity, insulin resistance
Insulin resistance, cardiovascular disease, hypertension, and dyslipidemia are all closely linked with obesity in the cluster of pathologies known as the Metabolic Syndrome (1). Obesity can be considered a chronic inflammatory state with adipose tissue secreting multiple cytokines, hormones, and lipids that have wide-ranging metabolic effects (2). At the core of this syndrome is the dysregulation of lipid metabolism. Serum fatty acid levels are elevated in the state of insulin resistance and are thought to contribute to the formation of obesity and diabetes by modifying glucose and lipid metabolism as well as inflammatory cascades (3,4). While the classic understanding of fatty acids describes their role in the basic maintenance of cell structure and energy metabolism, current research demonstrates that fatty acids are also crucial in cell signaling cascades (5-8).
Compared to protein mediated signaling cascades, comprehension of lipid-mediated signaling is in its infancy. Fatty acids and their metabolites can modulate the action or localization of proteins like G-protein coupled receptors, activate or inhibit kinases, and serve as ligands for transcription factors (4,9-12). For example, fatty acids may transmit a stress response through activation of multiple kinases such as inhibitor of kappa kinase (IKK),3 protein kinase C θ, and c-jun NH2-terminal kinase (JNK), all of which have been intimately linked to insulin resistance and other aspects of the Metabolic Syndrome (11-14). Yet, it is clear that lipids like cyclopentanone prostaglandins can also inhibit IKK and are beneficial in rodent models of inflammation (15-19). Additionally, many transcription factors are regulated by lipids including the families of peroxisome proliferator activated receptors (PPARs), liver X receptors (LXRs), hepatocyte nuclear factors, and sterol regulatory element-binding proteins, which play central roles in lipid metabolism, cell differentiation, and the inflammatory response (6,20-22).
The study of lipid-mediated signaling in metabolic and inflammatory pathways and subsequent biological consequences is essential to understand critical cellular responses as well as the fundamental role that these lipids have in the pathogenesis of a range of diseases associated with Metabolic Syndrome. Currently the import of lipids into the cell through transporters such as fatty acid translocase/CD36 and fatty acid transport protein is well characterized, but the intracellular modulation of lipid mediators is poorly understood (5,23). It is likely that protein chaperones are required to aid in the solubilization of intracellular fatty acids and other bioactive lipids. But what protein is capable of conducting this symphony of unconventional signals?
Intracellular lipid binding proteins called fatty acid binding proteins (FABPs) appear to serve as the ringleader in lipid signaling cascades. FABPs are small, abundantly expressed cytoplasmic proteins that reversibly bind hydrophobic ligands such as saturated and unsaturated long chain fatty acids, eicosanoids, and other lipids (24,25). FABPs are found across all species, from Drosophila melanogaster and Caenorhabditis elegans to mice and humans demonstrating a strong evolutionary conservation, yet little is known about their biological function. Each FABP has a binding affinity in the low micromolar range and is promiscuous in that they bind a multitude of lipid molecules, perhaps indicating a more passive, nonspecific role in the cell. But some tissues such as adipose and cells like macrophages express abundant levels of at least 2 FABPs, suggesting a functional specificity for individual family members (25-27). FABPs may maintain membrane integrity by protecting the cell from the detergent effects of excess non-protein bound fatty acids. They may also facilitate the transport of fatty acids and other lipid mediators throughout the cell. In this capacity, FABPs likely either sequester and/or distribute ligands to regulate signaling processes. Overexpression and anti-sense studies in cultured cells have suggested potential roles in fatty acid import, storage, and export, as well as cholesterol and phospholipid metabolism (28-33). In addition, one FABP has been shown to positively modify hormone sensitive lipase (HSL) activity, and may alter PPAR-mediated transcription (33-35). Clear evidence of how FABPs specifically affect cell biology and lipid metabolism generated in complex systems was lacking until FABP-deficient mice were created.
