

Abstract

A novel series of alkoxyimino derivatives as S1P1 agonists were discovered through de novo design using FTY720 as the chemical starting point. Extensive structure–activity relationship studies led to the discovery of (E)-1-(4-(1-(((4-cyclohexyl-3-(trifluoromethyl)benzyl)oxy)imino)ethyl)-2-ethylbenzyl)azetidine-3-carboxylic acid (32, BAF312, Siponimod), which has recently completed phase 2 clinical trials in patients with relapsing–remitting multiple sclerosis.
Keywords: S1P receptor, S1P1 agonist, lymphocytes
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid that regulates a diverse range of physiological processes such as lymphocyte trafficking, cardiac function, vascular development, and inflammation.1−1d S1P mediates these functions primarily through its interaction with five G-protein-coupled receptors known as S1P1–5.2,2b The S1P-mediated signaling has become the focus of intense research during the past decade, partly inspired by the therapeutic development of FTY720 (fingolimod) (Figure 1), which was recently approved as the first oral drug for relapsing–remitting multiple sclerosis.3 FTY720 effectively inhibits the lymphocyte egress from the thymus and lymph nodes and thereby leads to redistribution of lymphocytes away from peripheral inflammatory tissues.3 FTY720 is phosphorylated in vivo by sphingosine kinase to the active metabolite FTY720-phosphate (Figure 1).4,4b The latter binds with nanomolar affinity as an agonist at four of the five S1P receptors, namely, S1P1, S1P3, S1P4, and S1P5.4,4b Interestingly, FTY720-phosphate was shown to elicit its immunosuppressive effect through functionally antagonizing S1P1 signaling pathway.5,5b It induces rapid and persistent internalization of the S1P1 receptor that is required for lymphocyte egress from the secondary lymphoid organs.
Figure 1.

Structures of FTY720, FTY720-phosphate, and their analogues.
A transient, dose-dependent decrease of heart rate was observed with FTY720 in clinical studies.6 The S1P3 receptor is thought to be responsible for the bradycardia, based on the lack of S1P-induced heart rate reduction in S1P3 knockout mice.7,7b S1P3 agonism is also known to be associated with vaso- and bronchoconstriction and pulmonary epithelial leakage.8 Numerous research groups have been working toward discovering S1P3-sparing S1P1 agonists.9−10 However, our recent report suggested species-dependent bradycardia and a dominant role of S1P1 in mediating heart rate in humans, as the treatment with BAF312, a dual S1P1,5 agonist sparing S1P3 activity, caused GIRK channel activation in human atrial myocytes and bradycardia in healthy volunteers.11 In the same report, we presented the in vitro profile of BAF312, including its effects on S1P1 receptor internalization as well as its preclinical efficacy in a rat experimental autoimmune encephalomyelitis (EAE) model. Herein, we report our medicinal chemistry efforts that culminated in the discovery of (E)-1-(4-(1-(((4-cyclohexyl-3-(trifluoromethyl)benzyl)oxy)imino)ethyl)-2-ethylbenzyl)azetidine-3-carboxylic acid 32 (BAF312, Siponimod), a potent S1P3-sparing S1P1 agonist currently undergoing clinical trials in patients with multiple sclerosis.
One focus of our initial efforts was to develop FTY720 analogues that could (1) achieve selectivity against S1P3 and (2) reduce in vivo elimination half-life. We envisioned that modifications providing increased rigidity in the lipophilic alkyl chain of FTY720 may lead to S1P receptor subtype selectivity. One of the approaches led to the discovery of analogues containing substituted benzyloxy oximes that replace the n-octyl moiety, as exemplified in compound 1 (Figure 1). When dosed orally to mice, compound 1 was equally efficacious as FTY720 in inducing lymphocyte redistribution. However, the in vivo elimination half-life of the corresponding phosphate remained very long (t1/2 > 30 h), and the volume of distribution was large (Vss = 36.9 L/kg). One possible explanation for this pharmacokinetic behavior is that the charged phosphate moiety, which is generated in vivo, contributes to high nonspecific binding to lipoproteins in tissues. We decided to explore replacing the phosphate moiety with a carboxylic acid, which might reduce nonspecific binding and thereby reduce the volume of distribution and elimination half-life. An additional advantage of pursuing a nonpro-drug strategy is that we eliminated the complication of understanding two overlapping structure–activity relationships (SAR), namely, the SAR of the sphingosine kinase, whose action is required to generate the bioactive aminophosphate, and the SAR of the resulting aminophosphate for the S1P-receptor family. To efficiently identify an optimal amino carboxylate group, we utilized a reductive amination strategy whereby an aldehyde fragment corresponding to the “hydrophobic tail” of compound 1 could be efficiently derivatized with a collection of amino carboxylic acids (Figure 2). From this effort, compound 2 was identified as one of the most potent compounds with an EC50 value of 300 nM at the S1P1 receptor subtype in a GTPγS binding assay.12 This compound was then used as a new lead for further optimization.
