

Abstract
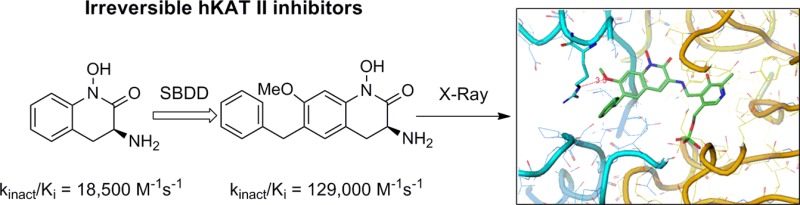
A series of aryl hydroxamates recently have been disclosed as irreversible inhibitors of kynurenine amino transferase II (KAT II), an enzyme that may play a role in schizophrenia and other psychiatric and neurological disorders. The utilization of structure–activity relationships (SAR) in conjunction with X-ray crystallography led to the discovery of hydroxamate 4, a disubstituted analogue that has a significant potency enhancement due to a novel interaction with KAT II. The use of kinact/Ki to assess potency was critical for understanding the SAR in this series and for identifying compounds with improved pharmacodynamic profiles.
Keywords: kynurenine amino transferase, kynurenic acid, aryl hydrocarbon receptor, irreversible inhibition, hydroxamic acid, dose response modeling, in vivo microdialysis, schizophrenia
The kynurenine amino transferase (KAT) class of pyridoxal phosphate (PLP)-dependent transaminases catalyzes the cyclization of l-kynurenine to kynurenic acid (KYNA) in the l-tryptophan metabolic pathway.1,2 Recently, KYNA has been shown to agonize the arylhydrocarbon receptor (AHR), a nuclear protein involved in gene transcription and other cellular regulatory functions.3 Additionally, a burgeoning body of research implicates KYNA in a range of neurological disorders due to its antagonism of the α7 nicotinic acetylcholine receptor and the N-methyl-d-aspartate (NMDA) receptor.4,5 Elevated levels of KYNA have been found in the cerebral spinal fluid (CSF) and postmortem brain tissue of schizophrenics.6−8 Furthermore, individuals with bipolar disorder have also been found to have increased levels of KYNA in the CSF.9 Decreasing central KYNA levels in afflicted individuals may therefore provide therapeutic benefit for treating these and other psychiatric and neurological disorders.10,11
Among the four homologous members of the KAT family, KAT II is largely responsible for centrally produced KYNA, thereby emerging as an attractive target for medicinal chemists.12 As such, recent articles have reported both reversible and irreversible KAT II inhibitors.13−16 One useful scaffold is hydroxamate 1, a centrally active and irreversible inhibitor of KAT II.14,17 Preliminary structure–activity relationship (SAR) indicated that substituents were better tolerated on C6 and C7, whereas potency losses were observed with substitution on C5 and C8 (Figure 1). This SAR was supported by the crystal structure, which showed that positions 6 and 7 are oriented toward a solvent-exposed region in the enzyme.14 In contrast, positions 5 and 8 are directed toward the walls of the binding pocket. As a consequence of these findings, a broad range of analogues were designed to assess the SAR of this series.14,15 This communication focuses on key analogues that led to the discovery of substantial potency-enhancing interactions with human KAT II.
Figure 1.
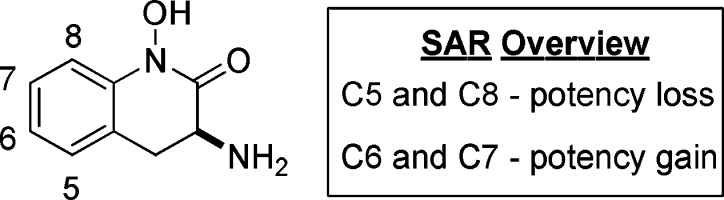
SAR trends on the aryl hydroxamate scaffold.
At the outset, a reliable method for assessing potency of irreversible inhibition was required. For example, analysis of IC50 values does not discriminate potency differences among these KAT II inhibitors (Tables 1 and 2, entries 1–4), and consistent with earlier accounts, kinact/Ki proved to be a critical measure that differentiates these compounds (Table 1).18,19 Indeed, these data led to the selection of 2 and 3 for X-ray analysis, which subsequently led to the design of 4.
Table 1. Key SAR of Hydroxamate Derivatives.