FABP-deficient models and the Metabolic Syndrome
To study the effects of altered fatty acid binding capacity in vivo several FABP null mouse models have been created, including the macrophage/adipocyte isoform aP2 (also known as FABP4), mal1 (epidermal-FABP, FABP5), heart-FABP (FABP3), intestine-FABP (FABP2), and recently liver-FABP (FABP1) (26,36-41). All of the mice display alterations in lipid metabolism, but only the aP2- and mal1- deficient mice have been extensively characterized for development of the Metabolic Syndrome and will be the focus of the remainder of this review.
aP2−/− mice
The first FABP mouse model created was the aP2-deficent mouse model (26). At baseline, the phenotype of aP2−/− mice was unremarkable. The mice were healthy with no defects in the adipose tissue, reproduction, or growth. It was surprising that the deletion of a protein that comprised up to 5% of total cellular protein in adipocytes had no effect on fat development (24,26). It became clear upon examination that another family member was upregulated to compensate for the lack of aP2, to be discussed below (26,29). To provoke stress on the lipid pathways in aP2−/− mice, obesity was induced by a high fat diet and a dramatic phenotype ensued. Despite weighing slightly more than aP2+/+ controls, aP2 null mice were significantly protected from developing high fat diet-induced insulin resistance. aP2−/− mice had slightly, but significantly, increased plasma free fatty acids and moderately decreased cholesterol and triglyceride levels compared to wildtypes (26). There were also alterations in β-adrenergic-induced lipolysis and associated insulin secretion, indicating that FABPs might play a role in systemic defects such as lipotoxicity (29,30,42). To determine if the protected phenotype of increased insulin sensitivity persisted in a more remarkable model of obesity and potentially involved leptin action, aP2-deficient mice were crossed into the leptin-deficient (ob/ob) background (42). The reduction in obesity-induced insulin resistance remained in this model as well, indicating that the protection was not mediated through leptin signaling. In fact, aP2−/− mice in the ob/ob background weighed 15% more, yet had plasma glucose and insulin concentrations that were significantly lower than aP2+/+ob/ob mice. Insulin and glucose tolerance tests demonstrated that aP2−/−ob/ob mice were substantially more insulin sensitive than aP2+/+ob/ob controls (42). From these 2 obesity models, it became clear that aP2 deficiency alters adipocyte biology and FA metabolism such that the animals are partially protected from developing systemic insulin resistance, dyslipidemia, and defects resulting from lipotoxicity.
The improvement in insulin sensitivity and alterations in systemic lipid metabolism in aP2−/− mice stimulated studies to examine the potential impact of aP2 deficiency on the development of cardiovascular disease, another key component of Metabolic Syndrome. This possibility was tested by generating aP2 deficiency in mouse models of atherosclerosis. These studies demonstrated that aP2−/− mice develop as much as 88% less atherosclerosis throughout the aorta when compared to controls in the apolipoprotein (apo)E−/− model on a chow diet, and 91% less on a high-fat Western diet, a more vigorous model of complex lesion formation (27,43). This dramatic protection occurred independent of alterations in serum lipids and systemic insulin sensitivity (27,43).
The overall impact of aP2 deficiency on atherosclerosis was so extensive that we investigated whether aP2 may act locally in the formation of lesions. Indeed, aP2 was observed in lesion macrophages, key cells in the initiation and promotion of atherogenesis (44,45). Until this point, aP2 was thought to be adipocyte-specific. In fact, we found that macrophages express both FABPs expressed in adipocytes, aP2 and mal1 (27). Our group and others have shown that differentiation of monocytes and activation of macrophages by lipopolysaccharide, phorbol esters, modified lipoprotein particle internalization, and ligands for PPARγ will upregulate the expression of aP2 (27,46,47). Furthermore, Fu et al. demonstrated that overexpression of aP2 drives the accumulation of cholesterol in macrophages (31). Importantly, macrophage-specific aP2 was established as primarily responsible for the protection from developing atherosclerosis through aP2−/−apoE−/− bone marrow transplantation into aP2+/+apoE−/− mice (27). These data suggest that local effects of macrophage-aP2 are critical to the formation of atherosclerotic lesions, rather than potential systemic effects mediated through adipocyte-aP2, at least in the context of the models studied. Our current work demonstrates that aP2 plays a role in both generating the local macrophage inflammatory response and in cholesterol trafficking [(27) and unpublished observations]. Observations described here illustrate the functional similarities between the macrophage and adipocyte in that both accumulate lipids, secrete cytokines, and express many of the same genes involved in lipid metabolism and inflammation (48,49). These results underscore that aP2 modulates the development of varied components of the Metabolic Syndrome through its actions in distinct cell types involved in the immune response, energy homeostasis, and metabolic equilibrium.
