

Abstract
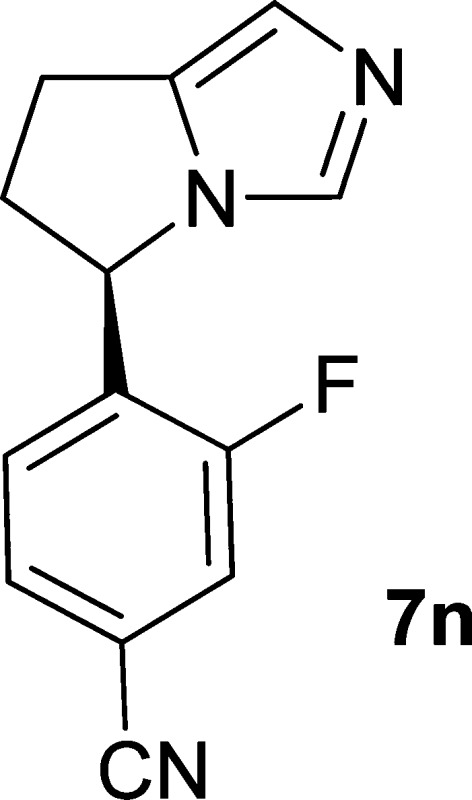
Aldosterone is a key signaling component of the renin-angiotensin-aldosterone system and as such has been shown to contribute to cardiovascular pathology such as hypertension and heart failure. Aldosterone synthase (CYP11B2) is responsible for the final three steps of aldosterone synthesis and thus is a viable therapeutic target. A series of imidazole derived inhibitors, including clinical candidate 7n, have been identified through design and structure–activity relationship studies both in vitro and in vivo. Compound 7n was also found to be a potent inhibitor of 11β-hydroxylase (CYP11B1), which is responsible for cortisol production. Inhibition of CYP11B1 is being evaluated in the clinic for potential treatment of hypercortisol diseases such as Cushing’s syndrome.
Keywords: Inhibitor, CYP11B2, aldosterone synthase, aldosterone, hypertension, enzyme, CYP11B1, Cushing’s syndrome, cortisol
One of the primary functions of aldosterone through the mineralocorticoid receptor (MR) is to effect retention of sodium and excretion of potassium by the kidney.1 The elevation of aldosterone causes an increase in blood pressure as well as facilitating other cardiac, renal, and vascular damage. Activation of the renin-angiotensin system (RAS) induces aldosterone production. As such, MR antagonists have been used in the treatment of heart failure.2−4 While both RAS inhibitors and MR antagonists have been shown to reduce some of the pathological effects of aldosterone, there are noted drawbacks. In the case of RAS inhibition, a reduction of aldosterone is realized initially; however, this is not maintained.5,6 Likewise, MR antagonists do not reduce the level of aldosterone, and in fact, they have been shown to induce aldosterone production.7 Thus, it was thought that there would be therapeutic benefit in directly inhibiting aldosterone production.
Aldosterone is produced in the zona glomerulosa of the adrenal gland by the enzymatic action of aldosterone synthase (CYP11B2) on deoxycorticosterone.8,8b Clinical observations suggested that the racemic aromatase (CYP19) inhibitor fadrazole affected aldosterone levels and subsequent preclinical studies demonstrated that the R-enantiomer (FAD286, Figure 1) was a potent inhibitor of CYP11B2.9 From this understanding we embarked on a program to investigate the structure–activity relationship of the FAD286 scaffold and to gain a more extensive understanding of the potential of aldosterone synthase inhibition to treat aldosterone-driven pathologies.
Figure 1.

Imidazole derived aldosterone synthase inhibitors.
Relatively little was known about the impact of substitution at R1, although we quickly realized that, as with FAD286, chirality at this point of attachment was important. The impact of altering the size of the saturated ring was not understood at the outset. Preliminary structure–activity relationships (SAR) around the phenyl ring indicated that R2 would be a most promising site for optimization.
In general the compounds from series I (n = 1), series II (n = 2), and series III (n = 3) were prepared as outlined in Scheme 1. Intermediates 3 could be prepared by straightforward methods from the corresponding starting materials 1a, 1b, or 2. The imidazole intermediates 3 underwent alkylation with the corresponding substituted benzyl bromide 4 upon heating in acetonitrile. Full removal and scavenging of the trityl group was accomplished by treatment with diethylamine and MeOH. Following alkylation, ring closure for series I and II was readily possible following removal of the TBS protecting group, chlorination, and then treatment with potassium tert-butoxide. The racemic product 6 then underwent separation by chiral HPLC to afford enantiomers 7 and 8.