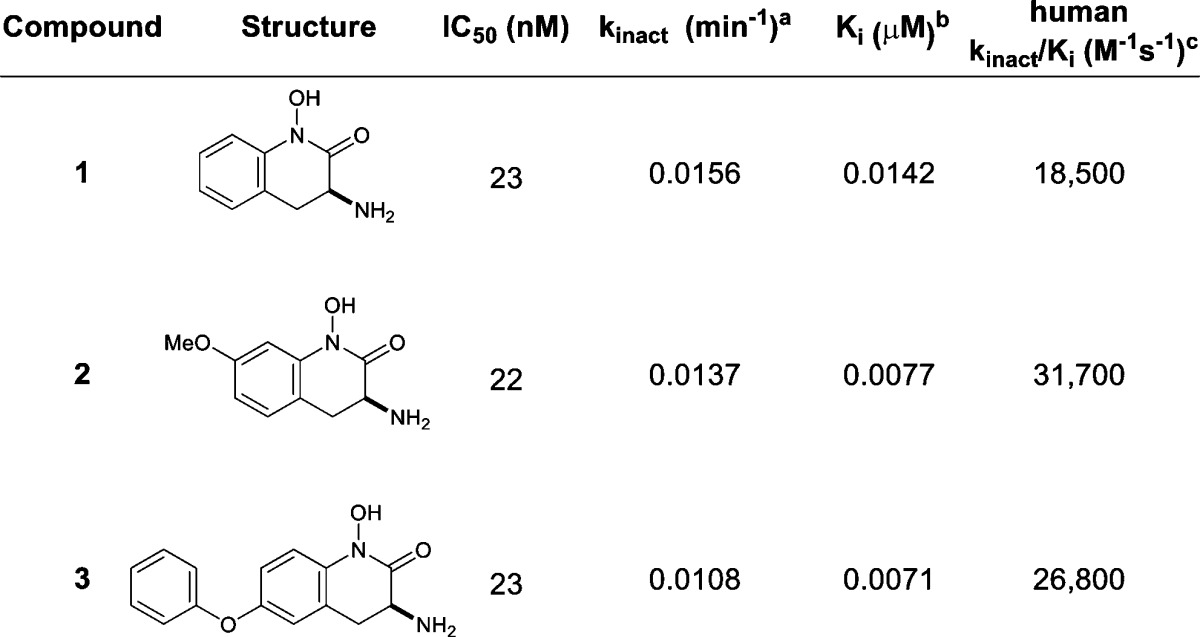
Values represent the geometric mean of at least three experiments.
Values represent the arithmetic mean of at least three experiments.
Additional statistical details are provided in the Supporting Information.
Table 2. Activity of Compound 4.

Values represent the geometric mean of at least three experiments.
Values represent the arithmetic mean of at least three experiments. Additional statistical details are provided in the Supporting Information.
Cocrystal structures of PLP bound to hydroxamates 2 and 3 in the KAT II active site show intriguing changes to the protein. In the case of PLP adduct of compound 2 bound to KAT II, the overlay with a crystal structure of 1 bound to the same components highlights discrete differences (Figure 2), in particular with a domain consisting of Ile-19 to Gly-29, located at the entrance of the binding pocket.20 In the structure containing hydroxamate 1, this domain is highly disordered. In contrast, the crystal structure containing 2 shows a highly ordered region.21 The 2-fold kinact/Ki enhancement of derivative 2 as compared to parent may be attributed to an additional van der Waals interaction with Ile-19.
Figure 2.
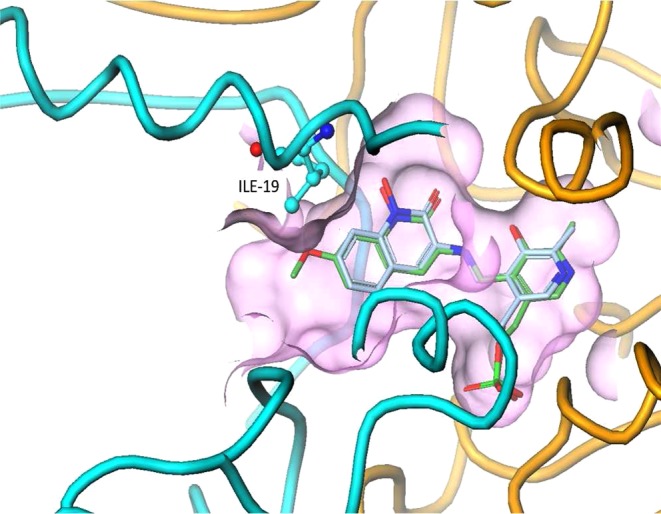
Superposition of the crystal structures of 1 and 2 with KAT II, showing the additional van der Waals interactions made by the methoxy substituent at C-7. The entire helix at the entrance of the binding pocket, consisting of residues Ile-19 to Gly-29, is disordered in the structure of PLP-bound 1 (yellow), whereas it is ordered in the structure of 2 (light blue).