mal1−/− mice
One caveat about the interpretation of the aP2−/− obesity studies is that another minor adipocyte isoform, mal1, was dramatically upregulated in the adipose tissue of aP2 null mice, but interestingly not to the same extent in macrophages (26,27). Therefore, the mal1-deficient model was created to investigate the contribution of this FABP to the Metabolic Syndrome and to the aP2 null phenotype. In contrast to the aP2−/− model, the obese mal1−/− mouse did not have a dramatic phenotype in terms of glucose or lipid metabolism [(38) and unpublished observations]. mal1-deficient mice on a high fat diet weighed slightly less than wildtype mice and demonstrated small improvements in insulin sensitivity (38). From these studies, we can conclude that the phenotype of the aP2−/− mice is not accounted for by a compensatory upregulation of mal1, because mal1 deficiency also improved components of the Metabolic Syndrome. Hence, our recent efforts have been directed at a full-scale analysis of combined FABP-deficient mouse models in the context of obesity, insulin resistance, diabetes, atherosclerosis, and other inflammatory diseases. These mice are valuable tools in the evaluation of FABP function and the contribution of lipid-mediated signaling to the pathogenesis of varied diseases.
Mice deficient for aP2 or mal1 have one notable feature associated with obesity: a minor but significant elevation of plasma fatty acids (26,38). As discussed above, increased fatty acids are correlated to the development of obesity and insulin resistance, but paradoxically, FABP−/− mouse models are more insulin sensitive (26,38,42). This observation challenges the current dogma in the mechanistic basis of fatty acid action in the Metabolic Syndrome. Is it that the individual fatty acid content is more relevant than the total serum fatty acid concentration? Alternatively, the distribution and availability of intracellular fatty acids (and derivatives) and the cellular responses generated, rather than the absolute amounts, may be more critical in pathological conditions. Although the exact mechanisms are under investigation, it is evident that the ability of FABPs to modify the intracellular and systemic lipid milieu places it proximal to any step in the coupling of lipid mediators to multiple lipid-sensitive metabolic and inflammatory pathways. Further studies of aP2 and mal1 should provide invaluable insights into these questions.
Discussion
Observations obtained from FABP null mice raise the concept of an interesting biological role for FABPs as a potential central regulator of common pathways controlling inflammatory and metabolic signaling. Mice under normal physiologic conditions do not have a compromised phenotype when FABPs are deleted, but they benefit enormously when faced with systemic pathologic stresses, particularly of metabolic and inflammatory origin. Why does this protein exist if its expression promotes such dysfunction? Evolutionary selection has clearly preserved the FABP from worms to humans, indicating that the close link between the inflammatory and metabolic responses underlies the conservation of FABP function. For survival of the most successful organism, efficiency in these responses is crucial to resist infection as well as starvation.