Figure 2.

Amino carboxylate to mimic amino phosphate.
We first turned our attention to the substitutions on the phenyl rings A and B (see Figure 2). The key SAR results for select analogues within this series are summarized in Table 1. On ring A, substitution at the 2-position is well tolerated. Both 2-fluoro 4 and 2-trifluoromethyl 6 show increased agonism in comparison to compound 2. Especially the latter reaches single digit nanomolar potency. In contrast, the 1-position is less preferred for substitution. 1-Fluoro 3 is 2-fold less potent than its corresponding 2-fluoro analogue 4, while 1-trifluoromethyl 5 shows very low activity at 10 μM. Substitutions on ring B (compounds 7–12) are in general less tolerated than those on ring A. Additional substitutions of ring B (compounds 13–16) fail to further increase the potency of compound 6.
Table 1. S1P1 Potencies with Different Biphenyl Substituents.
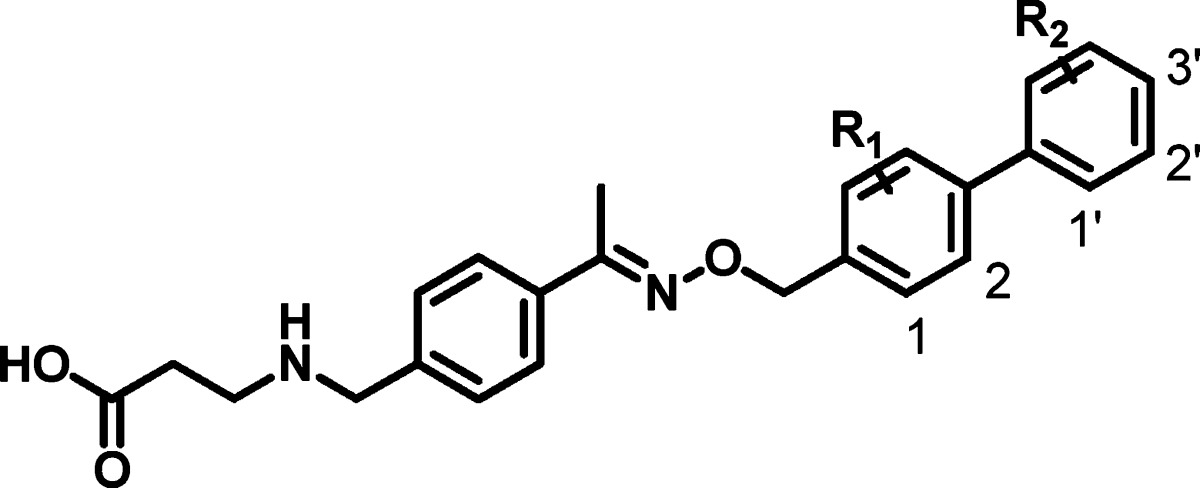
| compd | R1 | R2 | S1P1 EC50 (μM)a |
|---|---|---|---|
| 2 | H | H | 0.3 |
| 3 | 1-F | H | 0.12 |
| 4 | 2-F | H | 0.061 |
| 5 | 1-CF3 | H | 10 |
| 6 | 2-CF3 | H | 0.009 |
| 7 | H | 3′-F | 0.21 |
| 8 | H | 3′-Me | 0.64 |
| 9 | H | 3′-OMe | 1.5 |
| 10 | H | 1′-OMe | 1.4 |
| 11 | H | 3′-CF3 | 10 |
| 12 | H | 1′-CF3 | 1.7 |
| 13 | 2-CF3 | 1′-F | 0.008 |
| 14 | 2-CF3 | 3′-F | 0.011 |
| 15 | 2-CF3 | 3′-Me | 0.094 |
| 16 | 2-CF3 | 2′-F-3′-F | 0.016 |
GTPγS binding assay (see ref (12) for detailed assay descriptions).