A previously unknown lipophilic interaction was discovered by introducing a phenoxy side chain at C-6. In the crystal structure containing compound 3 with KAT II, the aryl group rotates into a hydrophobic pocket found at the entrance to the active site and bounded on one face by Leu-293 (Figure 3), leading to a 1.5-fold boost in potency over compound 1 (Figure 3).22
Figure 3.
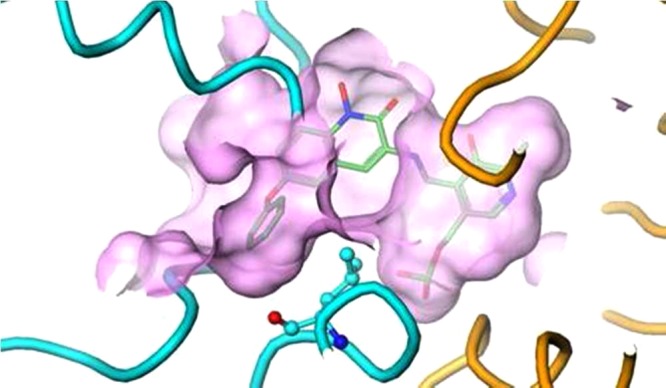
Crystal structure of 3 with KAT II. The C-6 phenoxy substituent of 3 orients into a shallow pocket at the mouth of the binding site, which is bounded on one face by Leu-293.
These unique interactions inspired the design of a disubstituted analogue that combined both key elements from compounds 2 and 3. To avoid the potential formation of a reactive metabolite characteristic of catechols, hydroxamate 4 was synthesized (Table 2). Interestingly, a 4–5-fold enhancement in potency was observed in the kinact/Ki, an improvement not evidenced by the IC50 value of 4.
In the crystal structure of 4 bound to KAT II, in analogy to the structure of compound 3, the benzyl group is oriented into a lipophilic pocket framed by Leu-293.23 Interestingly, an apparent cation−π interaction formed with Arg-20, the residue adjacent to Ile-19, and the phenyl ring (3.5 Å,24 Figure 4). This type of interaction is well-known in proteins, where it plays a role in secondary and tertiary structure stabilization.25,26 The previously disordered region now acts as a clamp that anchors the hydroxamic acid–PLP complex into the active site and, as a result, enhances potency.21
Figure 4.
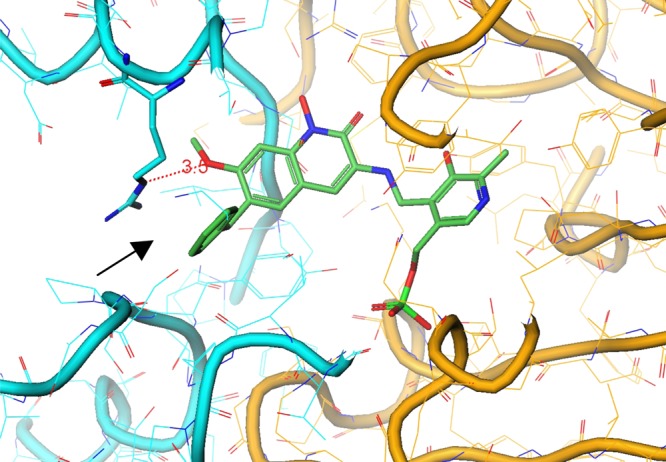
Crystal structure of 4 with KAT II. The combination of 7-OMe and 6-Bn substituents results in additional interactions with Arg-20. The arrow marks a potential cation−π interaction between the guanidium group in Arg-20 and the phenyl ring.