For many signaling systems acutely activated, such as during inflammation, regulatory mechanisms have evolved to amplify and/or attenuate the response. For example, FABPs appear necessary to evoke a strong inflammatory response, but the drawback is that too strong a response can be damaging, as in the case of macrophage infiltration of atherosclerotic lesions (27). Hence, FABPs may be necessary to fine-tune the balance between the availability of metabolic resources, a robust inflammatory response, and its resolution. Evidence obtained thus far indicates that FABPs might serve such a master role in the control of vital cellular responses through lipid-sensitive proteins, such as JNK and PPARγ (unpublished observations). First, aP2 might coordinate the lipid-mediated activation of stress kinases such as JNK or IKK under immune or metabolic stimuli, thus directly linking lipid signals to the established role of these kinases in pro-inflammation and anti-insulin action (11,12,14). Second, consider that the stimulation of the lipid-ligand activated nuclear hormone receptor PPARγ in adipocytes and macrophages strongly induces the expression of aP2 (47,50). This may serve as a negative regulatory mechanism where aP2 acts to sequester lipid ligands from PPARγ to attenuate the signal once activated. Therefore, a working model is that when aP2 is absent, there is no brake to slow the circuit, resulting in a hyperactive PPARγ transcriptional state. In fact, aP2-deficient mice display a phenotype similar to mice or humans being treated with drugs that activate PPARγ, the thiazolidinediones, including improved glucose and lipid metabolism, and decreased insulin resistance, atherosclerosis, and inflammatory response [(26,27,51-53) and unpublished observations]. When life for humans was dictated by feast or famine, the presence of aP2 may have been beneficial to a strong macrophage immune response or the maintenance of adipose as part of the “thrifty” phenotype to survive (54).
The molecular and functional overlaps between macrophages and adipocytes are a striking example of the link between common signaling pathways operating in “immune” and “metabolic” cells. Figure 1 is a model illustrating FABPs at the center of lipid-mediated signaling pathways potentially affecting enzymes and transcription factors involved in inflammation and metabolism. In fact, the foundation for this link is seen in simpler organisms like the fruit fly, where the fat body houses the homologs of the liver, hematopoeitic and immune systems, and adipose tissue in a single functional unit (55,56). Today, under the unnaturally excessive caloric intake, decreased energy expenditure, and high stress lifestyle of modern day humans, FABPs cannot maintain inflammatory or metabolic homeostasis. The presence of FABPs may aid in the formation of obesity, dyslipidemia, atherosclerosis, and overly active immune responses. Further understanding the mechanism of action and eventually modulating activity of FABPs creates exiting opportunities to regulate lipid-sensitive pathways. Interfering with FABP’s ability to orchestrate lipid signals may provide clinicians with pharmaceutical specificity in a highly cell type-restricted manner, based on the limited expression pattern of FABPs. Ideally, therapeutic options may be developed for a broad range of pathologies including obesity, insulin resistance, type 2 diabetes, atherosclerosis, and possibly other inflammatory conditions such as arthritis, asthma, or Alzheimer’s disease. Research is ongoing in our lab and others to determine the true nature of FABPs as master regulators. Are FABPs our friend or foe? That remains to be determined.
FIGURE 1.
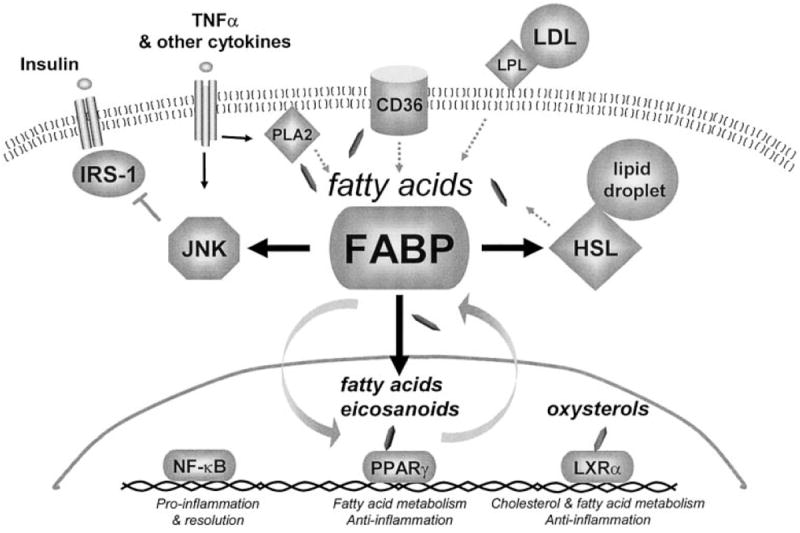
A model demonstrating FABPs at the crossroads of inflammatory and metabolic signaling pathways. Fatty acids are liberated by lipase activity from lipoprotein particles (LPL), membranes (PLA2), and lipid droplets (HSL), or delivered into the cell through membrane transporters like CD36. FABP may regulate 1) the generation, distribution, and coupling of these lipid signals to their biological targets through direct interaction with enzymes such as HSL, 2) the delivery of ligands to the nucleus, and/or 3) the sequestration of lipid mediators such as fatty acids, eicosanoids, or oxysterols. Potential lipid-sensitive targets linked to inflammation and insulin signaling include stress-activated kinases such as JNK and IKK/NFκB, and nuclear hormone receptors such as PPARγ and LXRα. Although the mechanisms are unclear, this model suggests that FABPs control the lipid balance in inflammatory and metabolic cells like macrophages and adipocytes to alter the formation of components of the Metabolic Syndrome.