Although these chemistry efforts were not informed by molecular modeling, they can now be rationalized using molecular modeling based upon the recently solved S1P1 crystal structure (PDB code: 3V2Y).13 The predicted binding mode of compound 2 is illustrated in Figure 3. The ligand binding pocket of S1P1 consists of a charged entrance and a large enclosed hydrophobic cavity. The carboxylic acid headgroup forms salt bridges with Lys34 and Arg120 and a hydrogen bond with Tyr29. In addition, the charged amine group forms a salt bridge with Glu121. Therefore, the polar headgroup involves multiple strong electrostatic interactions that anchor the ligand molecule in the binding site. The hydrophobic tail (i.e., biphenyl moiety) is predicted to fit nicely into the large hydrophobic cavity. On the basis of the binding mode, 2-F substitution (compound 4) adds additional hydrophobic interaction relative to compound 2 and leads to an increase in potency. In contrast, the fluorine at the 1-position in compound 3 does not interact with the protein residue as tightly as at the 2-position, and this is consistent with the observed SAR. A bulkier trifluoromethyl group at the 1-position in compound 5 introduces a potential steric clash with the receptor that leads to much reduced activity. The same group substituted at the 2-position in compound 6, however, engages improved nonpolar interactions with Leu276 and Leu272. In addition, it forces the terminal phenyl ring into near perpendicular position, which results into better van der Waals interactions at this orientation. As the terminal phenyl group is at the bottom of the hydrophobic pocket, substitutions at this ring are much less tolerated (compounds 7–12).
Figure 3.
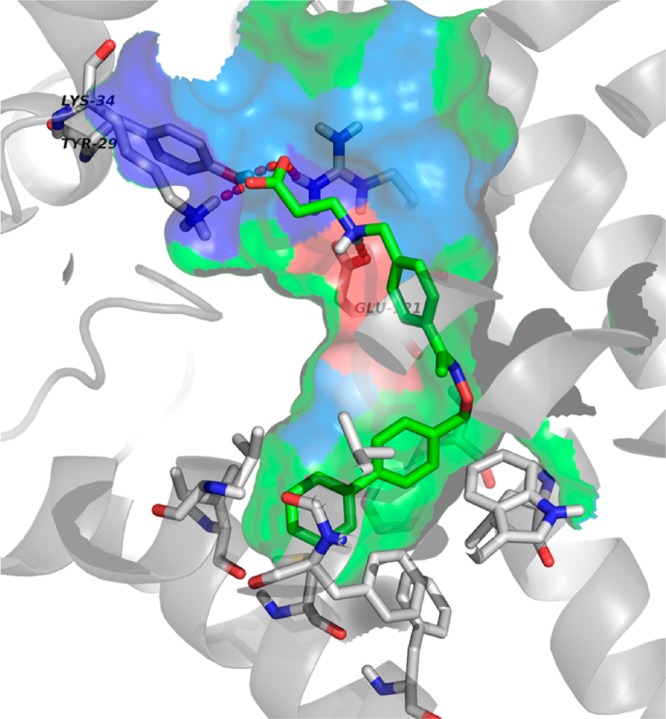
Binding mode of compound 2 in S1P1. The electrostatic surface is color coded: blue is positively charged, red is negatively charged, and green is hydrophobic.
To further optimize the interaction with the hydrophobic pocket that is tightly occupied by phenyl ring B, we explored several phenyl replacements as summarized in Table 2. Compounds containing five-membered heterocycles such as 2-furanyl 17 or 2-thienyl 18 showed comparable activity to the corresponding phenyl analogue 6. To our delight, the replacement of the phenyl moiety by either a cyclopentyl 19 or a cyclohexyl 20 group resulted into a greater than 10-fold increase in potency, while it had only marginal impact on the S1P3 activity.
Table 2. Structure–Activity Relationship of the Terminal Hydrophobic Groups.