It is intriguing to speculate that mechanistically, the 6-OMe may form lipophilic and/or hydrogen bond interactions with Ile-19 and Arg-20 on the α-helix that, upon orienting Arg-20 into a coplanar interaction with the benzyl group, is further stabilized by a hydrogen bond with the oxygen of the methoxy group. This bidentate interaction is only possible when both interactions are present, as this clamped architecture is not observed in the KAT II structures with 2 and 3 (Figure 5, compare to Figure 3). Additionally, this suggests that enthalpic stabilization overrides the entropic loss and leads to this greatly improved potency.
Figure 5.
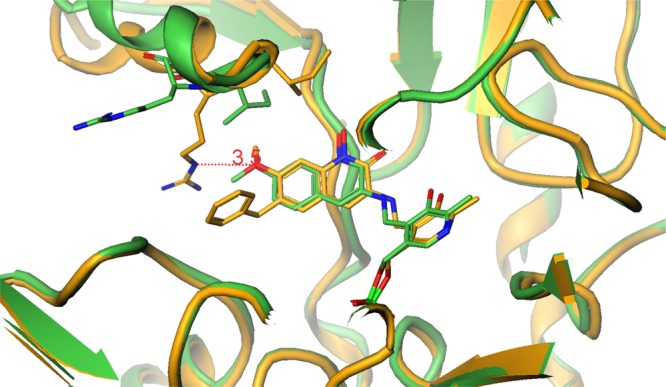
Overlay of crystal structures generated from 2 (green) and 4 (yellow). Two binding elements are necessary to interact with Arg-20.
Further analysis of the individual kinact and Ki values provided insight into the nature of the enhanced potency. In the overall scheme of enzyme (E) inhibition by an irreversible inhibitor (I), the first step is reversible binding characterized by equilibrium binding dissociation constant Ki followed by the irreversible covalent bond forming event characterized by first-order inactivation rate constant kinact (Scheme 1).20
Scheme 1. Schematic of Irreversible Inhibition.

For these analogues, kinact values fall within a close range from 0.0108 to 0.0156 min–1, indicating that the rates of covalent bond formation with PLP are similar. The difference lies in Ki, where there is a large discrepancy in values, indicating that the inhibition for these compounds is largely Ki driven (Tables 1 and 2, compare entries 1–4). In the event, analogue 4 binds to KAT II 5× more efficiently than 2 and 3.
Comparing entries 1 and 3 by in vivo microdialysis experiments illustrates the impact that increased potency has on lowering central KYNA. Subcutaneous administration of each compound at 10 mg/kg results in a 9-fold lower free brain exposure of 3 as compared to 1 (65 ng h/g vs 584 ng h/g), due in part to the high nonspecific binding of 3 to brain tissue (Table 3, 99% bound). Nevertheless, compound 3 reduces brain KYNA levels for a significantly longer period of time and to a greater degree than compound 1, at the same dose, as a result of its improved rat KAT II kinact/Ki. (Figure 6).
Table 3. PK of Compounds 1 and 3.
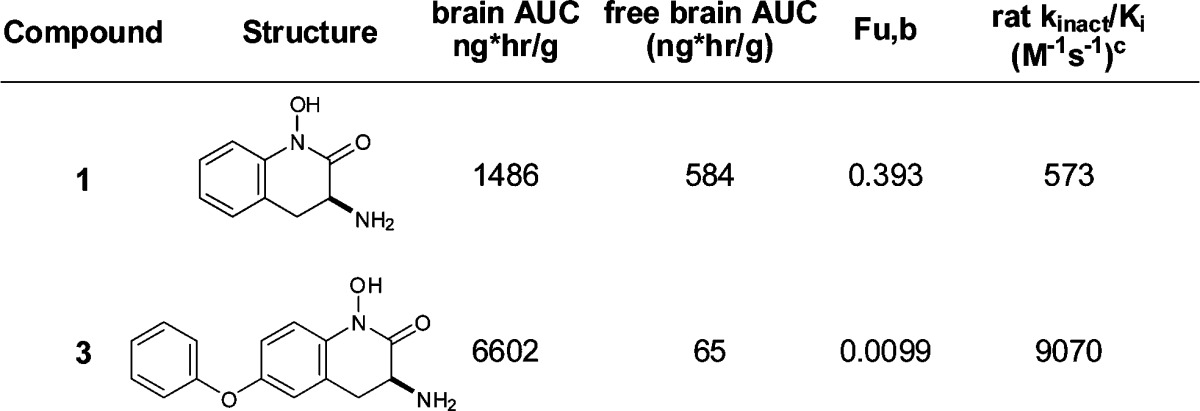
Figure 6.