Footnotes
Presented at the 6th Postgraduate Course on Nutrition entitled “Nutrition and Gene Regulation” Symposium at Harvard Medical School, Boston, MA, March 13–14, 2003. This symposium was orted by Conrad Taff Nutrition Educational Fund, ConAgra Foods, GlaxoSmithKline Consumer Healthcare, McNeil Nutritionals, Nestle Nutrition Institute, The Peanut Institute, Procter & Gamble Company Nutrition Science Institute, Ross Products Division–Abbott Laboratories, and Slim Fast Foods Company. The proceedings of this symposium are published as a lement to The Journal of Nutrition. Guest editors for the lement publication were: W. Allan Walker, Harvard Medical School, George Blackburn, Harvard Medical School, Edward Giovanucci, Harvard School of Public Health, Boston, MA, and Ian Sanderson, University of London, London, UK.
Abbreviations used: apo, apolipoprotein; JNK, c-jun NH2-terminal kinase; FABP, fatty acid binding protein; HSL, hormone sensitive lipase; IKK, inhibitor of kappa kinase; LXR, liver X receptor; PPAR, peroxisome proliferator activated receptor.
LITERATURE CITED
- 1.Saltiel AR. Series introduction: the molecular and physiological basis of insulin resistance: emerging implications for metabolic and cardiovascular diseases. J Clin Invest. 2000;106:163–164. doi: 10.1172/JCI10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hotamisligil GS. Inflammation, TNFalpha, and insulin resistance. In: LeRoith DTS, Olefsky JM, editors. Diabetes Mellitus—A Fundamental and Clinical Text. 3. Lippincott, Williams and Wilkins; New York, NY: 2004. pp. 953–962. [Google Scholar]
- 3.Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. 2000;106:453–458. doi: 10.1172/JCI10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 5.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 6.Moller DE, Berger JP. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int J Obes Relat Metab Disord. 2003;27(Suppl):S17–S21. doi: 10.1038/sj.ijo.0802494. [DOI] [PubMed] [Google Scholar]
- 7.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 8.Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jump DB. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr Opin Lipidol. 2002;13:155–164. doi: 10.1097/00041433-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 11.Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 13.Schmitz-Peiffer C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann N Y Acad Sci. 2002;967:146–157. doi: 10.1111/j.1749-6632.2002.tb04272.x. [DOI] [PubMed] [Google Scholar]
- 14.Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon M, Ye J. Serine phosphorylation of insulin receptor substrate 1 (IRS-1) by inhibitor KappaB kinase (IKK) complex. J Biol Chem. 2002;277:48115–48121. doi: 10.1074/jbc.M209459200. [DOI] [PubMed] [Google Scholar]
- 15.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 16.Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- 17.Cernuda-Morollon E, Pineda-Molina E, Canada FJ, Perez-Sala D. 15-Deoxy-Delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J Biol Chem. 2001;276:35530–35536. doi: 10.1074/jbc.M104518200. [DOI] [PubMed] [Google Scholar]
- 18.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem. 2002;277:10459–10466. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- 20.Muller-Wieland D, Kotzka J. SREBP-1: gene regulatory key to syndrome X? Ann N Y Acad Sci. 2002;967:19–27. doi: 10.1111/j.1749-6632.2002.tb04259.x. [DOI] [PubMed] [Google Scholar]