Scheme 1. General Synthetic Scheme.

Reagents and conditions: (a) 1a or 1b, MeOH, HCl. (b) TrCl, Et3N, CH3CN. (c) LiAlH4, THF, 0 °C. (d) TBSCl, imidazole, CH2Cl2. (e) 1a or 1b, TrCl, pyridine. (f) BH3·THF, THF, 0 °C. (g) Ethanolamine, 90 °C. (h) TBSCl, imidazole, CH2Cl2. (i) 2, TrCl, Et3N, DMF (j) Ph3P+–CH2CH2CH2OTBS, nBuLi, THF. (k) 5% Pd/C, H2, EtOH. (l) CH3CN, 80 °C; then MeOH/Et2NH, 80 °C. (m) 5b or 5c, 4 M HCl in dioxane, 0 °C. (n) SOCl2. (o) tBuOK, THF. (p) Chiral semiprep HPLC. (q) 5a, NCC(O)OMe, LiHMDS, THF, −78 °C. (r) MsCl, Et3N, CH2Cl2, 0 °C. (s) NaI, K2CO3, Et3N, DMF, 80 °C. (t) LiOH, THF/H2O (3:2); HCl neutralization. (u) 12, Et3N, DMSO, 100 °C. (v) Oxalyl chloride, cat. DMF, CH2Cl2; then Et3N, amine, or alcohol.
In the case of series I, the formation of the saturated ring in a similar manner to series II and III was problematic as the corresponding alkyl chloride derivative underwent elimination rather than cyclization. To overcome this problem, the benzylic position of 5a was activated by the installation of an ester moiety (10) prior to cyclization. This gave facile conversion to the desired five-membered ring. Compound 11 could then be separated by chiral HPLC to give 12 and provide the first data regarding the tolerance of substitution at R1. Alternatively, the ester functionality could be removed by hydrolysis followed by decarboxylation to give 6 (n = 1), which then underwent chiral HPLC separation to provide the corresponding enantiomers 7 and 8.
Following hydrolysis of ester 11, both amide and ester derivatives 14 could be prepared by treatment of the carboxylic acid with oxalyl chloride and then the corresponding amine or alcohol.
As had been noted with FAD286, chiral separation proved to be critical as it was shown early on that for the majority of compounds in all three series (I, II, and III), only one enantiomer inhibited CYP11B2 activity, while the opposite enantiomer inhibited CYP19 activity. Some exceptions to this trend are described below (Table 1).
Table 1. Inhibition of Cellular Aldosterone Production and Aromatase Enzymatic Function (CYP19)a.
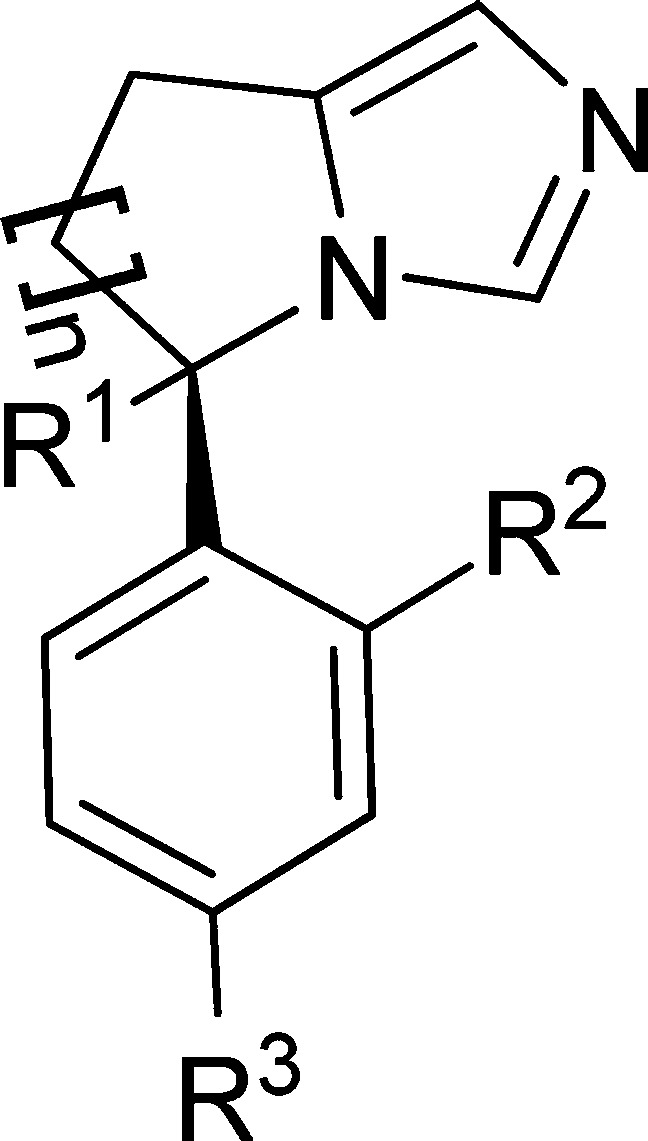
| compd | n | R1 | R2 | R3 | ASc cellular IC50 (nM) | CYP19d %inh @ 1 μM* or IC50 (μM)§ |
|---|---|---|---|---|---|---|
| FAD286 | 2 | H | H | CN | 30 | 6§ |
| 7a | 2 | H | Ph | CN | 56 | |
| 7b | 2 | H | Ph | H | 37 | |
| 7c | 2 | H | F | CN | 87 | 20* |
| 6ab | 2 | H | Cl | CN | 792 | 99* |
| 6bb | 2 | H | Me | CN | 464 | 100* |
| 6cb | 2 | H | –OMe | CN | 390 | 100* |
| 7d | 3 | H | 4-F–Ph– | CN | 1 | |
| 7e | 3 | H | 4-F–Ph– | H | 2 | |
| 7f | 3 | H | Ph | H | 2 | |
| 7g | 3 | H | F | CN | 42 | 1.1§ |
| 7h | 3 | H | Cl | CN | 16 | 100* |