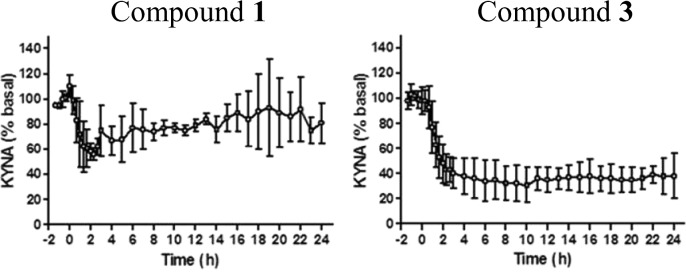
Prefrontal cortex extracellular levels of KYNA in freely moving rats measured using microdialysis. Compounds 1 and 3 were administered at 10 mg/kg sc at t = 0. Data are expressed as a percentage of preinjection baseline and are means ± SEMs (n = 4).
The lowering of KYNA is attributed to the selective inhibition of KAT II as both compounds are weakly potent at KAT I and KAT III isoforms. Inhibitors 2 and 4 exhibited a similar selectivity profile.27 While compound 4 was equipotent at rat KAT II (kinact/Ki = 763 M–1 s–1) as compared to compound 1, KYNA levels were not lowered at any dose due to high clearance. Currently, follow-up designs are focusing on lowering the clearance of compound 4, as the poor pharmacokinetics has a deleterious effect on in vivo efficacy.28
In summary, X-ray crystallography and a kinact/Ki assay led to the rational design of irreversible KAT II inhibitor 4. The crystal structure of this compound with KAT II led to the discovery of a novel interaction with a region previously found to be disordered. Ongoing efforts to optimize ligand interactions with the enzyme and improve pharmacokinetic properties will be reported in due course.
Acknowledgments
We thank Scot Mente and Ann Aulabaugh for helpful discussions. Katherine Brighty played a critical role in editing this manuscript. Zoe Hughes provided key insights into the analysis and depiction of the microdialysis data.
Supporting Information Available
Experimental data for the synthesis and characterization of compounds 3 and 4, assay protocols, and X-ray crystallography data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† 700 Main Street, Cambridge, Massachusetts 02139, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Han Q.; Cai T.; Tagle D. A.; Li J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. J. Cell. Mol. Life Sci. 2010, 67, 353–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot A. C.; Kirsch J. F. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [DOI] [PubMed] [Google Scholar]
- Moroni F.; Cozzi A.; Sili M.; Mannaioni G. Kynurenic acid: A metabolite with multiple actions and multiple targets in brain and periphery. J. Neural Transm. 2012, 119, 133–139. [DOI] [PubMed] [Google Scholar]
- Hilmas C.; Pereira E. F.; Alkondon M.; Rassoulpour A.; Schwarcz R.; Albuquerque E. X. The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons C. G.; Danysz W.; Quack G.; Hartmann S.; Lorenz B.; Wollenburg C.; Baran L.; Przegalinski E.; Kostowski W.; Krzascik P.; Chizh B.; Headley P. M. Novel systemically-active antagonists of the glycine site of the NMDA receptor—Electrophysiological, biochemical and behavioural characterization. J. Pharmacol. Exp. Ther. 1998, 283, 1264–1275. [PubMed] [Google Scholar]
- Erhardt S.; Blennow K.; Nordin C.; Skogh E.; Lindstrom L. H.; Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci. Lett. 2001, 313, 96–98. [DOI] [PubMed] [Google Scholar]
- Nilsson L. K.; Linderholm K. R.; Engberg G.; Paulson L.; Blennow K.; Lindstrom L. H.; Nordin C.; Karanti A.; Persson P.; Erhardt S. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr. Res. 2005, 80, 315–322. [DOI] [PubMed] [Google Scholar]
- Schwarcz R.; Rassoulpour A.; Wu H.-Q.; Medoff D.; Tamminga C. A.; Roberts R. C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521. [DOI] [PubMed] [Google Scholar]