- 21.Stride A, Hattersley AT. Different genes, different diabetes: lessons from maturity-onset diabetes of the young. Ann Med. 2002;34:207–216. [PubMed] [Google Scholar]
- 22.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 23.Abumrad N, Coburn C, Ibrahimi A. Membrane proteins implicated in long-chain fatty acid uptake by mammalian cells: CD36, FATP and FABPm. Biochim Biophys Acta. 1999;1441:4–13. doi: 10.1016/s1388-1981(99)00137-7. [DOI] [PubMed] [Google Scholar]
- 24.Coe NR, Bernlohr DA. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim Biophys Acta. 1998;1391:287–306. doi: 10.1016/s0005-2760(97)00205-1. [DOI] [PubMed] [Google Scholar]
- 25.Zimmerman AW, Veerkamp JH. New insights into the structure and function of fatty acid-binding proteins. Cell Mol Life Sci. 2002;59:1096–1116. doi: 10.1007/s00018-002-8490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 27.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, Parker RA, Suttles J, Fazio S, Hotamisligil GS, Linton MF. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolly CA, Murphy EJ, Schroeder F. Differential influence of rat liver fatty acid binding protein isoforms on phospholipid fatty acid composition: phosphatidic acid biosynthesis and phospholipid fatty acid remodeling. Biochim Biophys Acta. 1998;1390:258–268. doi: 10.1016/s0005-2760(97)00186-0. [DOI] [PubMed] [Google Scholar]
- 29.Scheja L, Makowski L, Uysal KT, Wiesbrock SM, Shimshek DR, Meyers DS, Morgan M, Parker RA, Hotamisligil GS. Altered insulin secretion associated with reduced lipolytic efficiency in aP2-/- mice. Diabetes. 1999;48:1987–1994. doi: 10.2337/diabetes.48.10.1987. [DOI] [PubMed] [Google Scholar]
- 30.Coe NR, Simpson MA, Bernlohr DA. Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. J Lipid Res. 1999;40:967–972. [PubMed] [Google Scholar]
- 31.Fu Y, Luo N, Lopes-Virella MF, Garvey WT. The adipocyte lipid binding protein (ALBP/aP2) gene facilitates foam cell formation in human THP-1 macrophages. Atherosclerosis. 2002;165:259–269. doi: 10.1016/s0021-9150(02)00305-2. [DOI] [PubMed] [Google Scholar]
- 32.Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci U S A. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helledie T, Antonius M, Sorensen RV, Hertzel AV, Bernlohr DA, Kolvraa S, Kristiansen K, Mandrup S. Lipid-binding proteins modulate ligand-dependent trans-activation by peroxisome proliferator-activated receptors and localize to the nucleus as well as the cytoplasm. J Lipid Res. 2000;41:1740–1751. [PubMed] [Google Scholar]
- 34.Shen WJ, Liang Y, Hong R, Patel S, Natu V, Sridhar K, Jenkins A, Bernlohr DA, Kraemer FB. Characterization of the functional interaction of adipocyte lipid-binding protein with hormone-sensitive lipase. J Biol Chem. 2001;276:49443–49448. doi: 10.1074/jbc.M104095200. [DOI] [PubMed] [Google Scholar]
- 35.Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, Wahli W, Noy N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol Cell Biol. 2002;22:5114–5127. doi: 10.1128/MCB.22.14.5114-5127.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vassileva G, Huwyler L, Poirier K, Agellon LB, Toth MJ. The intestinal fatty acid binding protein is not essential for dietary fat absorption in mice. FASEB J. 2000;14:2040–2046. doi: 10.1096/fj.99-0959com. [DOI] [PubMed] [Google Scholar]