| 7ia | 3 | H | –OMe | CN | 520 | |
| 12a | 1 | –C(O)OMe | Cl | CN | 17 | 0.050§ |
| 12b | 1 | –C(O)OMe | –OMe | CN | 3 | |
| 12c | 1 | –C(O)OMe | F | CN | 351 | |
| 14a | 1 | 4-F-Ph–CH2–O(O)C- | Cl | CN | 1.5 | 95* |
| 14b | 1 | 4-F–Ph–CH2(Me)N(O)C– | –OMe | CN | 5 | 45* |
| 7j | 1 | H | Ph | H | 441 | |
| 7k | 1 | H | 4-F–Ph– | H | 140 | |
| 7l | 1 | H | Cl | CN | 13 | |
| 7m | 1 | H | OMe | CN | 5 | 80* |
| 7n | 1 | H | F | CN | 9 | 6§ |
| 7o | 1 | H | Me | CN | 8 | 87* |
| 7p | 1 | H | Br | CN | 10 | 47* |
All compounds in the table are single enantiomers except where noted. With the exception of 7n, the absolute configuration was not determined.
Racemic compound.
Aldosterone production assay in human adrenocortical carcinoma (NCI-H295R) cells.
Enzymatic aromatase (CYP19) assay. See Supporting Information for more detail.
A number of modifications were made to the R2 position of series II, and the potency was assessed in an assay measuring inhibition of aldosterone secretion in NCI-H295R cells.11 The inclusion of a phenyl group at this position such as 7a (56 nM) and 7b (37nM) provided good potency. These compounds also demonstrate that the inclusion of a pheny group at R2 allows for removal of the nitrile at R3 without loss of activity. However, much to our surprise, other very simple modifications at R2 such as fluoro (7c; 87 nM), chloro (6a; 792 nM), methyl (6b; 464 nM), and methoxy (6c; 390 nM) led to loss in potency.
In parallel with SAR studies on series II, the exploration of the effect of the saturated ring size on activity was pursued. Because of the synthetic challenges with forming the five-membered ring analogues (vide supra), the more readily prepared seven-membered analogues were explored first. In this case, as was seen with series II, substitution of the phenyl ring at R2 with a substituted (7d and 7e) or unsubstituted phenyl ring (7f) provided potent inhibition of CYP11B2. In fact, these analogues were among the most potent compounds both in vivo and in vitro, yet they tended to be less selective for other P450 enzymes. As with series II, a phenyl substituent at R2 allowed for removal of the nitrile without a loss in potency (7d vs 7e). Encouraged by the tolerance for expansion of ring size, we pursued additional modifications at the R2 position in series III. In contrast to series II, nonphenyl substitution with small groups such as fluorine (7g) or chlorine (7h) were well tolerated with IC50 values of 42 and 16 nM, respectively. The methoxy derivative (7i), however, provided much lower inhibition. More troubling for series III was significant CYP19 inhibition with both 7g and 7h being >6-fold more potent than FAD286.