- Olsson S. K.; Samuelsson M.; Saetre P.; Lindstrom L.; Jonsson E. G.; Nordin C.; Engberg G.; Erhardt S.; Landen M. Elevated levels of kynurenic acid in the cerebrospinal fluid of patients with bipolar disorder. J. Psychiatry Neurosci. 2010, 35, 195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S.; Olsson S. K.; Engberg G. Pharmacological manipulaton of kynurenic acid: Potential in the treatment of psychiatric disorders. CNS Drugs 2009, 23, 91–101. [DOI] [PubMed] [Google Scholar]
- Potter M. C.; Elmer G. I.; Bergeron R.; Albuquerque E.; Guidetti P.; Wu Hx-Q.; Schwarcz R. Reduction of endogenous kynuirenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt W.; Guidetti P.; Okuno E.; Schwarcz R. Characterization of human brain kynurenine aminotransferases using [3H] kynurenine as a substrate. Neuroscience 1993, 55, 177–184. [DOI] [PubMed] [Google Scholar]
- Dounay A. B.; Anderson M.; Bechle B. M.; Campbell B. M.; Claffey M. M.; Evdokimov A.; Evrard E.; Fonseca K. R.; Gan X.; Ghosh S.; Hayward M. M.; Horner W.; Kim Ji.Y.; McAllister L. A.; Pandit J.; Paradis V.; Parikh V. D.; Reese M. R.; Rong S.; Salafia M. A.; Schuyten K.; Strick C. A.; Tuttle J. B.; Valentine J.; Wang H.; Zawadzke L. E.; Verhoest P. R.. Discovery of brain-penetrant, irreversible kynurenine aminotransferase II inhibitors for schizophrenia. ACS Med. Chem. Lett. 2012, 3, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claffey M. M.; Dounay A. B.; Gan X.; Hayward M. M.; Rong S.; Tuttle J. B.; Verhoest P. R.. Bicyclic and Tricyclic Compounds as KAT II inhibitors. WO2010146488.
- Pellicciari R.; Rizzo R. C.; Costantino G.; Marinozzi M.; Amori L.; Guidetti P.; Wu H.-Q.; Schwarcz R. Modulators of the kynurenine pathway of tryptophan metabolism: synthesis and preliminary biological evaluation of (S)-4-(ethylsulfonyl)benzoylalanine, a potent and selective kynurenine aminotransferase II (KAT II) inhibitor. ChemMedChem 2006, 1, 528–531. [DOI] [PubMed] [Google Scholar]
- Schwarcz R.; Kajii Y.; Ono S.. Preparation of aminopiperazinylquinolonecarboxylates as kynurenine-amino-transferase inhibitors. WO2009064836.
- In the event, when measuring KYNA in dialysates from rat prefrontal cortex, a 10 mg/kg dose, sc, showed a 50% decrease from basal levels that returned to baseline approximately 20 h postdose.
- Mileni M.; Johnson D. S.; Wang Z.; Everdeen D. S.; Liimatta M.; Pabst B.; Bhattacharya K.; Nugent R. A.; Kamtekar S.; Cravatt B. F.; Ahn K.; Stevens R. C. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12820–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; Wiley: Hoboken, NJ, 2005; pp 1–265. [PubMed] [Google Scholar]
- The coordinates have been deposited in the Protein Data Bank, with ID 4GE4.
- Average B values (in A2) for Arg-20 in each of the structures are as follows: 1 = no value, too disordered; 2 = 84.90; 3 = 103.35; and 4 = 27.30
- The coordinates have been deposited in the Protein Data Bank, with ID 4GE7.
- The coordinates have been deposited in the Protein Data Bank, with ID 4GE9.
- See the Supporting Information for the distance measurement method and corresponding figure.
- Gallivan J. P.; Dougherty D. A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley P. B.; Golovin A. Cation-π interactions in protein-protein interfaces. Proteins: Struct., Funct., Bioinf. 2005, 59, 231–239. [DOI] [PubMed] [Google Scholar]
- Compound 1: hKAT I IC50 = 30.4 μM and hKAT III IC50 = 9.04 μM. Compound 2: hKAT I IC50 = 15.3 μM and hKAT II IC50 = 6.4 μM; for comparison purposes, rat KAT II kinact/Ki = 1840 M–1 s–1. Compund 3: hKAT I IC50 = 2.04 μM and hKAT III IC50 = 1.23 μM. Compound 4: hKAT I IC50 > 36 μM and hKAT III IC50 = 13.6 μM.
- Dosing compound 4 in male rats at 32 mg/kg sc had no lowering effect on PFC kynurenic acid levels as measured by microdialysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.