- 37.Martin GG, Danneberg H, Kumar LS, Atshaves BP, Erol E, Bader M, Schroeder F, Binas B. Decreased liver fatty acid binding capacity and altered liver lipid distribution in mice lacking the liver fatty acid-binding protein gene. J Biol Chem. 2003;278:21429–21438. doi: 10.1074/jbc.M300287200. [DOI] [PubMed] [Google Scholar]
- 38.Maeda K, Uysal KT, Makowski L, Gorgun CZ, Atsumi G, Parker RA, Bruning J, Hertzel AV, Bernlohr DA, Hotamisligil GS. Role of the fatty acid binding protein mal1 in obesity and insulin resistance. Diabetes. 2003;52:300–307. doi: 10.2337/diabetes.52.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owada Y, Suzuki I, Noda T, Kondo H. Analysis on the phenotype of E-FABP-gene knockout mice. Mol Cell Biochem. 2002;239:83–86. [PubMed] [Google Scholar]
- 40.Binas B, Danneberg H, McWhir J, Mullins L, Clark AJ. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999;13:805–812. doi: 10.1096/fasebj.13.8.805. [DOI] [PubMed] [Google Scholar]
- 41.Schaap FG, Binas B, Danneberg H, van der Vusse GJ, Glatz JF. Impaired long-chain fatty acid utilization by cardiac myocytes isolated from mice lacking the heart-type fatty acid binding protein gene. Circ Res. 1999;85:329–337. doi: 10.1161/01.res.85.4.329. [DOI] [PubMed] [Google Scholar]
- 42.Uysal KT, Scheja L, Wiesbrock SM, Bonner-Weir S, Hotamisligil GS. Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology. 2000;141:3388–3396. doi: 10.1210/endo.141.9.7637. [DOI] [PubMed] [Google Scholar]
- 43.Boord JB, Maeda K, Makowski L, Babaev VR, Fazio S, Linton MF, Hotamisligil GS. Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:1686–1691. doi: 10.1161/01.atv.0000033090.81345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 45.Glass CK, Witztum JL. Atherosclerosis. The road ahead Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 46.Fu Y, Luo N, Lopes-Virella MF. Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res. 2000;41:2017–2023. [PubMed] [Google Scholar]
- 47.Pelton PD, Zhou L, Demarest KT, Burris TP. PPAR-gamma activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochem Biophys Res Commun. 1999;261:456–458. doi: 10.1006/bbrc.1999.1071. [DOI] [PubMed] [Google Scholar]
- 48.Cousin B, Munoz O, Andre M, Fontanilles AM, Dani C, Cousin JL, Laharrague P, Casteilla L, Penicaud L. A role for preadipocytes as macrophage-like cells. FASEB J. 1999;13:305–312. doi: 10.1096/fasebj.13.2.305. [DOI] [PubMed] [Google Scholar]
- 49.Charriere G, Cousin B, Arnaud E, Andre M, Bacou F, Penicaud L, Casteilla L. Preadipocyte conversion to macrophage. Evidence of plasticity. J Biol Chem. 2003;278:9850–9855. doi: 10.1074/jbc.M210811200. [DOI] [PubMed] [Google Scholar]
- 50.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 51.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 53.Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med. 1994;331:1188–1193. doi: 10.1056/NEJM199411033311803. [DOI] [PubMed] [Google Scholar]
- 54.Auwerx J. PPARgamma, the ultimate thrifty gene. Diabetologia. 1999;42:1033–1049. doi: 10.1007/s001250051268. [DOI] [PubMed] [Google Scholar]
- 55.Rehorn KP, Thelen H, Michelson AM, Reuter R. A molecular aspect of hematopoiesis and endoderm development common to vertebrates and Drosophila. Development. 1996;122:4023–4031. doi: 10.1242/dev.122.12.4023. [DOI] [PubMed] [Google Scholar]
- 56.Tong Q, Dalgin G, Xu H, Ting CN, Leiden JM, Hotamisligil GS. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290:134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]