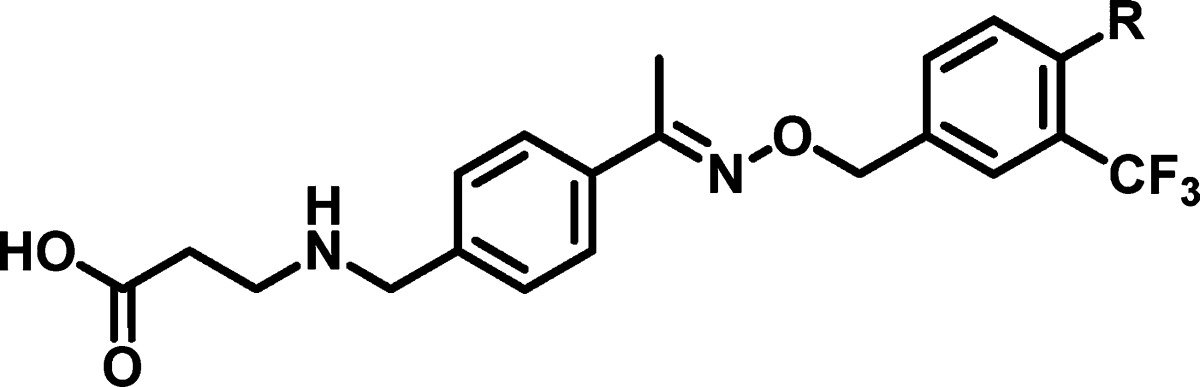
| EC50 (μM)a |
|||
|---|---|---|---|
| compd | R | S1P1 | S1P3 |
| 17 | 2-furanyl | 0.015 | 3.0 |
| 18 | 2-thienyl | 0.018 | 5.9 |
| 19 | cyclopentyl | 0.0006 | 1.6 |
| 20 | cyclohexyl | 0.0003 | 1.5 |
GTPγS binding assay (see ref (12) for detailed assay descriptions).
After successfully optimizing the hydrophobic tail to reach subnanomolar potency, we then turned our attention to the phenyl ring C and the hydrophilic head groups. The SAR of select compounds is summarized in Table 3. Replacing phenyl ring C by heterocyles, such as pyridines 21 and 22 or a thiophene 23, only led to marginal decreases in S1P1 activity while maintaining good selectivity against S1P3. However, the furanyl analogue 24 showed significant loss of activity (>20-fold) presumably due to the smaller ring size that leads to an increased dihedral angle between the head and the tail groups. Because there was no clear improvement in potency or selectivity achieved by these modifications, we next examined the effects of substitutions on the phenyl ring. Substitutions at the R2 position with halogens (compounds 25–27) or small alkyl groups (compounds 28–30) are very well tolerated with comparable activity to the phenyl analogue 20. Interestingly, a methyl group (28) or an ethyl group (29) leads to weaker activity on S1P3. As the azetidine carboxylic acid group showed comparable activity to the β-alanine analogue in our survey summarized in Figure 2, we re-evaluated this headgroup with the optimized lipophilic moieties with compounds 31 and 32. Both compounds showed comparable activity and selectivity.
Table 3. Structure–Activity Relationship of the Head Groups.