Given that the general SAR for CYP11B2 inhibition thus far strongly suggested that analogues with a saturated seven-membered ring were more tolerant of substitution than those with a six-membered ring, it would be expected that the five-membered analogues, if they fit the trend, would be less active than the six. With a reasonable synthesis for the five-membered analogues now in hand, it was decided to determine if this indeed was the case.
As mentioned above, an ester moiety needed to be incorporated into 5a to facilitate the eventual ring closure. Following resolution of the enantiomers, the simple chloro-substituted compound 12a provided good inhibition of CYP11B2 with IC50 of 17 nM. Unfortunately, 12a also demonstrated very potent inhibition of CYP19. However, the surprising tolerance for substitution at R1 in series I encouraged us to pursue the exploration of the phenyl ring. In contrast to series II, the substitution of R2 with a methoxy (12b) provided an increase in potency, whereas fluoro substitution (12c) caused a loss in potency. Additional ester and amide derivatives such as 14a and 14b were prepared and found to provide good inhibition of CYP11B2, yet most had an undesirable level of CYP19 inhibition.
The apparent increase in tolerance for substitution within series I vs series II and III warranted further exploration of analogues of type 7. As phenyl substitution at the R2 in both series II and III provided good potency in vitro and in vivo, we prepared such analogues in series I. Once again, we observed a disconnect in the SAR as the corresponding analogues 7j and 7k showed a substantial loss in potency against CYP11B2. Gratifyingly, 7l, obtained by decarboxylation of 12a followed by chiral separation, was very potent against CYP11B2 (IC50 13 nM). Again, no translation of SAR between the series was observed, as the direct analogues of 7l in the six-membered series (6a) had low activity (792 nM), while the seven-membered analogue (7h) provided similar inhibition (16 nM).
Building on this result, additional R2 analogues were prepared including methoxy (7m, 5 nM), fluoro (7n, 9 nM), methyl (7o, 8 nM), and bromo (7p, 10 nM), among others. All maintained potency against CYP11B2. Equally important was the fact that several of these analogues had attenuated potency against CYP19, with 7n inhibiting CYP19 comparable to FAD286.
Compound 7n was found to have good selectivity against other P450 isoforms (IC50s: CYP3A 4.8 μM; CYP2D6 >10 μM; CYP2C9 >10 μM). CYP11B1, which is highly homologous to CYP11B2 and is responsible for cortisol production, was a notable exception.10 As shown in Table 2, some compounds in this series, such as 14a, demonstrated greater inhibition of CYP11B1 enzymatic activity than that seen for CYP11B2. However, compound 7n along with most other compounds in the series provided modest selectivity over CYP11B1. On this basis, it was desirable to determine how well the compounds reduced aldosterone in vivo and to evaluate the impact of CYP11B1 inhibition.
Table 2. Inhibition of Recombinant Human and Rat CYP11B2 and CYP11B1 Enzyme Activity.
An efficient pharmacokinetic–pharmacodynamic (PK–PD) model in Sprague–Dawley (SD) rat was established to enable rapid compound screening and the development of in vivo SAR for the CYP11B2 inhibitors.12 Analogous to the cellular assay, an angiotensin-II (Ang-II) infusion was used to stimulate an increase in aldosterone production and establish an experimental baseline plasma aldosterone concentration (PAC). Following oral administration of various CYP11B2 inhibitors the PAC was measured at given time points. As shown in Table 3, compounds in series I (e.g., 7l and 7n) provided better oral exposure in general than either series II or III. Compound 7n provided both good exposure after oral dosing (%F = 42) and strong reduction of PAC (65%) over the duration of the study. Compounds 7d, 7g, and 12a provided good reduction in PAC (66 and 81%, respectively) despite having very low oral exposure. Given that the cellular and enzymatic potency for these compounds are in line with the others, the in vivo efficacy for 7d, 7g, and 12a may be in part due to the generation of active metabolites.