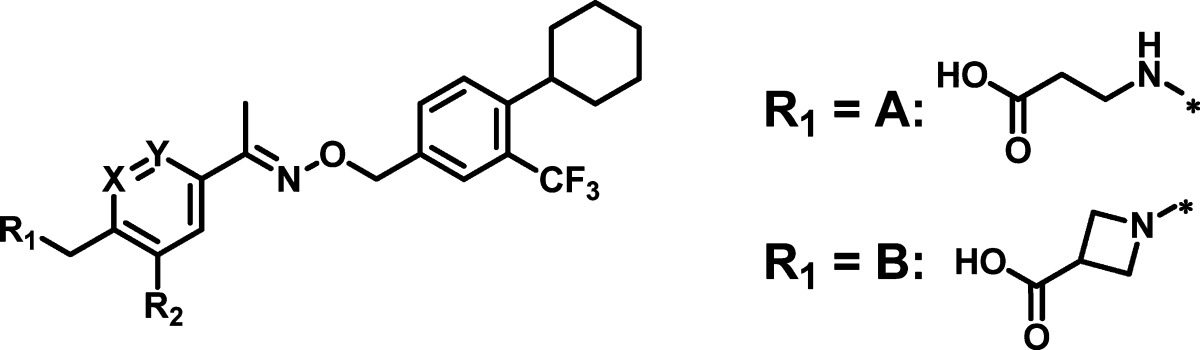
| EC50 (μM)a |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | X=Y | S1P1 | S1P3 |
| 21 | A | H | N=CH | 0.0026 (86) | 4.3 (44) |
| 22 | A | H | CH=N | 0.0014 (92) | 2.6 (53) |
| 23 | A | H | S | 0.0022 (104) | 4.0 (79) |
| 24 | A | H | O | 0.017 (100) | 2.5 (62) |
| 25 | A | F | CH=CH | 0.0003 (105) | 2.2 (61) |
| 26 | A | Cl | CH=CH | 0.0006 (106) | 1.9 (50) |
| 27 | A | Br | CH=CH | 0.0004 (85) | 1.1 (65) |
| 28 | A | Me | CH=CH | 0.002 (94) | 5.9 (159) |
| 29 | A | Et | CH=CH | 0.0007 (92) | 7.2 (43) |
| 30 | A | cyclo-Pr | CH=CH | 0.0004 (98) | 1.3 (63) |
| 31 | B | Me | CH=CH | 0.0016 (80) | 3.2 (127) |
| 32 | B | Et | CH=CH | 0.0004 (91) | 5.0 (55) |
GTPγS binding assay (see ref (12) for detailed assay descriptions); values in parentheses represent the Emax, % of agonism at 10 μM as compared to S1P.
On the basis of favorable potency, selectivity, and other druglike properties, compound 32 was chosen for further in vitro and in vivo testing. Compound 32 is extensively bound to rat, dog, monkey, and human plasma proteins (>99.9%). It shows high and passive permeability in a Caco-2 human cell permeability assay with no transporter-mediated efflux observed at the test concentrations of 0.5–50 μM. In vitro metabolism studies with liver microsomes indicate that the metabolic clearance of compound 32 is low in mouse and dog microsomes, low to moderate in monkey and human, but high in rat. In the rat, the fraction of the dose absorbed is 58%, which is calculated from the total amount of metabolites detected in feces and urine. The absolute bioavailability is 50 and 71% in the rat and monkey, respectively, indicating no major presystemic first pass metabolism. Consistent with the low metabolic clearance in vitro, the systemic clearance (CL) is low in both rat and monkey relative to respective hepatic blood flow (Table 4). The terminal half-life is approximately 6 and 19 h in rat and monkey, respectively, and therefore is considerably shorter than those of FTY720 and FTY720-phosphate. Compound 32 is moderately distributed to tissues as compared to total body water following intravenous administration to rat (Vss = 2.15 ± 0.31 L/kg) and monkey (Vss = 2.12 ± 0.12 L/kg). On the basis of the ratio of metabolites versus parent compound detected in feces and urine, oxidative metabolism is determined as the predominant mechanism of elimination for compound 32 in rat after intravenous and oral administration. Direct renal excretion of compound 32 is not observed in the rat.
Table 4. In Vivo Pharmacokinetics in Preclinical Species.
| mean ± SD |
||||
|---|---|---|---|---|
| CL (L/h/kg) | Vss (L/kg) | T1/2 (h) | F (%) | |
| rat | 0.36 ± 0.023 | 2.15 ± 0.311 | 6 ± 1.3 | 50 |
| monkey | 0.098 ± 0.9 | 2.12 ± 0.12 | 19 ± 0.5 | 71 |
For S1P1 agonists, the reduction of peripheral lymphocyte count has been used as a pharmacodynamic biomarker for efficacy in autoimmune diseases. On the basis of the favorable PK profile, compound 32 was evaluated in an in vivo PK/PD model in Lewis rats, where lymphocyte count is measured in blood at different time points. At a dose of 1 mg/kg per oral, the rapid increase in the systemic compound concentration coincides with a marked decrease in peripheral lymphocyte counts as illustrated in Figure 4. At the Tmax of 8 h postadministration, the lymphocyte counts were decreased by 88%. In contrast to FTY720, after 48 h, the peripheral lymphocyte counts returned to normal levels in animals treated with compound 32 accompanying compound elimination from the systemic circulation, suggesting it has a shorter duration of action than FTY720. As determined in a separate experiment, the effective dose for achieving 50% reduction of peripheral lymphocyte counts at the 6 h time point postdose was 0.14 mg/kg.
Figure 4.

PK/PD relationship of compound 32 after oral administration of 1 mg/kg to Lewis rats (n = 3).
In cardiac repolarization assays (hERG patch clamp), compound 32 showed minimal inhibition of less than 10% at 25 μM. It did not exhibit genotoxic potential in the standard battery of in vitro and in vivo tests. Its selectivity was evaluated by screening against a large panel of receptors, ion channels, transporters, and kinases. No appreciable activities at concentrations below 1 μM were identified, suggesting a low potential for off-target effects. This was also confirmed by the absence of unexpected adverse findings in the toxicology studies performed.
In summary, a novel chemical series was developed using FTY720 as an initial lead structure. Compounds were optimized for potency at S1P1, selectivity against S1P3, safety, and pharmacokinetics. This led to the discovery of the clinical candidate 32 (BAF312, Siponimod). Treatment with compound 32 in Lewis rats led to a dose-dependent reduction of peripheral lymphocyte counts that rapidly returned to normal levels upon compound clearance from systemic circulation. This compound is now in late-stage clinical trials in patients with multiple sclerosis.
Supporting Information Available
General scheme and analytical data of compounds 2–31; synthetic scheme, procedure, and analytical data for compound 32; and experimental procedure for PK/PD studies of 32. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Spiegel S.; Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster J. G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 2005, 23, 127–159. [DOI] [PubMed] [Google Scholar]
- Mutoh T.; Rivera R.; Chun J. Insights into the pharmacological relevance of lysophospholipid receptors. Br. J. Pharmacol. 2012, 165, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H. Sphingosine-1-phosphate and immune regulation trafficking and beyond. Trends Pharmacol. Sci. 2011, 32, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [DOI] [PubMed] [Google Scholar]
- Rosen H.; Gonzalez-Cabrera P. J.; Sanna M. G.; Brown S. Sphingosine-1-phosphate receptor signaling. Annu. Rev. Biochem. 2009, 78, 743–768. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Billich A.; Baumruker T.; Heining P.; Schmouder R.; Francis G.; Aradhye S.; Burtin P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discovery 2010, 9, 883–897. [DOI] [PubMed] [Google Scholar]
- Mandala S.; Hajdu R.; Bergstrom J.; Quackenbush E.; Xie J.; Milligan J.; Thornton R.; Shei G.; Card D.; Keohane C.; Rosenbach M.; Hale J.; Lynch C. L.; Rupprecht K.; Parsons W.; Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [DOI] [PubMed] [Google Scholar]
- Brinkmann V.; Davis M. D.; Heise C. E.; Albert R.; Cottens S.; Hof R.; Bruns C.; Prieschl E.; Baumruker T.; Hiestand P.; Foster C. A.; Zollinger M.; Lynch K. R. The immune modulator FTY720 targets sphingosine-1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [DOI] [PubMed] [Google Scholar]
- Matloubian M.; Lo C. G.; Cinamon G.; Lesneski M. J.; Xu Y.; Brinkmann V.; Allende M. L.; Proia R. L.; Cyster J. G. Lymphocyte dgress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [DOI] [PubMed] [Google Scholar]
- Cyster J. G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 2005, 23, 127–159. [DOI] [PubMed] [Google Scholar]
- Schmouder R.; Serra D.; Wang Y.; Kovarik J. M.; DiMarco J.; Hunt T. L.; Bastien M.-C. FTY720: Placebo-controlled study of the effect on cardiac rate and rhythm in healthy subjects. J. Clin. Pharmacol. 2006, 46, 895–904. [DOI] [PubMed] [Google Scholar]