Table 3. Pharmacokinetic–Pharmacodynamic Parameters for Selected Compoundsa.
| compd | dose (mg/kg)b | TWA0–8 [C] (nM)d | TWA0–8 % reduction of PACe | %Ff |
|---|---|---|---|---|
| 7d | 1b | 6 ± 1 | 66 ± 4 | 1 |
| 7f | 1b | 4 ± 2 | 27 ± 17 | <1 |
| 7e | 1b | 152 ± 72 | 73 ± 8 | 8 |
| 7g | 1b | BQLg | 69 ± 3 | n.d.h |
| 12a | 1c | 6 ± 2 | 81 ± 1 | 1 |
| 7l | 1c | 152 ± 21 | 51 ± 7 | 59 |
| 7n | 1c | 377 ± 27 | 65 ± 2 | 42 |
| 7m | 1b | 33 ± 8 | 50 ± 15 | 18 |
| 14b | 1c | 15 ± 8 | 35 ± 10 | 8 |
Sprague–Dawley rat (n = 3).
Compound dosed in corn starch/water.
Compound dosed in HCl (1.5 equiv of 1 N/cornstarch/water).
Time-weighted average (TWA) compound concentration from 0 to 8 h.
TWA % reduction in plasma aldosterone concentration (PAC) from baseline.
Calculated from 0.3 mg/kg i.a. dose.
Below quantitation limit.
Not calculated since oral exposure was BQL.
As noted above, one of the key questions was how the modest in vitro CYP11B2/CYP11B1 selectivity would translate to an effect on corticosterone levels in vivo. To address this question, a second PK–PD model in SD rat was developed to evaluate the effect of 7n on plasma corticosterone concentrations (PCC; unlike in humans, corticosterone is the primary corticosteroid in rats).12 In this model, an increase in baseline corticosterone level was stimulated with ACTH, followed by treatment with compound. Although compound 7n showed a dose-dependent reduction in PCC following ACTH stimulation, the effects on PAC levels were consistently greater on both a dose and exposure basis.13 On the basis of the ability of 7n to effectively reduce aldosterone levels in vivo and its generally favorable profile, the compound was selected for initial human proof-of-concept studies and to understand any limitations of the potential concurrent cortisol reduction.
In human studies, treatment with 7n was well tolerated and effective in reducing aldosterone levels to provide sustained lowering of blood pressure in patients with primary aldosteronism,14 primary hypertension,15 and resistant hypertension.16 It was found that 7n provided selective reduction of plasma aldosterone levels without an effect on baseline morning cortisol levels.14,15 However, suppression of stimulated cortisol levels was seen at doses above 0.5 mg, which can be attributed to the modest selectivity for CYP11B2 over CYP11B1.
While the inhibition of cortisol synthesis by 7n has limited its development to indications where this effect is either desired or neutral, it provided a valuable initial proof-of-concept for the ability of a CYP11B2 inhibitor to lower blood pressure in patients. In addition, the extensive profiling of 7n in hypertensive patients afforded an opportunistic approach to safely and effectively lower cortisol levels, which has led to investigation of the compound as a potential therapy for Cushing’s syndrome,17 a disease characterized by elevated levels of cortisol.
Acknowledgments
We acknowledge the support of the NIBR Analytical Sciences group for help in the characterization of the compounds herein.
Glossary
Abbreviations
- CYP11B2 or AS
aldosterone synthase
- CYP11B1
11β-hydroxylase
- CYP19
aromatase
- MR
mineralocorticoid receptor
- RAS
renin-angiotensin system
- PAC
plasma aldosterone concentration
- PCC
plasma corticonsterone concentration
- SAR
structure–activity relationship
- TWA
time-weighted average
- ACTH
adrenocorticotropic hormone
- PK–PD
pharacokinetic–pharmacodynamic
- SD
Sprague–Dawley
- [C]
compound concentration
Supporting Information Available
Procedures for the preparation of 7n and associated analytical data for 7n and other representative compounds described herein. Protocols for the aldosterone synthase cellular and CYP19 enzymatic assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Williams G. H. Essential hypertension as an endocrine disease. Endocrinol. Metab. Clin. North. Am. 1994, 23, 429–444. [PubMed] [Google Scholar]
- Zannad F.; McMurray J. J.; Krum H.; van Veldhuisen D. J.; Swedberg K.; Shi H.; Vincent J.; Pocock S. J.; Pitt B. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [DOI] [PubMed] [Google Scholar]