- Forrest M.; Sun S.-Y.; Hajdu R.; Bergstrom J.; Card D.; Doherty G.; Hale J.; Keohane C.; Meyers C.; Milligan J.; Mills S.; Nomura N.; Rosen H.; Rosenbach M.; Shei G.-J.; Singer I. I.; Tian M.; West S.; White V.; Xie J.; Proia R. L.; Mandala S. Immune cell regulation and cardiovascular effects of sphingosine-1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J. Pharmacol. Exp. Ther. 2004, 309, 758–768. [DOI] [PubMed] [Google Scholar]
- Sanna M. G.; Liao J.; Jo E.; Alfonso C.; Ahn M.-Y.; Peterson M. S.; Webb B.; Lefebvre S.; Chun J.; Gray N.; Rosen H. Sphingosine-1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004, 279, 13839–13848. [DOI] [PubMed] [Google Scholar]
- Murakami A.; Takasugi H.; Ohnuma S.; Koide Y.; Sakurai A.; Takeda S.; Hasegawa T.; Sasamori J.; Konno T.; Kayashi K.; Watanabe Y.; Mori K.; Sato Y.; Takahashi A.; Mochizuki N.; Takakura N. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713. [DOI] [PubMed] [Google Scholar]
- For recent examples:; Harrington P. E.; Croghan M. D.; Fotsch C.; Frohn M.; Lanman B. A.; Penninton L. D.; Pickrell A. J.; Reed A. B.; Sham K. K. C.; Tasker A.; Arnett H. A.; Fiorino M.; Lee M. R.; McElvain M.; Morrison H. G.; Xu H.; Xu Y.; Zhang X.; Wong M.; Cee V. J. Optimization of a potent, orally active S1P1 agonist containing a quinolinone core. ACS Med. Chem. Lett. 2012, 3, 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F.; Deng G.; Wang H.; Luan L.; Meng Q.; Xu Q.; Xu H.; Xu X.; Zhang H.; Zhao B.; Li C.; Guo T. B.; Yang J.; Zhang W.; Zhao Y.; Jia Q.; Lu H.; Xiang J.-N.; Elliott J. D.; Lin X. Discovery of novel 1,2,4-thiadiazole derivatives as potent, orally active agonists of sphingosine 1-phosphate receptor subtype 1 (S1P1). J. Med. Chem. 2012, 55, 4286–4296. [DOI] [PubMed] [Google Scholar]
- Buzard D. J.; Han S.; Lopez L.; Kawasaki A.; Moody J.; Thoresen L.; Ullman B.; Lehmann J.; Calderon I.; Zhu X.; Gharbaoui T.; Sengupta D.; Krishnan A.; Gao Y.; Edwards J.; Barden J.; Morgan M.; Usmani K.; Chen C.; Sadeque A.; Thatte J.; Solomon M.; Fu L.; Whelan K.; Liu L.; Al-Shamma H.; Gatlin J.; Le M.; Xing C.; Espinola S.; Jones B. M. Fused tricyclic indoles as S1P1 agonists with robust efficacy in animal models of autoimmune disease. Bioorg. Med. Chem. Lett. 2012, 22, 4404–4409. [DOI] [PubMed] [Google Scholar]
- For a recent review:Bolli M. H.; Lescop C.; Nayler O. Synthetic sphingosine 1-phosphate receptor modulators—Opportunities and potential pitfalls. Curr. Top. Med. Chem. 2011, 11, 726–757. [DOI] [PubMed] [Google Scholar]
- Gergely P.; Nuesslein-Hildesheim B.; Guerini D.; Brinkmann V.; Traebert M.; Bruns C.; Pan S.; Gray N. S.; Hinterding K.; Cooke N. G.; Groenewegen A.; Vitality A.; Sing T.; Luttringer O.; Yang J.; Gardin A.; Wang N.; Crumb W. J. Jr; Saltzman M.; Rosenderg M.; Wallström E. The selective S1P receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate: translation from preclinical to clinical studies. Br. J. Pharmacol. 2012, 167, 1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Mi Y.; Pally C.; Beerli C.; Chen A.; Guerini D.; Hinterding K.; Nuesslein-Hildesheim B.; Tuntland T.; Lefebvre S.; Liu Y.; Gao W.; Chu A.; Brinkmann V.; Bruns C.; Streiff M.; Cannet C.; Cooke N.; Gray N. Chem. Biol. 2006, 13, 1227–1234. [DOI] [PubMed] [Google Scholar]
- Hanson M. A.; Roth C. B.; Jo E.; Griffith M. T.; Scott F. L.; Reinhart G.; Desale H.; Clemons B.; Cahalan S. M.; Schuerer S. C.; Sanna M. G.; Han G. W.; Kuhn P.; Rosen H.; Stevens R. C. Crystal structure of a lipid G-protein-coupled receptor. Science 2012, 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.