- Pitt B.; Remme W.; Zannad F.; Neaton J.; Martinez F.; Roniker B.; Bittman R.; Hurley S.; Kleiman J.; Gatlin M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [DOI] [PubMed] [Google Scholar]
- Pitt B.; Zannad F.; Remme W. J.; Cody R.; Castaigne A.; Perez A.; Palensky J.; Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N. Engl. J. Med. 1999, 341, 709–717. [DOI] [PubMed] [Google Scholar]
- Struthers A. D. Aldosterone escape during ACE inhibitor therapy in chronic heart failure. Eur. Heart J. 1995, 16Suppl N103–106. [DOI] [PubMed] [Google Scholar]
- Staessen J.; Lijnen P.; Fagard R.; Verschueren L. J.; Amery A. Rise in plasma concentration of aldosterone during long-term angiotensin II suppression. J. Endocrinol. 1981, 91, 457–465. [DOI] [PubMed] [Google Scholar]
- Ramsay L. E.; Hettiarachchi J.; Fraser R.; Morton J. J. Amiloride, spironolactone, and potassium chloride in thiazide-treated hypertensive patients. Clin. Pharmacol. Ther. 1980, 27, 533–543. [DOI] [PubMed] [Google Scholar]
- a Rainey W. E. Adrenal zonation: clues from 11beta-hydroxylase and aldosterone synthase. Mol. Cell Endrocrinol. 1999, 151, 151. [DOI] [PubMed] [Google Scholar]; b Bureik M.; Lisurek M.; Bernhardt R. The human steroid hydroxylases CYP11B1 and CYP11B2. Biol. Chem. 2002, 383, 1537. [DOI] [PubMed] [Google Scholar]
- Bureik M.; Lisurek M.; Bernhardt R. The human steroid hydroxylases CYP11B1 and CYP11B2. Biol. Chem. 2002, 383, 1537. [DOI] [PubMed] [Google Scholar]
- Ménard J.; Pascoe L. Can the dextroenantiomer of the aromatase inhibitor fadrozole be useful for clinical investigation of aldosterone synthase inhibition. J. Hypertens. 2006, 24, 993. [DOI] [PubMed] [Google Scholar]
- LaSala D.; Shibanaka Y.; Jeng A. Y. Coexpression of CYP11B2 or CYP11B1 with adrenodoxin and adrenodoxin reductase for assessing the potency and selectivity of aldosterone synthase inhibitors. Anal. Biochem. 2009, 394, 56–61. [DOI] [PubMed] [Google Scholar]
- Ksander G. M.; Meredith E. M.; Monovich L. G.; Papillon J.; Firooznia F.; Hu Q.-Y.. Preparation of condensed imidazole derivatives for the inhibition of aldosterone synthase and aromatase. WO 2007024945, Mar 1, 2007.
- Rigel D. F.; Fu F.; Beil M.; Hu C. W.; Liang G.; Jeng A. Y. Pharmacodynamic and pharmacokinetic characterization of the aldosterone synthase inhibitor FAD286 in two rodent models of hyperaldosteronism: comparison with the 11beta-hydroxylase inhibitor metyrapone. J. Pharmacol. Exp. Ther. 2010, 334, 232–243. [DOI] [PubMed] [Google Scholar]
- Ménard J.; Rigel D. F.; Watson C.; Jeng A. Y.; Fu F.; Beil M.; Liu J.; Chen W.; Hu C.-W.; Leung-Chu J.; LaSala D.; Liang G.; Rebello S.; Zhang Y.; Dole W. P.. Aldosterone Synthase Inhibtion: Blood Pressure Independent Protection Against Cardiorenal Injury and Death in Animal Disease Models and Translation of Experimental Hormonal Effects to Human Subjects. In press. [DOI] [PMC free article] [PubMed]
- Amar L.; Azizi M.; Ménard J.; Peyrard S.; Watson C.; Plouin P. F. Aldosterone synthase inhibition with LCI699: a proof-of-concept study in patients with primary aldosteronism. Hypertension 2010, 831–838. [DOI] [PubMed] [Google Scholar]
- Calhoun D. B.; White W. B.; Krum H.; Guo G.; Bermann G.; Trapani A.; Leftkoowitz M. P.; Ménard J. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: Results of a randomized, double-blind, placebo. Circuation 2011, 124, 1945–1955. [DOI] [PubMed] [Google Scholar]
- Kams A. D.; Bral J. M.; Hartman D.; Peppard T.; Schumacher C. Study of aldosterone synthase inhibition as an add-on therapy in resistant hypertension. J. Clin. Hypertens. 2013, 15, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivonello R.; Fleseriu M.; Guignat L.; Zhang Y.; Robinson P.; Taylor A.; Watson C.; Maldonado M.; Hamrahian A. H.; Boscaro M.; Biller B. M. K. Patients with Cushing disease achieve normal urinary cortisol with LCI699, a potent 11β-hydroxylase inhibitor: Preliminary results from a multicenter, proof-of-concept study. Endocr. Rev. 2012, 33, OR49-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.